

Overexpression of MYC is widespread in human cancer, often as a consequence of mutation in upstream mitogenic pathways such as EGFR-RAS and WNT-βCatenin. De-regulated MYC perturbs many aspects of cell function but its influence on cellular metabolism may impart specific vulnerabilities that can be exploited for cancer therapy [1]. Two new studies shine the spotlight on oxidative stress as a specific vulnerability in MYC-dependent cancers and pinpoint NUAK1 and AMPK as key participants in how cancer cells cope with oxidative stress [2, 3].
Our previous work identified NUAK1 (aka ARK5) and AMPKα1 as synthetic lethal interactors with overexpressed MYC [4]. Colorectal cancer (CRC) is characterised by near uniform overexpression of MYC [5], suggesting that CRC may be particularly dependent upon these kinases. Supporting this hypothesis, we have now found that genetic deletion of Nuak1 suppresses tumour formation in a mouse model of CRC, while acute depletion of Nuak1 drives regression of pre-existing colonic tumours [2]. Transcriptomic analysis revealed that loss of NUAK1 suppressed expression of a host of anti-oxidant genes, focusing our attention on the anti-oxidant transcriptional master regulator NRF2 (NFE2L2). In unstressed cells, NRF2 is targeted for degradation in the following ways: (a) by the cytosolic ubiquitin ligase KEAP1 and (b) via GSK3β-dependent phosphorylation [6, 7]. Reactive Oxygen species (ROS) drive nuclear stabilisation of NRF2 by directly modulating KEAP1 and by indirectly suppressing GSK3β, the latter via inhibition of PTEN, allowing inhibitory phosphorylation of GSK3β by AKT. We found that this latter pathway requires concomitant suppression of the protein phosphatase PP1β, which otherwise counteracts AKT-dependent GSK3β phosphorylation [8]. NUAK1 was previously shown to inhibit PP1β activity by phosphorylating the substrate-targeting subunit, MYPT1 [9]. Strikingly, we found that ROS activates NUAK1 kinase activity, possibly via direct oxidation of multiple NUAK1 Cysteines, resulting in increased inhibitory phosphorylation of the MYPT1/PP1β complex. Depletion or inhibition of NUAK1 thus allows unrestrained PP1β-dependent de-phosphorylation of the AKT site on GSK3β, with the consequence that nuclear NRF2 fails to accumulate and ROS levels breach the threshold for loss of viability. Crucially, provision of exogenous anti-oxidants protected autochthonous CRC tumours from NUAK1 depletion, confirming the functional requirement for NUAK1 to suppress oxidative stress [2]. (Fig. 1)
Fig. 1.
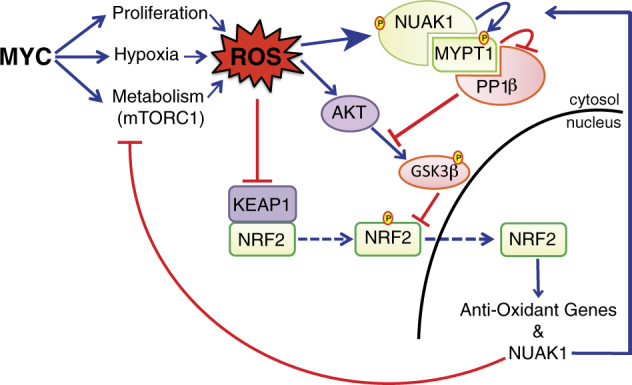
The role of NUAK1 in anti-oxidant signalling. Reactive Oxygen species mobilise the anti-oxidant master regulatory transcription factor NRF2 by inhibiting KEAP1 and indirectly suppressing GSK3β. Inhibitory phosphorylation of GSK3β by AKT requires concomitant suppression of the PP1β phosphatase by NUAK1. NRF2 moreover drives expression of NUAK1, firmly placing NUAK1 in the oxidative stress response pathway. MYC has many influences on ROS homeostasis and establishes an ectopic dependency on the NUAK1/NRF2 pathway to sustain tumour cell viability
Relating the genetic model to human disease, multiple human CRC cell lines were exquisitely sensitive to pharmacological inhibition of NUAK1 and again were rescued by provision of exogenous anti-oxidants. Tellingly, the human CRC cell lines were also rescued by depletion of cMYC, whereas forced expression of MYC in drug-resistant cells conferred sensitivity to the NUAK1 inhibitor, firmly supporting our previous demonstration of a synthetic lethal requirement for NUAK1 in MYC-de-regulated cells [4].
A separate study by Radtke and colleagues has now revealed a similar role for AMPK, or rather, for a specific subset of AMPK complexes, in MYC-dependent Melanoma [3]. Although often discussed as though it were a single entity, the AMP-activated protein kinase is a trimeric complex comprised of 1 of 2α, 1 of 2β and 1 of 3γ subunits, and there is evidence that multiple distinct complexes can assemble and co-exist in cells, presumably responding to distinct cues and/or regulating specific downstream substrates [10]. The α1 and β2 subunits of AMPK are selectively amplified across a spectrum of human cancers, suggesting a specific enrichment for complexes containing these subunits in a cancer setting [11]. Using a mouse model of Melanoma driven by NRAS activation in the absence of Ink4a (Cdkn2a), the Radtke group found that elevated expression of both of these AMPK subunits correlated with high MYC expression at the cellular level in FACS-sorted disaggregated tumour cells. Suppression of MYC specifically reduced expression of both of these subunits, whereas the β1 and γ1 subunits were unaffected, providing the first evidence that MYC may selectively enrich for a specific subset of AMPK complexes. Significantly, depletion of either AMPKα1 or β2 resulted in pronounced apoptosis of MYC positive melanoma cells [3].
In contrast with our analysis of CRC cells, wherein elevated MYC correlated with increased cytosolic ROS, the Radtke group reported increased ROS upon depletion of MYC in their Melanoma model and indeed, genetic deletion of MYC alone resulted in pronounced cell death. The reason for this difference is unclear and may relate to the presence of distinct oncogenic drivers (RAS activation in Melanoma versus APC loss in CRC) in different tissues. However, treatment of MYC-deleted cells with 991, a highly selective pharmacological activator of AMPK [12], completely rescued both the elevated ROS and the cytotoxic effect of MYC deletion, mirroring the cytoprotective role of NUAK1 in counteracting ROS. Furthermore, as is the case for high expression of NUAK1 in CRC, high expression of AMPK in Melanoma correlates with significantly reduced overall survival of human patients [2, 3].
Mitochondrial ROS production is intimately linked to Calcium signalling and we recently demonstrated a specific role for NUAK1 in Calcium/CamKKβ-dependent activation of AMPK [13], suggesting that the activity of these kinases in response to oxidative stress is likely coordinated. Cellular sensitivity to Calcium flux appears to increase with MYC levels, providing another potential link to regulation of these kinases. From MYC’s multiple roles in mitochondrial perturbation to the promotion of nutrient uptake, much of which are diverted for generation of anti-oxidants (e.g., glutathione and NADPH), MYC likely contributes both positively and negatively to oxidative stress [14]. The precise level of MYC expression coupled with the context of co-incident mutations and varying exposure to oxidative stress will all contribute to determine the influence of MYC from one instance to another. Targeting the defence mechanisms employed by cancer cells to counteract oxidative stress and maintain ROS homeostasis may well improve therapeutic outcomes for many cancer patients.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Li B, et al. Clin Cancer Res. 2013;19:5835–41. doi: 10.1158/1078-0432.CCR-12-3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Port JLF, et al. Cancer Discov. 2018. 10.1158/2159-8290.CD-17-0533.
- 3.Kfoury A, et al. EMBO J. 2018. 10.15252/embj.201797673
- 4.Liu L, et al. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. Nature. 2014;511:543–50.
- 6.McMahon M, et al. J Biol Chem. 2003;278:21592–600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 7.Rojo AI, et al. J Neurochem. 2008;105:192–202. doi: 10.1111/j.1471-4159.2007.05124.x. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez F, et al. Mol Cell Biochem. 2010;344:211–5. doi: 10.1007/s11010-010-0544-0. [DOI] [PubMed] [Google Scholar]
- 9.Zagorska A, et al. Sci Signal. 2010;3:ra25. doi: 10.1126/scisignal.2000616. [DOI] [PubMed] [Google Scholar]
- 10.Ross FA, et al. FEBS J. 2016;283:2987–3001. doi: 10.1111/febs.13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteverde T, et al. FEBS J. 2015;282:4658–71. doi: 10.1111/febs.13534. [DOI] [PubMed] [Google Scholar]
- 12.Xiao B, et al. Nat Commun. 2013;4:3017. doi: 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monteverde T, et al. Oncogene. 2018;37:982–92. [DOI] [PMC free article] [PubMed]
- 14.Dang CV. Cold Spring Harb Perspect Med. 2013;3:a014217. [DOI] [PMC free article] [PubMed]