

Abstract
Non-symmetric 1,3-substituted imidazopyridin-2-ones are a common structural scaffold found among many biologically active molecules. Herein we report an efficient, mild, and transition-metal free C–H amidation strategy to access such a pyrido-fused cyclic urea framework in good yields and with a broad functional group tolerance.
The imidazo[4,5-b]pyridin-2-one ring system, which is structurally related to purine, constitutes the heterocyclic core of a wide range of pharmaceutically important molecules. For example, compounds within the imidazo[4,5-b]pyridin-2-one structural class have demonstrated antidepressant,1 antimigraine,2-4 cardiotonic,5 hypotensive and antiarrhythmic,6 antiulcer and antisecretory activity.7 In addition, they have been shown to act as nonsteroidal anti-inflammatory and analgesic agents,8, 9 as well as to exhibit antiviral,10 and antibacterial11, 12 properties. Telcagepant, a CGRP (calcitonin gene-related peptide) receptor antagonist, was an investigational drug developed by Merck & Co. for the acute treatment and prevention of migraine headaches (Scheme 1a). Notably, the 1-(piperidin-4-yl)-1H-imidazo[4,5-b]pyridin-2(3H)-one core of Telcagepant is such a privileged structure that it constitutes the backbone of more than 1000 unique small molecule antagonist of CGRP receptors.13 Other prominent examples of pharmacologically relevant derivatives of imidazo[4,5-b]pyridin-2-ones examples include WO2011021678A1 (p38 MAPK inhibitor)14 and GW808990 (a CRF1 receptor antagonist).15
Scheme 1.

Selected examples of biologically active derivatives and selected syntheses of imidazo[4,5-b]pyridin-2-ones.
Due to their high pharmaceutical importance, general synthetic strategies that allow easy access to imidazo[4,5-b]pyridin-2-ones and facile SAR studies within this important class of heterocyclic compounds are highly desired. While 1,3-unsubstituted imidazo[4,5-b]pyridin-2-ones are readily prepared from 2,3-diaminopyridines with acylating reagents16 or carbon monoxide,17 the synthesis of non-symmetrical 1,3-disubstituted imidazo[4,5-b]pyridin-2-ones (R2 ≠ R3) from the same precursor is challenging and requires laborious protecting group strategies.18 In addition, differently substituted 2,3-diaminopyridines have limited commercial availability, which further hinders facile preparation and SAR investigations of imidazo[4,5-b]pyridin-2-ones bearing two differently substituted N atoms. To address this challenge, several routes to non-symmetric 1,3-substituted imidazopyridin-2-ones utilizing transition-metal catalysed coupling reactions have recently been reported (Scheme 1b).13, 19-23 Keuthe et al. synthesized such scaffolds from 3-iodo-2-chloropyridines utilizing a tandem chemo- and regioselective palladium-catalysed amination sequence.19 Scott and co-workers prepared the desired pyrido-fused cyclic urea framework from tert-butyl (2-chloropyridin-3-yl)carbamates utilizing a one-pot Buchwald-Hartwig amination/intramolecular amidation strategy.21 Furthermore, Youn has recently reported an elegant route for the synthesis of N, N′-disubstituted imidazopyridinones containing two differently substituted N atoms from 3-aminopyridines and isocyanates via an intramolecular C–H amidation reaction.23
We envisioned that an alternative approach to imidazo[4,5-b]pyridin-2-ones involving a C–H amidation step could commence with N-protected N-pyridyl-N-hydroxylamines and exploit the inherent weakness of the N-O bond (bond dissociation energy: ~50 kcal/mol).24, 25 We anticipated that O-cyanation of N-pyridyl-N-hydroxylamides followed by a hetero oxy-Cope rearrangement would afford the desired imidazo[4,5-b]pyridin-2-ones.26, 27 The so-formed pyrido-fused cyclic urea derivatives would have one of the nitrogen atoms protected with an acetyl or a methoxycarbonyl group, which would enable further selective elaborations on the other nitrogen atom, thus providing access to more complex molecular frameworks containing differently-substituted nitrogen atoms. Our approach would (i) obviate the need for pre-halogenation at the 2-position, thus greatly simplifying the synthesis of the target scaffold, and (ii) provide a greener alternative to the Pd(II)/Ag(I)-promoted reaction, as the use of transition metals becomes unnecessary. In addition, we anticipated that N-protected N-pyridyl-N-hydroxylamines could be utilized in the synthesis of 2-chloro-3-aminopyridines derivatives, which are precursors for Buchwald-Hartwig’s amination strategy (Scheme 1b–ii).
The N-pyridyl-N-hydroxylamine derivatives required for the synthesis of imidazo[4,5-b]pyridin-2-ones and chlorinated 3-pyridinyl acetamides/carbamates were prepared via a one-pot two-step reaction sequence using 3-nitropyridines.28-30 To examine the feasibility of our proposed strategies, we subjected N-hydroxy carbamate 1a (1.00 equiv) to cyanogen bromide (1.20 equiv) in the presence of Hünig’s base (1.20 equiv) in THF (0.300 M) at room temperature, and were pleased to isolate the desired product 2a in 96% yield and with 8:1 regioselectivity (Table 1). With these reaction conditions in hand, we then explored the generality of this transformation utilizing structurally diverse N-protected N-pyridyl-N-hydroxylamines. Notably, the reaction was insensitive to the electronic character of 1 as both electron-poor and electron-rich substrates reacted with high efficiency and high regioselectivity. In addition, halogen functionalities (2a–2c, 2f, 2h, 2k) remained intact during the reaction, providing useful synthetic handles for further structural elaborations. Moreover, substrates with a substituent at the 4-position of the pyridine ring were suitable for this transformation (2d, 2e, 2h). Other functional groups such as alkyl- and aryl-ethers (2c, 2d, 2g–2k), ketals (2i), enolizable ketones (2j), and amides (2l) were compatible under the reaction conditions. The synthetic advantage of this transformation was further exemplified by the successful formation of the desired pyrido-fused cyclic urea derivatives from complex organic molecules. For instance, substrates derived from protected glucose (2i), estrone (2j), Triclosan (2k), and Tadalafil (2l) reacted smoothly to afford the desired imidazo[4,5-b]pyridin-2-ones in high yields and high levels of regioselectivity. Therefore, our strategy allows a late-stage functionalization of drug-like molecules.
Table 1.
Selected examples of the O-cyanation/hetero oxy-Cope rearrangement.a
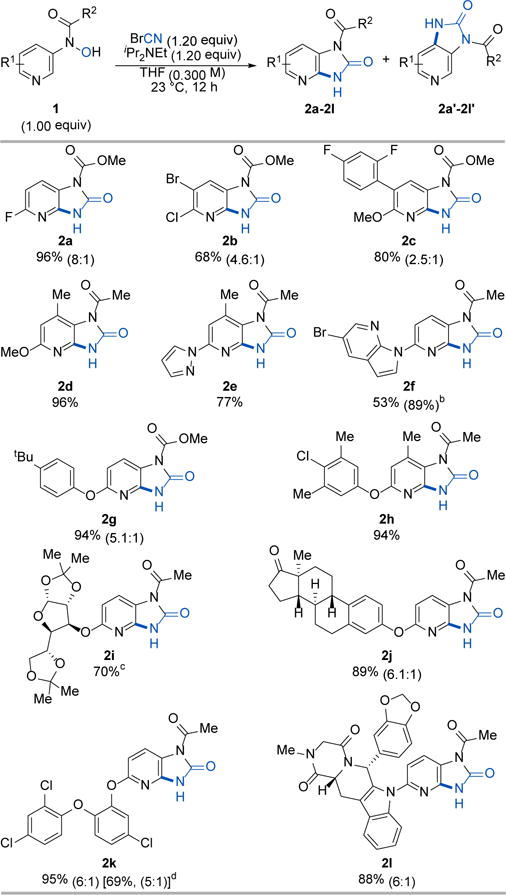
|
Cited yields and isomeric ratios are of isolated material by column chromatography.
Yield in parenthesis was determined by 1H NMR analysis of the crude reaction mixture with CH2Br2 an internal standard.
The reaction exhibited 6.4:1 selectivity, but the minor product came with inseparable impurities. The yield reported here is of the major isomer only.
Yield in the bracket is the isolated yield of a gram-scale reaction.
While our approach provides a general and mild protocol for the preparation of 1-unsubstituted imidazo[4,5-b]pyridin-2-ones, Scott’s amination/cyclization process provides a complementary strategy for the synthesis of the 3-unsubstituent analogs (Scheme 1b-ii). We envision that N-pyridyl-N-hydroxylamine derivatives could also serve as versatile starting materials to gain access to the precursors for the amination/cyclization reaction. Indeed, upon exposure of 1 (1.00 equiv) to thionyl chloride (1.20 equiv) in CH2Cl2 (0.100 M) in the presence of potassium carbonate (1.20 equiv), the desired chlorinated products 3a–3j were formed in good to excellent yields (up to 99%) and with high levels of selectivity at the 2-position (Table 2). Examination of the reaction scope showed that the reaction tolerates a wide array of functionalities including halides (3a–c, 3i), ethers (3b–c, 3f–i), amides (3d, 3j), ketals (3g), ketones (3h), and heterocycles such as pyrazole (3e) and indole (3j). More importantly, N-pyridyl-N- hydroxylamine derivatives bearing more elaborated molecular architectures such as protected fructose (3g), Triclosan (3h), estrone (3l), and Tadalafil (3j) were all successfully chlorinated with excellent yield and regioselectivity. These results demonstrated the synthetic utility and versatility of substrates 1.
Table 2.
Selected examples of synthesis of chlorinated 3-pyridyl acetamides/carbamates.a

|
Cited yields and isomeric ratios are of isolated material by column chromatography.
Yield in the bracket is the isolated yield of a gram-scale reaction.
In order to demonstrate both the practicality and effectiveness of our strategies for large-scale synthesis, we performed the cyanation/hetero oxy-Cope rearrangement and the reaction with sulfonyl chloride on a gram scale. Triclosan® derivatives 2k and 3i were both obtained in synthetically useful yields under standard reaction conditions (Table 1 and Table 2).
To gain insight into the mechanism of the cyanation/hetero oxy-Cope rearrangement, we performed the reaction (i) under standard conditions, (ii) in the absence of light and (iii) in the presence of a stoichiometric amount of a radical trap, TEMPO (Scheme 2a). We anticipated that if a photo-induced homolytic cleavage of the N–OCN bond takes place leading to the formation of N-amidyl and •OCN radicals, then the yield of the process should decrease without photo-irradiation or upon addition of TEMPO. However, similar yields were obtained under the above-mentioned three conditions, which suggest that the reaction unlikely involves any long-lived radical species. Thus, we hypothesize a reaction mechanism involving the heterolytic cleavage of the N–O bond as shown in Scheme 2b.26, 27 Deprotonation of N-protected N-pyridyl-N-hydroxylamine (1) gives N-hydroxide (Ia) which reacts with cyanogen bromide to afford O-cyanated intermediate Ib. Subsequently, Ib undergoes [3,3]-hetero oxy-Cope rearrangement resulting in isocyanate Ic followed by rearomatization and cyclization to form the final imidazo[4,5-b]pyridin-2-one (2). With regard to the synthesis of chlorinated 3-pyridyl acetamides/carbamates, the transformation proceeds through rearrangement of chlorosulphite Id to Ie, the isomerization of which affords to the chlorinated pyridine derivative 3 (Scheme 2c).25, 31 The lack of 6-chlorinated products and products arising from a reaction with other nucleophiles indicates that the chlorine transfer step is most likely an intramolecular process.
Scheme 2.

Mechanistic studies and proposed reaction mechanisms for the synthesis of imidazo[4,5-b]pyridin-2-ones and chlorinated 3-pyridyl acetamides/carbamates.
In conclusion, we have developed an efficient synthetic route to imidazo[4,5-b]pyridin-2-ones, in which one of the nitrogen atoms is protected with an acetyl or methoxycarbonyl group, thus enabling further selective structural elaborations. Our approach to imidazo[4,5-b]pyridin-2-ones does not require 2-halo-functionalized aminopyridines but starts with readily accessible N-pyridyl-N-hydroxylamine derivatives. Our method is transition metal-free, affords the desired products in good to excellent yields and with high levels of regioselectivity, proceeds at room temperature, and tolerates broad functional groups and complex molecular scaffolds. In addition, we have demonstrated that the versatile N-protected N-pyridyl-N-hydroxylamines can be employed in the synthesis of chlorinated 3-pyridinyl acetamides/carbamates, which are valuable intermediates in the synthesis of pyrido-fused cyclic urea framework using Buchwald-Hartwig’s amination reaction. We expect that our protocols will provide easy access to imidazo[4,5-b]pyridin-2-ones and facilitate SAR studies within this important class of heterocyclic compounds.
Supplementary Material
Acknowledgments
This work was partially supported by National Institute of General Medical Sciences (R35GM119652) and start-up funds from SUNY Stony Brook University (SBU). K.N.L. received the graduate fellowship from the NIH Chemical-Biology training grant (T32GM092714). J.W.L. received the 2017 Chemistry Graduate Fellowship from the Department of Chemistry at SBU.
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.US3719683A. United States Pat. 1973
- 2.Xu F, Zacuto M, Yoshikawa N, Desmond R, Hoerrner S, Itoh T, Journet M, Humphrey GR, Cowden C, Strotman N, Devine P. J Org Chem. 2010;75:7829–7841. doi: 10.1021/jo101704b. [DOI] [PubMed] [Google Scholar]
- 3.Luo GL, Chen L, Conway CM, Denton R, Keavy D, Gulianello M, Huang Y, Kostich W, Lentz KA, Mercer SE, Schartman R, Signor L, Browning M, Macor JE, Dubowchik GM. ACS Med Chem Lett. 2012;3:337–341. doi: 10.1021/ml300021s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell IM. J Med Chem. 2014;57:7838–7858. doi: 10.1021/jm500364u. [DOI] [PubMed] [Google Scholar]
- 5.FR2478637A1. France Pat. 1981
- 6.Kuczynski L, Mrozikiewicz A, Banaszkiewicz W, Respond S. Pol J Pharmacol Pharm. 1982;34:223–228. [PubMed] [Google Scholar]
- 7.US4195088A. United States Pat. 1980
- 8.Clark RL, Pessolano AA, Shen TY, Jacobus DP, Jones H, Lotti VJ, Flataker LM. J Med Chem. 1978;21:965–978. doi: 10.1021/jm00207a023. [DOI] [PubMed] [Google Scholar]
- 9.Gustin DJ, Ma ZH, Min XS, Li YH, Hedberg C, Guimaraes C, Porter AC, Lindstrom M, Lester-Zeiner D, Xu GF, Carlson TJ, Xiao SH, Meleza C, Connors R, Wang ZL, Kayser F. Bioorg Med Chem Lett. 2011;21:2492–2496. doi: 10.1016/j.bmcl.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 10.US5763469. United States Pat. 1998
- 11.Choi JY, Plummer MS, Starr J, Desbonnet CR, Soutter H, Chang J, Miller JR, Dillman K, Miller AA, Roush WR. J Med Chem. 2012;55:852–870. doi: 10.1021/jm201349f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WO1994029321A1. Korea Pat. 1994
- 13.Leahy DK, Desai LV, Deshpande RP, Mariadass AV, Rangaswamy S, Rajagopal SK, Madhavan L, Illendula S. Org Process Res Dev. 2012;16:244–249. [Google Scholar]
- 14.Popkin ME, Bellingham RK, Hayes JF. Synlett. 2006;2716–2718 [Google Scholar]
- 15.WO2011021678. Japan Pat. 2011;(A1)
- 16.Liu YH, Zhang WH, Sayre LM. J Heterocycl Chem. 2010;47:683–686. [Google Scholar]
- 17.Yoshida T, Kambe N, Murai S, Sonoda N. Bull Chem Soc Jpn. 1987;60:1793–1799. [Google Scholar]
- 18.Meanwell NA, Sit SY, Gao JN, Wong HS, Gao Q, Stlaurent DR, Balasubramanian N. J Org Chem. 1995;60:1565–1582. [Google Scholar]
- 19.Kuethe JT, Wong A, Davies IW. J Org Chem. 2004;69:7752–7754. doi: 10.1021/jo048887v. [DOI] [PubMed] [Google Scholar]
- 20.McLaughlin M, Palucki M, Davies IW. Org Lett. 2006;8:3311–3314. doi: 10.1021/ol061233j. [DOI] [PubMed] [Google Scholar]
- 21.Scott JP. Synlett. 2006;2083–2086 [Google Scholar]
- 22.Lach F, Koza P. ACS Comb Sci. 2012;14:491–495. doi: 10.1021/co300078f. [DOI] [PubMed] [Google Scholar]
- 23.Youn SW, Kim YH. Org Lett. 2016;18:6140–6143. doi: 10.1021/acs.orglett.6b03151. [DOI] [PubMed] [Google Scholar]
- 24.Luo YR. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton: 2007. [Google Scholar]
- 25.Tabolin AA, Ioffe SL. Chem Rev. 2014;114:5426–5476. doi: 10.1021/cr400196x. [DOI] [PubMed] [Google Scholar]
- 26.Almeida PS, Lobo AM, Prabhakar S. Heterocycles. 1989;28:653–656. [Google Scholar]
- 27.Saczewski F, Debowski T. Tetrahedron Lett. 1993;34:2843–2846. [Google Scholar]
- 28.Hojczyk KN, Feng PJ, Zhan CB, Ngai MY. Angew Chem Int Ed. 2014;53:14559–14563. doi: 10.1002/anie.201409375. [DOI] [PubMed] [Google Scholar]
- 29.Feng PJ, Lee KN, Lee JW, Zhan CB, Ngai MY. Chem Sci. 2016;7:424–429. doi: 10.1039/c5sc02983j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JW, Spiegowski DN, Ngai MY. Chem Sci. 2017;8:6066–6070. doi: 10.1039/c7sc01684k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayyangar NR, Kalkote UR, Nikrad PV. Tetrahedron Lett. 1982;23:1099–1102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.