

Abstract
1,4-Dihydropyridine (1,4-DHP) derivatives have been synthesized and characterized by 1H, 13C, 15N nuclear magnetic resonance (NMR) spectroscopy, secondary proton/deuterium 13C isotope shifts, variable temperature 1H NMR experiments and quantum-chemical calculation. The intramolecular hydrogen bonds NH⋯O=C and CH⋯O=C in these compounds were established by NMR and quantum-chemical studies The downfield shift of the NH proton, accompanied by the upfield shift of the 15N nuclear magnetic resonance signals, the shift to the higher wavenumbers of the NH stretching vibration in the infrared spectra and the increase of the 1J(15N,1H) values may indicate the shortening of the N–H bond length upon intramolecular NH⋯O=C hydrogen bond formation.
Keywords: NMR, DFT, 1, 4-dihydropyridines, hydrogen bond, H/D 13C isotope effects, IR
1. Introduction
Since the original discovery of amlodipine, the 2-substituted 1,4-dihydropyridines have attracted considerable attention owing to their various biological activities [1]. For the aimed synthesis of novel therapeutic agents, it is important to establish what structural factors influence their biological activity. To this end, the modification of 2,6-Me groups was performed. The original method was elaborated for obtaining both alkyl 4-substituted 2-acetoxymethyl-(3) and 2,6-bis-acetoxymethyl-1,4-dihydropyridine-3,5-dicarboxylates (5) (scheme 1).
Scheme 1.

Synthesis of compounds 2–5. 1a–5a R = Me; R1 = Et; 1a′–3a′ R = Me; R1 = Me; 1b–5b R = Ph; R1 = Et; 1c–5c R = o-CHF2-Ph, R1 = CH2CH2O-Pr-n; 3d R = o-F-Ph, R1 = Et; 3e R = m-NO2-Ph, R1 = Et.
Non-covalent interactions have fundamental roles in supramolecular chemistry, drug design, protein folding, crystal engineering and other areas of molecular science. Hydrogen bond interactions are principal forces, which determine the molecular recognition and self-assembly processes as well as the structure of a great variety of chemical and biological systems [2]. The structures of the novel compounds 3 and 5 are interesting because there could be different types of intramolecular interactions.
The investigated compounds 1, 3 and 5 can be divided into three groups according to their structure. In the first group (1) any intramolecular hydrogen NH⋯O bonds are absent, the second one (3)—could have one NH⋯O bond and the third group (5) may have two such bonds. The NH chemical shift (CS) in 1H nuclear magnetic resonance (NMR) spectra in general reflects the strength of the intramolecular hydrogen bond [3]. Additionally, intramolecular hydrogen bonding can be characterized by means of secondary deuterium isotope effects NH(D) on 13C NMR chemical shifts [4] as well as Fourier transform infrared (FTIR) spectroscopy [5]. Quantum-chemical calculations should give insight for energetic and geometrical parameters of the intramolecular hydrogen bond in the optimal conformations of the molecules.
2. Material and methods
2.1. General
All reagents were purchased from Acros, Aldrich, Alfa Aesar or Merck and used without further purification. Thin layer chromatography was performed on silica gel 60 F254 aluminium sheets 20 × 20 cm (Merck), as eluent using (20% EtOAc/hexane). The purities of compounds were determined by high performance liquid chromatography on a Waters Alliance 2695 system and Waters 2489 ultraviolet–vis detector equipped with Alltima C18 column (5 µm, 4.6 × 150 mm, Grace) using a gradient elution with acetonitrile/phosphoric acid (0.1%) in water, at a flow rate of 1 ml min−1. Peak areas were determined electronically with a Waters Empower 2 chromatography data system.
2.2. Nuclear magnetic resonance spectroscopic experiments
The one-dimensional 1H- and 13C- and two-dimensional 1H-1H nuclear overhauser effect spectroscopy (NOESY), 13C-1H heteronuclear multiple bond correlation (HMBC), 13C-1H heteronuclear single-quantum correlation (HSQC) spectra of compounds 1–5 were recorded on a Varian-Mercury 400 MHz, Varian-400mr 400 MHz, Varian UNITY INOVA 600 MHz and Bruker Avance III HD 800 MHz, in CDCl3, at temperature 25°C.
15N NMR spectra were recorded at 60.81 MHz (Varian UNITY INOVA 600 MHz) spectrometer equipped with a cryoprobe and at 81.10 MHz (Bruker Avance III HD 800 MHz). Spectra were acquired using HSQC standard pulse sequences provided by Varian or Bruker Biospin spectrometer libraries. The magnetization from 1H to 15N in HSQC spectra were transferred using coupling constants 1J(15N,1H) = 95. 15N spectra were indirectly referenced to liquid ammonia.
2.3. Infrared spectroscopic experiments
The infrared (IR) spectra were obtained in the fine films on the Shimadzu IR Prestige 21 FTIR spectrometer.
2.4. Elemental analysis experiments
Instrumental elemental analysis is based on method of ‘flash combustion’ Elemental analyzer Carlo Erba mod. EA-1108.
2.5. Compounds
Dialkyl 4-aryl-2-acetoxymethyl-6-methyl-1,4-dihydropyridine-3,5-dicarboxylates are scarcely investigated; they were mainly obtained by Hantzsch cyclization using commercially not available ethyl 4-acetoxy-3-oxobutanoate [6] or in a four-step procedure from appropriate 2-dietoxymethyl-1,4-DHP [7].
We propose the two steps procedure from dimethyl 4-substituted 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate via bromination of 2-methyl- or 2,6-dimethyl groups.
The necessary 2-bromomethyl-1,4-DHP 2 and 2,6-bis-bromomethyl-1,4-DHP 4 were obtained by our elaborated method including the bromination of the methyl groups in the position 2- (or 2,6-) with N-bromosuccinimide (NBS) in methanol according to the literature [8,9]. The further reaction of appropriate monobromomethyl-1,4-dihydropyridine (DHP) 2 or bis-bromomethyl-1,4-DHP 4 with dry potassium acetate in dry dimethylformamide (DMF) lead to target compounds 3 and 5 in medium-to-good yields (figure 1).
Figure 10.

(a–f) The most stable conformations of 5a.
2.6. General procedure for the synthesis of 4-substituted diethyl(dimethyl) 2-acetoxymethyl-6-methyl-1,4-dihydropyridine-3,5-dicarboxylates 3
Appropriate 4-substituted ethyl(methyl) 2-bromomethyl-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate 2 (0.9 mmol, 1 eq) was dissolved in dry DMF (3 ml) under argon. Anhydrous potassium acetate (97 mg, 0.99 mmol, 1.1 eq) was added at 0°C. The reaction mixture was stirred at room temperature for 20 h. The mixture was diluted with water (10 ml) and extracted with EtOAc, the organic layer washed with water and brine and dried over Na2SO4. After filtration and evaporation, the crude residue was purified by column chromatography (silica gel, eluent—chloroform/petroleum ether (bp 40–60°C)/acetone (9 : 7 : 1), giving 2-acetoxymethyl-1,4-DHP 3 as powder or oil.
2.6.1. Diethyl 2-acetoxymethyl-4,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3a)
Yield: 0.22 g 74%; yellowish oil. Mass spectrometry (MS) (+electrospray ionization (ESI)) m/z (relative intensity) 326 ([M + H]+ 60).
2.6.2. Dimethyl 2-acetoxymethyl-4,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3a′)
Yield: 0.15 g 56%; white powder; m.p. 78–83°C. MS (+ESI) m/z (relative intensity) 298 ([M + H]+ 45). Anal. Calc. for C14H19NO6: C, 56.56; H, 6.44; N, 4.71; found: C, 56.48; H, 6.31; N, 4.69.
2.6.3. Diethyl 2-acetoxymethyl-6-methyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (3b)
Yield: 0.23 g 67%; white powder; m.p. 104–106°C ([10] 106°C. MS (+ESI) m/z (relative intensity) 388 ([M + H]+ 50). Anal. Calc. for C21H25NO6: C, 65.10; H, 6.50; N, 3.62; found: C, 64.99; H, 6.53; N, 3.60.
2.6.4. Bis-(2-propoxyethyl) 2-acetoxymethyl-4-(2-difluoromethoxyphenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate (3c)
Yield: 0.37 g 72%; yellow oil. MS (+ESI) m/z (relative intensity) 570 ([M + H]+ 72).
2.6.5. Diethyl 2-acetoxymethyl-4-(2-fluorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate (3d)
Yield: 0.22 g 61%; white powder; m.p. 102–104°C. MS (+ESI) m/z (relative intensity) 406 ([M + H]+ 82). Anal. Calc. for C21H24 FNO6: C, 62.21; H, 5.97; N, 3.45; found: C, 62.02; H, 6.05; N, 3.37.
2.6.6. Diethyl 2-acetoxymethyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3e)
Yield: 0.20 g 52%; light yellow powder; m.p. 105–107°C ([11] 106°C. MS (+ESI) m/z (relative intensity) 433 ([M + H]+ 100). Anal. Calc. for C21H24 N2O8: C, 58.33; H, 5.59; N, 6.48; found: C, 58.12; H, 5.65; N, 6.21.
2.7. General procedure for the synthesis of 4-substituted diethyl 2,6-bis-acetoxymethyl-1,4-dihydropyridine-dicarboxylates 5
Appropriate 4-substituted diethyl 2,6-bis-bromomethyl-1,4-dihydropyridine-3,5-dicarboxylate 4 (1.2 mmol, 1 eq) was dissolved in dry DMF (4 ml) under argon. Anhydrous potassium acetate (283 mg, 2.88 mmol, 2.4 eq) was added at 0°C. The reaction mixture was stirred at room temperature for 16 h. The mixture was diluted with water (20 ml) and extracted with EtOAc, the organic layer washed with water and brine and dried over Na2SO4. After filtration and evaporation, the crude residue was crystallized from dilute methanol giving 2,6-bis-acetoxymethyl-1,4-DHP 5.
2.7.1. Diethyl 2,6-bis-acetoxymethyl-4-methyl-1,4-dihydropyridine-3,5-dicarboxylate (5a)
Yield: 0.37 g 81%; light yellow crystals; m.p.91°C. MS (+ESI) m/z (relative intensity) 385 ([M + H]+ 100). Anal. Calc. for C18H25NO8: C, 56.39; H, 6.57; N, 3.65; found: C, 56.08; H, 6.60; N, 3.49.
2.7.2. Diethyl 2,6-bis-acetoxymethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (5b)
Yield: 0.40 g 75%; light yellow crystals; m.p.128–130°C. MS (+ESI) m/z (relative intensity) 446 ([M + H]+ 82). Anal. Calc. for C23H27NO8: C, 62.01; H, 6.11; N, 3.14; found: C, 61.85; H, 6.25; N, 3.10.
2.7.3. Bis-(2-propoxyethyl) 2,6-bis-acetoxymethyl-4-(2-difluoromethoxyphenyl)-1,4-dihydropyridine-3,5-dicarboxylate (5c)
Yield: 0.44 g 58%; white crystals; m.p.76–78°C. MS (+ESI) m/z (relative intensity) 628 ([M + H]+ 100). Anal. Calc. for C30H39 F2 NO11: C, 57.41; H, 6.26; N, 2.23; found: C, 57.11; H, 6.35; N, 2.08.
2.8. Computational details
All calculations were performed in Gaussian 09 [12] using the hybrid density functional theory (DFT) B3LYP functional with basis set 6–311++G(d,p) with empirical dispersion D3 version of Grimme with Becke-Johnson damping (GD3BJ) [13]. All geometries were optimized using the polarizable continuum model (PCM) solvation model for chloroform. For all optimized conformers, vibrational harmonic frequencies were calculated at the same level of theory, all the obtained harmonic frequencies were positive. For chemical shift calculation a gauge-independent atomic orbital (GIAO) method was used.
3. Results and discussion
The structures of 1, 3, 5 were analysed by multinuclear 1H, 13C, 15N NMR spectroscopy (tables 1 and 2) and quantum-chemical methods.
Table 1.
1H,15N NMR data for 1,3,5 in CDCl3.
| δ(1HN), ppm | δ(HA), ppm | 1J(13C,1HA), Hz | δ(HB), ppm | 1J(13C,1HB), Hz | 2J(H,H), Hz | Δδ(1HN)/T, ppb K−1 | δ(15N), ppm | 1J(15N,1H), Hz | |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 5.54 | −5.6 | 134.53 | 92.4 | |||||
| 1b | 5.67 | −6.1 | 134.01 | 93.6 | |||||
| 1c | 5.73 | −10.4 | 134.36 | 93.6 | |||||
| 3a | 6.48 | 5.21 | 153 | 5.27 | 155 | 14.7 | −3.2 | 122.90 | 93.3 |
| 3a′ | 6.53 | 5.25 | 153 | 5.29 | 155 | 14.7 | −3.0 | 125.42 | 93.0 |
| 3b | 6.55 | 5.27 | 153 | 5.40 | 156 | 14.8 | −2.9 | 124.12 | 93.6 |
| 3c | 6.64 | 5.26 | 154 | 5.32 | 155 | 14.8 | −2.8 | 122.69 | 93.6 |
| 3d | 6.57 | 5.27 | 154 | 5.39 | 156 | 14.9 | −2.8 | 123.31 | 94.8 |
| 3e | 6.67 | 5.32 | 154 | 5.37 | 156 | 14.9 | −2.5 | 124.25 | 94.2 |
| 5a | 7.55 | 5.23 | 153 | 5.36 | 156 | 15.3 | −3.6 | 113.14 | 94.8 |
| 5b | 7.65 | 5.24 | 154 | 5.44 | 157 | 15.2 | −3.6 | 114.18 | 94.8 |
| 5c | 7.77 | 5.25 | 154 | 5.35 | 156 | 15.2 | −3.6 | 115.38 | 95.6 |
Table 2.
13C Chemical shifts, (ppm) in 1, 3, 5.
| COO···H | C6 | C2 | C5 | C3 | C6-Me | C2-CH2 | C4 | C4Me | |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 144.28 | 144.28 | 104.63 | 104.63 | 19.44 | 28.49 | 22.22 | ||
| 1b | 144.39 | 144.39 | 103.79 | 103.79 | 19.28 | 39.62 | |||
| 1c | 144.96 | 144.96 | 102.35 | 102.35 | 19.22 | 35.88 | |||
| 3a | 170.82 | 144.10 | 141.76 | 105.81 | 104.49 | 19.50 | 61.37 | 28.54 | 22.05 |
| 3a′ | 170.85 | 144.43 | 142.07 | 105.46 | 104.25 | 19.48 | 61.28 | 28.43 | 22.00 |
| 3b | 170.76 | 143.47 | 141.35 | 105.05 | 104.15 | 19.67 | 61.41 | 39.60 | |
| 3c | 170.71 | 144.16 | 142.03 | 103.28 | 102.52 | 19.70 | 61.33 | 36.22 | |
| 3d | 170.71 | 143.88 | 141.82 | 103.76 | 103.01 | 19.60 | 61.31 | 34.18 | |
| 3e | 170.72 | 144.36 | 142.27 | 103.99 | 103.32 | 19.76 | 61.24 | 39.83 | |
| 5a | 170.75 | 141.95 | 141.95 | 105.21 | 105.21 | 61.25 | 28.58 | 22.08 | |
| 5b | 170.70 | 141.44 | 141.44 | 104.52 | 104.52 | 61.26 | 39.54 | ||
| 5c | 170.59 | 142.23 | 142.23 | 102.83 | 102.83 | 61.23 | 36.19 |
3.1. 1H, 15N nuclear magnetic resonance and infrared spectra
The X-ray structures of 3,5-mono- and 3,5-dicarboxylates of 1,4-DHP as well as quantum-chemical calculations [6] show that the carbonyl groups adopt the s-cis, s-cis conformation relatively to C3=C2 or C6=C5 double bonds. This is unusual, because it is well known that for any dienes s-trans orientation is preferable, owing to the better conjugation. The reason for the peculiar s-cis conformation of the fragment –C=C–C=O is not clear. However, in the case of 1,4-DHP derivatives it was proposed that the s-cis orientation is favoured by weak intramolecular CH⋯O=C interaction [7]. Such interactions are possible in 1, 3, 5 too.
The signals of the methylene groups of the alkoxy carbonyl chains in position 3 and 5 for compounds 3 and 5 show a rather complex splitting pattern, owing to the presence of a prochiral centre on C4 leading to the diasteretopicity of the two hydrogens of the methylene group. The rest of the signals in 1H NMR spectra are in agreement with the nature of aromatic or aliphatic hydrogen atoms (table 1).
The two methylene hydrogens of the substituents on C2 and C6 in 3 and 5 are also diastereotopic and they give an AB system at 5.24–5.44 ppm in the 1H NMR spectra. The non-equivalence of these protons is characterized by the 1H chemical shift difference (δ(HA) − δ(HB)) which may be affected by such factors as the conformation of the 1,4-DHP ring, the anisotropic influence of the substituents, the hindered rotation of the substituents at C2,6 and the intramolecular hydrogen bond of the type CH⋯O. The geminal constants 2J(HA,HB) slightly increase from 14.8 Hz in 3 to 15.2 Hz in 5 (table 1). For the compounds 3 the difference in the chemical shifts of the AB methylene protons varies in the range of 0.043–0.130 ppm depending on the R substituents at C4.
1H NOESY spectra of compounds 3 and 5, allow us to determine the relative position of the AB protons for the methylene groups—the NOE from NH to the more shielded protons HA is more intense than the NOE to HB. This means that the HA proton is located closer to the NH proton and cannot form a CH⋯O = C hydrogen bond.
The 1H NMR signals of these protons show unusual temperature dependence (figure 1) similarly to 4-aryl-2,6-bis-(bromo-methyl)-1,4-DHP [14]. The temperature dependence of the AB quartet excludes an ordinary exchange process, because the signals do not exhibit exchange line broadening prior to coalescence and the chemical shifts of the both protons vary simultaneously. This is in contradiction with classical dynamic NMR theories [15].
Figure 1.

Temperature dependence of the AB system 1H chemical shifts for 3a in CDCl3.
At temperature variation, the CS of the HA proton in 3 and 5 changes linearly with the same coefficient as the CS of 2-CH3 methyl protons in 1 (table 1). So, the HA proton should be available to the solvent (chloroform), to the same extent as in 1. At the same time the signal of HB changes nonlinearly and the curve is close to a negative parabolic (figure 2). The nonlinear dependence (δ(HB) versus T) can point to the fact that HB is engaged in a weak intramolecular hydrogen bond C–H⋯O=C.
Figure 2.
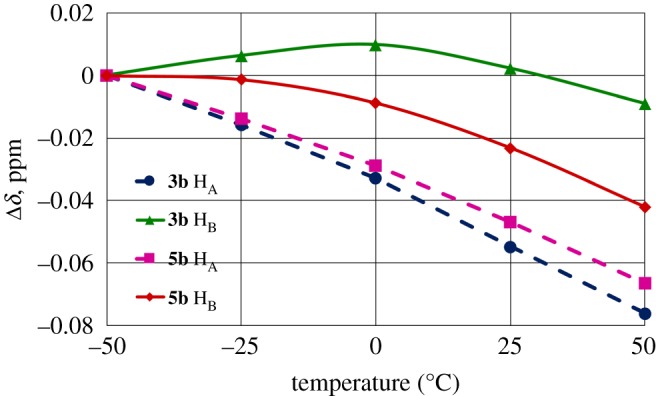
Temperature dependence of the C2,6-CH2 methylene protons chemical shifts for 3b and 5b in CDCl3. Solid and dashed lines represent the values for HB and HA protons, respectively. The chemical shift at 0°C is taken as zero point for each proton. 3b HA (filled circles), 3b HB (filled triangles), 5b HA (filled squares) and 5b HB (filled diamonds).
The unusual temperature influence on the diastereotopic methylene proton signals could be explained by changes in the conformer ratio owing to two conformational processes: the rotation of the substituent around the C2,6─CH2 and around the C3,5─CO2 bonds. These processes have relatively high barriers for transitions between the possible conformations (approximately 16–18 kcal mol−1 (66.94–75.31 kJ mol−1) and approximately 8–10 kcal mol−1 (33.47–41.84 kJ mol−1), respectively) and unusual temperature effects on the methylene proton signals are caused by changes in the conformer ratio. So, the observed AB proton resonances are the statistically averaged resonance signals of the various conformations, the populations of which are temperature dependent [14].
The measured coupling constants 1J(13C,1H) for each of the methylene AB protons are different (table 1). The larger 1J(13C,1H) value corresponds to the less shielded proton HB. This is consistent with the literature data, because if the CH proton is involved in a hydrogen bond the 1J(13C,1H) value usually increases [16]. Indeed, in our previous work [9] the hydrogen bonded protons were characterized by the larger value of the 1J(13C,1H). On the basis of the data obtained it can be concluded that the intramolecular CH⋯O=C hydrogen bond could be one of the reasons for the non-equivalence of the methylene protons in these systems and conformations with s-cis-s-cis carboxyl group orientation relative to the double bond in the dihydropyridine cycle.
The 1H NMR spectra show a broad signal of the NH proton in the intervals, 5.54–5.70 ppm (1), 6.55–6.68 ppm (3) and 7.65–7.77 ppm (5) (table 1).
The low field shift of the N1H proton signal in the sequence 1, 3, 5, may reflect the formation of an intramolecular N–H⋯O hydrogen bond in compounds 3 and 5 between the N1H proton and the oxygen atoms of the OCOMe substituents at C2 and C6 carbons [17,18]. On the other side the low-field shift of N1H resonance could correspond partly to the decrease of the electron density on N1H atom as calculated by the B3LYP-NBO method in the sequence 1, 3, 5 (figure 3).
Figure 3.
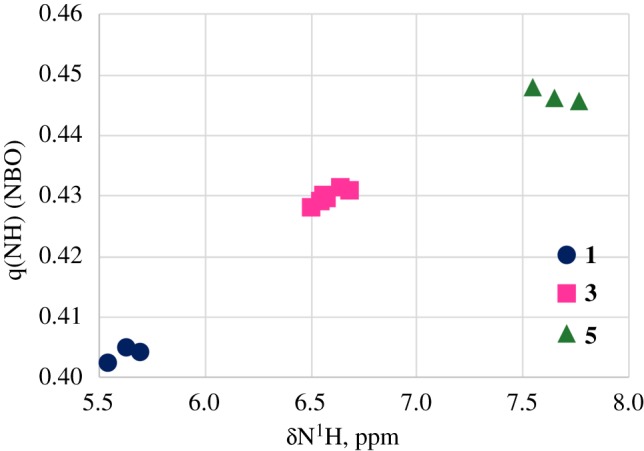
A plot of the NBO charges (q) versus δ(1HN) in 1(filled circles)–3(filled squares)–5(filled triangles).
The temperature coefficient (Δδ/ΔT) of the N1H signal is usually used to detect the presence of intramolecular H-bonds [18,19]. The breaking of hydrogen bonds, induced by increasing the temperature, moves the resonance signal of N1H upfield. The temperature coefficients with the values more positive than −4.5 ppb K−1 are strongly indicative of intramolecular H-bonds.
The temperature coefficients of the NH proton resonance in 1, 3, 5 have been measured recording the 1H NMR spectra in CDCl3 in the temperature range 223–323 K (table 1). The obtained values fall between −2.5 and −10.4 ppb K−1. The NH protons of 3 have the Δδ/ΔT values more positive than −4.5 ppb K−1 (approx. −3 ppb K−1) which indicates the involvement of these NH protons in the intramolecular H-bonding. In compounds 5, that have two acetoxymethyl substituents and, consequently, may have two hydrogen bonds, the Δδ/ΔT values are lower than in 3 (approx. −3.6 ppb K−1). This could be the consequence of the weaker NH⋯O=C hydrogen bond in case of 5 when compared to 3.
The absolute values of the measured coupling constants 1J(15N,1H) 92–96 Hz slightly increase in the sequence 1, 3, 5 (figure 4) and suggest that the hydrogen is stably bonded to the nitrogen atom and there is little intermolecular exchange, this is in contradiction, because increased positive charge on the NH proton (deshielding) should decrease the absolute value of the 1J(15N,1H) [20,21]. However, when the nitrogen of the proton donor is sp2 hybridized the changes in 1J(15N,1H) are quadratically related to the length of the corresponding NH distances of nitrogen bases: the shorter d(NH) corresponds to the larger 1J(15N,1H) value [22]. This could point to the slight decrease of the NH distance in the sequence 1–3–5.
Figure 4.
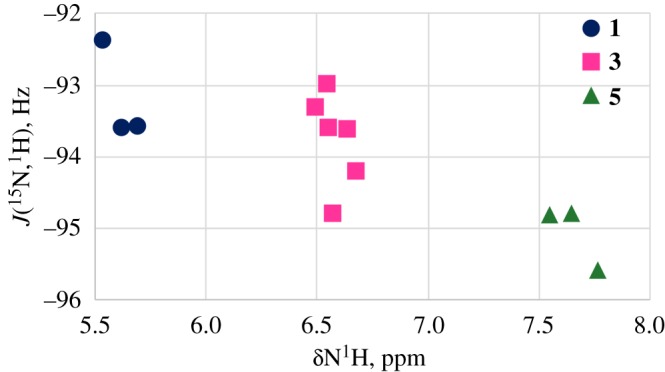
A plot of 1J(15N,1H) versus N1H chemical shifts in 1(filled circles)–3(filled squares)–5(filled triangles).
According to [23] if NH participate in the N–H⋯X hydrogen bond, the 1J(15N,1H) coupling is affected by mainly two factors, the electrostatic effect and the n(X) → (N–H)* charge transfer interaction. The first one is known to cause an increase in the absolute value of 1J(15N,1H) and the second leads to a decrease in the absolute value of 1J(15N,1H) coupling. Their influence on the IR stretching frequency of the N–H bond are also opposite to each other, so the charge transfer interactions correspond to a red shift while the electrostatic effect corresponds to a blue shift. Usually, hydrogen bonds lead to an elongation of the X–H covalent bond and its stretching vibration νXH shifts to the lower wavenumbers (red shift) [5,18]. The shift of ν(NH) to the higher wavenumbers on hydrogen bonding probably is accompanied by the shortening of the NH distance (so-called blue shift). Such a kind of effect has been registered for the C–H and N–H bonds previously [24].
The IR spectral data of 1a–c with no hydrogen bonds are characterized by the stretching ν(NH) bands in the range 3344–3336 cm−1. The NH groups engaged in one hydrogen bond in compounds 3a–c display the NH-stretching vibration in the interval 3347–3348 cm−1 and the NH groups engaged in two hydrogen bonds in compounds 5a–c display the ν(NH) bands in the range 3363–3437 cm−1 (figure 5). The ν(NH) band moves up to 8 cm−1 on going from 1b to 3b, and up to 64 cm−1 on going from 1b to 5b. The IR stretching ν(NH) band frequencies and the 1J(15N,1H) values in compounds 1–5 indicate that both of these parameters (ν(NH), 1J(15N,1H)) could be partly caused by the same reason—the difference in the length of the NH bond [25].
Figure 5.

N–H and C–H stretching frequencies for 1b, 3b and 5b, cm−1.
The 15N HMQC spectra with the value of the direct NH coupling constants (1J(15N,1H) = 95 Hz) were used to measure the chemical shift for the nitrogen nucleus [26,27]. The 15N resonance signals appear in three distinct regions: 134.53–134.01 ppm in 1, 123.31–124.42 ppm in 3 and 114.18–115.58 ppm in 5 ppm (table 1).
The downfield shift of the NH proton signal is accompanied by the upfield shift of the 15N signal in the sequence 1, 3, 5 (figure 6). This is surprising because usually the shielding of the N1H proton is accompanied by the shielding of the adjacent nitrogen atom [28]. Commonly the NH bond elongates on hydrogen bond formation [29]. According to [30] the observed 15N CS of amide nitrogens move upfield with a shortening of the N–H bond in the C=O···H–N hydrogen bond. So, the reverse dependence δ(15N)/δ(1H) in the sequence 1, 3, 5 (figure 7) may probably indicate the shortening of the N–H bond, but cannot quantitatively explain a significant upfield shift of the 15N resonance signal on going from 1–3–5
Figure 6.
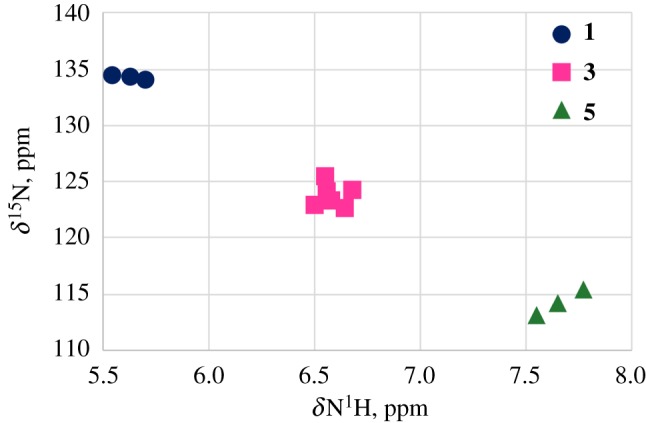
The relationship between the 15N chemical shifts and 1H chemical shift in 1(filled circles)–3(filled squares)–5(filled triangles).
Figure 7.
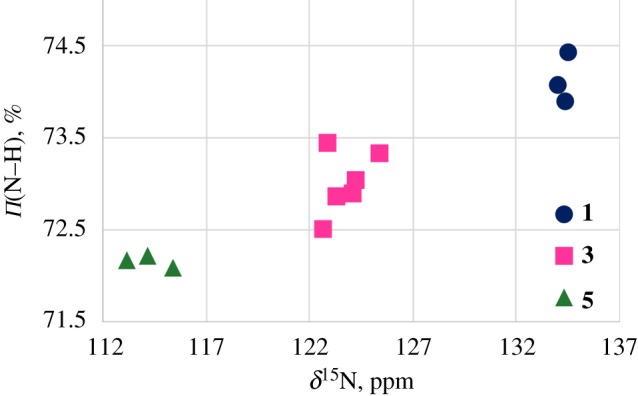
The relationship between 15N chemical shifts and the π-components of the N–H bonds calculated by NBO method for 1(filled circles)–3(filled squares)–5(filled triangles).
In the data obtained by the natural bond orbital (NBO) method, the average π-orbital character of the N atom in NH-bond decreases in the sequence 1(74.2%)–3(73.0%)–5(72.2%) (figure 7). That should lead to a diamagnetic shift of the 15N-resonance signal.
A relationship between the 15N chemical shifts and the 1J(15N,1H) values in compounds 1, 3 and 5 (figure 8), reflects that both of these parameters (δ(15N), 1J(15N,1H)) could be caused partly by the same reason—the difference in the length of the NH bond [25].
Figure 8.

The correlation between 1J(15N,1H) values and δ15NH chemical shifts in compounds 1, 3 and 5.
So, the δ(1H) downfield shift and the upfield shift for δ(15N) in the sequence 1–3–5 partly can be accompanied by the decrease of the N–H bond length. This is supported by the increase of the 1J(15N,1H) absolute value in compounds 1, 3 and 5 (figure 8). A very popular explanation of diamagnetic shift of heavy nuclei is sterical perturbation of the electronic cloud nearby. In our case this may be owing to the spatial affinity between the acetoxy substituent and 15N nucleus. According to the literature data [31] such effect could shift 15N resonance signal up to 10 ppm to higher fields.
3.2. 13C nuclear magnetic resonance spectra
The 13C NMR spectra of compounds 1, 3, 5 display signals in the carboxyl, aromatic and aliphatic regions (table 2). The 13C resonances in the 1,4-DHP ring (table 2) are relatively insensitive to the nature of the substituents on C3, C4 and C5. The 13C NMR spectra of compounds 1, 3, 5 show the signals for C3 and C5 atoms at the lower values (ca 102–104 ppm), than those expected for the typical olefinic carbons atoms, while the C2 and C6 signals appear at higher values (approx. 141–145 ppm). These findings have been accounted for by the strong push–pull effect of the groups linked to the olefinic double bonds like ones previously observed in other related systems. The carbon signal of C4 is in agreement with the previous data [32,33] and appears at 28.5 ppm for the aliphatic R and in the interval 34.2–39.8 ppm when R is aromatics.
3.3. Proton/deuterium isotope effects on 13C chemical shifts
Measurement of the secondary nΔ13C(proton/deuterium (H/D)) deuterium isotope effects (IE) is known to be a very good tool in studying intramolecular hydrogen bonds [4]. In compounds 1, 3, 5 the replacement of H by D at the nitrogen atom produces only intrinsic isotope effects, because the values of 1J(15N,1H), approximately 92–96 Hz, allow us to neglect any equilibrium processes. Intrinsic isotope effects are of vibrational origin and are owing to the anharmonicity of the potential curve of the N–H⋯X bond. Hydrogen bonding increases this anharmonicity. The changes in δ(13C) are the result of the shortening of the NH/D bond upon deuteration [34].
The values of IE (nΔ13C(H/D) = δ(13C(NH)) − δ(13C(ND))) have been measured from the 13C spectra of partly deuterated samples where the signals of the two isotopomers could be easily identified on the basis of their relative intensity. All measured isotope effects in 1, 3, 5 were positive (table 3).
Table 3.
N(H/D) isotope effects on 13C atoms for 1–3–5, ppb.
| COO···H | C6 | C2 | C5 | C3 | C6-Me | C2-CH2 | C4Me | |
|---|---|---|---|---|---|---|---|---|
| 1a | 85 | 85 | 50 | 50 | 81 | — | 26 | |
| 1b | 84 | 84 | 51 | 51 | 82 | — | — | |
| 1c | 90 | 90 | 47 | 47 | 84 | — | — | |
| 3a | 86 | 78 | 59 | 44 | 84 | 37 | 30 | |
| 3a′ | 88 | 79 | 59 | 45 | 84 | 37 | 28 | |
| 3b | 3 | 87 | 80 | 59 | 42 | 84 | 36 | — |
| 3c | 5 | 96 | 88 | 58 | 41 | 84 | 36 | — |
| 3d | 5 | 94 | 87 | 58 | 41 | 84 | 35 | — |
| 3e | 4 | 91 | 85 | 57 | 43 | 84 | 37 | — |
| 5a | 15 | 82 | 82 | 56 | 56 | — | 32 | 27 |
| 5b | 18 | 85 | 85 | 55 | 55 | — | 33 | — |
| 5c | 18 | 91 | 91 | 53 | 53 | — | 34 | — |
The substituent effects on IE values are mainly described by two factors: the hydrogen bond and the transmission of the electronic effects of substituents [35].
The N(H/D) isotope effects on the acetoxymethyl carbonyl 13C atoms have been registered (table 3). For compounds 3 where the NH atom is involved in one hydrogen bond, the 5Δ13CCOO (H/D) values are in the range 3–5 ppb. At the same time the 5Δ13CCOO (H/D) values at the carboxyl carbon for 5 3–5 times exceed (approx. 15–18 ppb) them for 3. Probably, this is owing to some difference in NH⋯O hydrogen bonding in 3 and 5.
If we consider the influence of the N(H/D) replacement on the 13C resonances inside the 1,4-DHP cycle (table 3) there is little change in the sequence 1–3–5. This corresponds to the small change of the 13C chemical shifts (table 2). Interestingly, that the N(H/D) IE on the C2,6-13CH3 atom of the methyl groups separated from the N1H proton by three bonds, is similar to the effect on the 13C2,6 carbons separated by only two bonds. At the same time, the N(H/D) IE values for the 13C3,5 atoms are much smaller (table 3). The 13C isotope shifts of the methyl groups at C2 and C6 in 1 and 3 are considerably larger than the corresponding values of the methylene groups at the same atoms in compounds 3 and 5 (table 3).
No 13C(NH/D) isotope effects are observed on the C4 carbon atom in the 1,4-DHP cycle, however in 4-Me substituted compounds 1a, 3a and 5a the positive long-range isotope effects 5ΔC4−Me (H/D) approximately 26–28 ppb are observed. The observation of such a long-range isotope effect at the methyl group C4-CH3 is rather unusual. Since there is no general theory about the long-range IE we have problems in interpretation of the long ranges 13C IE observed in 1, 3, 5 compounds.
3.4. Density functional theory calculations
In compounds 3 and 5 there could be four possible types of the intramolecular H-bonds (NH⋯O=, NH⋯-O–, CH⋯O = and CH⋯–O–) defining the optimal configuration of the molecules.
To establish which of the available types of bonds is dominant in stabilizing the optimal geometry of compounds 3 and 5, the quantum-chemical calculations of the possible conformations of compounds 3a′ and 5a were performed.
The most stable conformations of 3a′ and 5a and the calculated geometric parameters of the intramolecular hydrogen bonds CH⋯O and NH⋯O are presented in figures 9 and 10 and table 4.
Figure 9.

(a–f) The most stable conformations of 3a’.
Table 4.
Intramolecular H-bonds parameters for the preferred conformations of 3a′ and 5a.
| compound (conformation) | C3–C = O, C5C = O orientation | energy, kcal mol−1 (kJ mol−1) | d, Å (α,°) NH⋯O = C | d, Å (α,°) NH⋯–O– | d, Å (α,°) CH⋯O = C | d, Å (α,°) CH⋯–O– |
|---|---|---|---|---|---|---|
| 3a′ (A) | s-cis, s-cis | 0.00 (0.00) | 2.035 (141.3) | 2.149 (123.4) | ||
| 3a′ (B) | s-trans, s-cis | 0.68 (2.85) | 2.053 (140.1) | 2.170 (115.8) | ||
| 3a′ (C) | s-trans, s-trans | 1.48 (6.19) | 2.065 (139.9) | 2.172 (115.8) | ||
| 3a′ (D) | s-cis, s-cis | 0.72 (3.01) | 2.042 (109.0) | 2.512 (88.8) | ||
| 2.471 (91.0) | ||||||
| 3a′ (E) | s-trans, s-cis | 1.39 (5.82) | 2.031 (109.5) | 2.446 (88.4) | ||
| 2.426 (89.4) | ||||||
| 3a′ (F) | s-trans,s-trans | 2.31 (9.67) | 2.034 (109.5) | 2.482 (86.8) | ||
| 2.408 (90.7) | ||||||
| 5a (A) | s-cis, s-cis | 0.00 (0.00) | 2.163 (130.9) | 2.183 (119.2) | ||
| 2.306 (119.7) | 2.177 (120.9) | |||||
| 5a (B) | s-cis, s-trans | 0.45 (1.88) | 2.195 (128.9) | 2.182 (119.3) | 2.194 (112.8) | |
| 2.289 (120.8) | ||||||
| 5a (C) | s-trans, s-trans | 1.09 (4.56) | 2.171 (130.5) | 2.193 (111.3) | ||
| 2.420 (113.7) | 2.200 (113.2) | |||||
| 5a (D) | s-cis, s-cis | 8.32 (34.81) | 2.058 (107.2) | 2.464 (90.8) | ||
| 2.469 (90.6) | ||||||
| 2.058 (107.2) | 2.501 (89.0) | |||||
| 2.437 (92.3) | ||||||
| 5a (E) | s-cis, s-trans | 9.32 (38.99) | 2.062 (107.3) | 2.468 (90.7) | 2.472 (86.6) | |
| 2.058 (107.2) | 2.468 (90.7) | 2.363 (92.4) | ||||
| 5a (F) | s-trans, s-trans | 10.46 (43.76) | 2.044 (107.9) | 2.410 (89.5) | ||
| 2.408 (89.6) | ||||||
| 2.045 (107.8) | 2.442 (88.0) | |||||
| 2.387 (91.0) |
In compound 3a′ it is possible to form two intramolecular hydrogen bonds of the type NH⋯O: one with the carbonyl oxygen (conformations A, B, C) and the other with the ether oxygen (conformations D, E, F) of the adjacent 2-acetoxymethyl substituent.
The interaction between the NH hydrogen and the carbonyl oxygen of the carboxylic group (conformations A–C) when compared to ones with the alkoxy oxygen (structures D–F) is characterized by the similar geometric and energetic characteristics (table 4.). The most energy preferable conformation of 3a′ is A. But the energy difference of D and A is small and equal to 0.72 kcal mol−1 (3.01 kJ mol−1). The similar values are observed for the energy difference between E and B (0.71 kcal mol−1 (2.97 kJ mol−1)) as well as for F and C (0.83 kcal mol−1 (3.47 kJ mol−1)).
The longer distances dNH⋯O (2.04–2.07 Å) and the larger angles αNH⋯O (139.9–141.3°) in A–C are counterbalanced by the shorter values of dNH⋯O (2.03–2.04 Å) and the smaller values of αNH⋯O (109.0–109.5°) in D–F. It is known that the shorter the hydrogen bond length and the larger angle the stronger the hydrogen bond is. Thus, it can be argued that in 3a′ both NH⋯O = and NH⋯–O– hydrogen bonds are about equally probable.
In 5a the conformations A, B, C in NH⋯O interactions with O=C groups are energetically more preferable than the corresponding D, E, F with NH interacting with alkoxy oxygen. The energy difference is in the interval 8.32–9.37 kcal mol−1 (34.81–39.20 kJ mol−1).
The geometric characteristic of conformations A–C of 5a are close to each other and are characterized by the longer distances dNH⋯O (2.16–2.42 Å) and the larger angles αNH⋯O (113.7–130.9°) as compared to D–F with the shorter values of dNH⋯O (2.04–2.06 Å) and the smaller values of αNH⋯O (107.1–107.9°).
The stability of the NH⋯O hydrogen bond depends on the 3-(5-) side chain orientation and decrease in the following sequence: s-cis, s-cis (A, D); s-cis, s-trans (B, E), s-trans, s-trans (C, F).
The optimal conformations of the side chain 3,5-carbomethoxy groups were shown before to be s-cis, s-cis [9,26]. The obtained results reveal that in 3a’ the energy minima correspond to the conformations with s-cis-s-cis carboxyl group orientation (A, D) relative to the double bond in the dihydropyridine cycle and there is a possibility to form the C2,6H···O bonds with the oxygens of the carboxyl groups at C3 and C5. These additional H-bonds stabilize the molecule, as the structures with s-cis, s-trans (B, E) and s-trans, s-trans (C, F) carboxyl groups orientation have the higher energy.
According to the calculations, the distances d(CH⋯O) increase, and the angles α(C-H⋯O) decrease on going from A–C to D–F, indicating weakening of the CH⋯O hydrogen bonds between the methylene protons and the oxygens of the carboxyl groups. At the same time in conformations A–C only one methylene proton participates in the CH⋯O hydrogen bond, and in D–E conformations both methylene protons form CH⋯O bonds. Although the first one is stronger because it is characterized by the shorter distance dCH⋯O and the larger angle αCH⋯O, two weaker CH⋯O bonds in D–E might have the same stabilizing effect for the molecule optimal conformations. This tendency is true for any orientations of the carbomethoxy groups at C3, C5 positions.
In 5a the optimal conformation A is characterized by s-cis, s-cis O=C 3,5-carboxyl group orientation relative to the double bond in the dihydropyridine cycle and the NH⋯O hydrogen bond with two O=C oxygens (A).
The structures with s-cis-s-trans (B) and s-trans-s-trans (C) orientation of the 3,5-carboxyl groups in 5a similarly to 3a′ have the higher energy.
The calculations of 3,5-side chain conformations of 5a show that, the distances d(CH⋯O) increase, and the angles (C–H⋯O) decrease on going from –-C to D–F, indicating a weakening of the CH⋯O hydrogen bonds between the methylene protons and the oxygens of the carboxyl groups. Similarly to 3a’ in conformations A–C of 5a only one methylene proton participates in the CH⋯O = hydrogen bond, and in D–E conformations of 5a both methylene protons form approximately equal CH⋯–O– bonds. Although the first hydrogen bond is stronger because it is characterized by the shorter distance dCH⋯O and the larger angle αCH⋯O, two weaker CH⋯O bonds in D–E of 5a might have the same stabilizing effect on the optimal conformations of the molecule. This tendency is true for any orientations of the carbomethoxy groups at C3, C5 positions.
In conformations A–C of the compounds 3a′ and 5a, the methylene protons at C2, C6 are nonequivalent, because only one participates in the CH⋯O hydrogen bond. In conformations D–F of 3a′ and 5a, both methylene protons are equivalent because they form two equal CH⋯O hydrogen bonds (table 4). Owing to the energy advantage of the conformation A over D of compounds 5 and energetic equivalence of A and D conformations in 3, the difference of the CS of the methylene protons (δ(HA)-δ(HB)) in 5 is twice as high than in 3.
The calculations performed demonstrate that in case of compounds 3, the NH proton could preferably form a 5-membered H-chelate cycle with the alkoxy oxygen. In compounds 5 two 7-membered H-chelate cycles with the C=O oxygens in 2 and 6 substituents are formed. Different types of the NH⋯O bonds in 3 and 5 can be the reason for the increase of the values of the secondary nΔ13C(NH/D) isotope shifts in 13C NMR spectra on the carboxyl group carbon in substituents at C2,6 on going from 3 to 5. The greater isotope effect of the NH/D substitution through 6 bonds to the 13C CS of the carboxylic carbons in substituents at positions 2 and 6 in compounds 5 (6Δ13C(NH/D)), than through 4 bonds (4Δ13C(NH/D)) in 3 proves that the NH···O hydrogen bond in the seven-membered H-chelate cycle formed by the C=O group is stronger than the one in the five-membered H-chelate formed by the alkoxy group.
4. Conclusion
The 1H,13C,15N NMR, FTIR spectral data and quantum chemical calculations show that alkyl 4-substituted 2-acetoxymethyl- (3) and 2,6-bis-acetoxymethyl-1,4-dihydro-pyridine-3,5-dicarboxylates (5) form four types of the intramolecular H-bonds (NH⋯O and CH⋯O) defining the optimal structure of the molecule.
13C isotope effects induced by N(H/D) substitution registered on the acetoxymethyl carbonyl carbon in compounds 5 are considerably larger than in 3. That demonstrates that the NH proton interacts differently with oxygens in 3 and 5.
In monosubstituted compounds 3, the NH proton preferably forms a five-membered H-chelate cycle with the alkoxy oxygen, while in disubstituted compounds 5, two 7-membered H-chelate cycles with the carbonyl oxygens of the carboxyl groups in 2- and 6-substituents are preferred.
The downfield shift of the 1H NMR signal of the NH proton, accompanied by the upfield shift of the 15N NMR signals and the increase of the 1J(15N,1H) values in the sequence from dimethyl 4-substituted 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1) to 3 and 5, may indicate the shortening of the N–H bond length upon hydrogen bond formation. This is supported by the shift of NH stretching band to the higher wavenumbers in the FTIR spectra of 3 and 5
The optimal conformers of 3 and 5 with the lowest energy have the 3,5-carbonyl groups in s-cis, s-cis position relative to the double bonds of the DHP cycle (C3=C2 and C6=C5). They are stabilized by weak intramolecular hydrogen bonds of the CH⋯O type. The diastereotopic protons of the methylene groups at positions C2 and C6 show unusual temperature dependence due to the changes in the populations of the optimal conformations.
Acknowledgements
We thank Prof. Brenno A. D. Neto (Associate Editor, RSC Advances) who recommended us to submit our paper to Royal Society Open Science. The authors are very grateful to the reviewers for the careful and meticulous reading of the paper and very helpful comments.
Data accessibility
Supporting data are available at Dryad (http://dx.doi.org/10.5061/dryad.j3m73) [36].
Authors' contributions
M.P and E.L. designed the study. B.V. and G.D. prepared all samples for analysis. R.M. and M.P. collected and analysed the data. M.P, R.M, B.V., G.D. and E.L. interpreted the results and wrote the manuscript. All authors gave final approval for publication.
Competing interests
The authors declare no competing interests.
Funding
Financial support came from the Latvian State Research Programme BIOMEDICINE.
References
- 1.Hyvönen Z, Plotniece A, Reine I, Chekavichus B, Duburs G, Urtti A. 2000. Novel cationic amphiphilic 1,4-dihydropyridine derivatives for DNA delivery. Biochim. Biophys. Acta 1509, 451–466. (doi:10.1016/S0005-2736(00)00327-8) [DOI] [PubMed] [Google Scholar]
- 2.Kuhn B, Mohr P, Stahl M. 2010. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 53, 2601–2611. (doi:10.1021/jm100087s) [DOI] [PubMed] [Google Scholar]
- 3.Del Bene JE, Perera SA, Bartlett RJ. 1999. Hydrogen bond types, binding energies, and 1H NMR chemical shifts. J. Phys. Chem. A 103, 8121–8124. (doi:10.1021/jp9920444) [Google Scholar]
- 4.Dziembowska T, Hansen PE, Rozwadowski Z. 2004. Studies based on deuterium isotope effect on 13C chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc. 45, 1–29. (doi:10.1016/j.pnmrs.2004.04.001) [Google Scholar]
- 5.Joseph J, Jemmis ED. 2007. Red-, blue-, or no-shift in hydrogen bonds: a unified explanation. J. Am. Chem. Soc. 129, 4620–4632. (doi:10.1021/ja067545z) [DOI] [PubMed] [Google Scholar]
- 6.Meyer H, Scherling D, Karl W. 1983. Nitrendipine: identification and synthesis of main metabolites. Arzneimittelforschung. 33, 1528–1534. [PubMed] [Google Scholar]
- 7.Chang C-C, et al. 2010. Antagonism of 4-substituted 1,4-dihydropyridine-3,5-dicarboxylates toward voltage-dependent L-type Ca2+ channels CaV1.3 and CaV1.2. Bioorg. Med. Chem. 18, 3147–3158. (doi:10.1016/j.bmc.2010.03.038) [DOI] [PubMed] [Google Scholar]
- 8.Petrova M, Muhamadejev R, Cekavicus B, Vigante B, Plotniece A, Sobolev A, Duburs G, Liepinsh E. 2014. Experimental and theoretical studies of bromination of diethyl 2,4,6-trimethyl-1,4-dihydropyridine-3,5-dicarboxylate. Heteroat. Chem. 25, 114–126. (doi:10.1002/hc.21145) [Google Scholar]
- 9.Petrova M, Muhamadejev R, Vigante B, Cekavicus B, Plotniece A, Duburs G, Liepinsh E. 2011. Intramolecular C-H⋯O hydrogen bonding in 1,4-dihydropyridine derivatives. Molecules 16, 8041–8052. (doi:10.3390/molecules16098041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bossert FDCD, Wehinger EDCD, Meyer HDCD, Heise AD, Kazda SD, Stoepel KD, Towart RD, Vater WD, Schlossmann KDCD. 1978. In 2-position substituierte 1,4- dihydropyridin-derivate, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel In the 2-position substituted 1,4-dihydropyridine derivatives, process for their production and their use as drug. Patent no. DE2658183A1.
- 11.Satoh Y, Ichihashi M, Okumura K. 1991. Studies on Nilvadipine. I. Synthesis and structure-activity relationships of 1,4-Dihydropyridines containing novel substituents at the 2-Position. Chem. Pharm. Bull. (Tokyo) 39, 3189–3201. (doi:10.1248/cpb.39.3189) [DOI] [PubMed] [Google Scholar]
- 12.Frisch MJ, et al. 2013. Gaussian 09, revision D.01. Gaussian 09, revis. Wallingford, CT: D.01, Gaussian, Inc. [Google Scholar]
- 13.Grimme S, Ehrlich S, Goerigk L. 2011. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465. (doi:10.1002/jcc.21759) [DOI] [PubMed] [Google Scholar]
- 14.Petrova M, Muhamadejev R, Chesnokov A, Vigante B, Cekavicus B, Plotniece A, Duburs G, Liepinsh E. 2014. Spectral and quantum-chemical study of nonequivalence of methylene protons in 1,4-Dihydropyridine derivatives*. Chem. Heterocycl. Compd 49, 1631–1639. (doi:10.1007/s10593-014-1414-6) [Google Scholar]
- 15.Günther H. 2013. NMR spectroscopy: basic principles, concepts and applications in chemistry. New York, NY: John Wiley & Sons. [Google Scholar]
- 16.Marshall JL. 1983. Carbon-Carbon and carbon-proton Nmr couplings: applications to organic stereochemistry and conformational analysis. Deerfield Beach, FL: Verlag Chemie International. [Google Scholar]
- 17.Becker ED. 2007. Hydrogen bonding. In Encyclopedia of Magnetic Resonance, Chichester, UK: John Wiley & Sons, Ltd. [Google Scholar]
- 18.Gellman SH, Dado GP, Liang GB, Adams BR. 1991. Conformation-directing effects of a single intramolecular amide-amide hydrogen bond: variable-temperature NMR and IR studies on a homologous diamide series. J. Am. Chem. Soc. 113, 1164–1173. (doi:10.1021/ja00004a016) [Google Scholar]
- 19.Baxter NJ, Williamson MP. 1997. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369. (doi:10.1023/A:1018334207887) [DOI] [PubMed] [Google Scholar]
- 20.Dingley AJ, Masse JE, Peterson RD, Barfield M, Feigon J, Grzesiek S. 1999. Internucleotide scalar couplings across hydrogen bonds in Watson–Crick and Hoogsteen base pairs of a DNA triplex. J. Am. Chem. Soc. 121, 6019–6027. (doi:10.1021/ja9908321) [Google Scholar]
- 21.Ishikawa R, Kojima C, Ono A, Kainosho M. 2001. Developing model systems for the NMR study of substituent effects on the N-H⋯N hydrogen bond in duplex DNA. Magn. Reson. Chem. 39, S159–S165. (doi:10.1002/mrc.941) [DOI] [PubMed] [Google Scholar]
- 22.Del Bene JE, Elguero J. 2006. Systematic ab Initio Study of 15 N– 15 N and 15 N– 1H spin–spin coupling constants across N–H+ –N hydrogen bonds: predicting N–N and N–H coupling constants and relating them to hydrogen bond type. J. Phys. Chem. A 110, 7496–7502. (doi:10.1021/jp0613642) [DOI] [PubMed] [Google Scholar]
- 23.Contreras R. 2000. Angular dependence of spin–spin coupling constants. Prog. Nucl. Magn. Reson. Spectrosc. 37, 321–425. (doi:10.1016/S0079-6565(00)00027-3) [Google Scholar]
- 24.Hobza P, Havlas Z. 2000. Blue-shifting hydrogen bonds. Chem. Rev. 100, 4253–4264. (doi:10.1021/cr990050q) [DOI] [PubMed] [Google Scholar]
- 25.Contreras RH, Peralta JE, Giribet CG, Ruiz de azúa MC, Facelli JC. 2000. Advances in theoretical and physical aspects of spin-spin coupling constants. Ann. Rep. NMR Spectrosc 41, 55–184. (doi:10.1016/S0066-4103(00)41009-4) [Google Scholar]
- 26.Goba I, Turovska B, Belyakov S, Liepinsh E. 2014. Synthesis, spectroscopic and conformational analysis of 1,4-dihydroisonicotinic acid derivatives. J. Mol. Struct. 1074, 549–558. (doi:10.1016/j.molstruc.2014.06.044) [Google Scholar]
- 27.Goba I, Liepinsh E. 2013. 15 N NMR of 1,4-dihydropyridine derivatives. Magn. Reson. Chem. 51, 391–396. (doi:10.1002/mrc.3959) [DOI] [PubMed] [Google Scholar]
- 28.Kyogoku Y. 1981. Application of 15 N NMR spectroscopy to studies of the intermolecular interaction of biomolecules. Appl. Spectrosc. Rev. 17, 279–335. (doi:10.1080/05704928108060407) [Google Scholar]
- 29.Bagheri S, Masoodi HR, Abadi MN. 2015. Estimation of individual NH⋯X (X = N, O) hydrogen bonding energies in some complexes involving multiple hydrogen bonds using NBO calculations. Theor. Chem. Acc. 134, 127 (doi:10.1007/s00214-015-1738-z) [Google Scholar]
- 30.Kuroki S, Ando S, Ando I, Shoji A, Ozaki T, Webb GA. 1990. Hydrogen-bonding effect on 15N NMR chemical shifts of the glycine residue of oligopeptides in the solid state as studied by high-resolution solid-state NMR spectroscopy. J. Mol. Struct. 240, 19–29. (doi:10.1016/0022-2860(90)80492-3) [Google Scholar]
- 31.Witanowski M, Stefaniak L, Webb GA.. 1982. Nitrogen NMR Spectroscopy. In Annual reports on NMR spectroscopy (ed. Webb GA.), pp. 1–486. Cambridge, MA: Academic Press. [Google Scholar]
- 32.Suárez M, Molero D, Salfrán E, Rodríguez H, Coro J, Sáez E, Martínez-Álvarez R, Martín N. 2011. NMR study of 1,4-dihydropyridine derivatives endowed with long alkyl and functionalized chains. J. Braz. Chem. Soc. 22, 166–171. (doi:10.1590/S0103-50532011000100022) [Google Scholar]
- 33.Liepin'sh EE, Zolotoyabko RM, Chekavichus BS, Sausin’ AE, Lusis VK, Dubur GY. 1989. 13C-NMR spectra of substituted 1,4-dihydropyridines. Chem. Heterocycl. Compd. 25, 1032–1037. (doi:10.1007/BF00487304) [Google Scholar]
- 34.Sobczyk L, Obrzud M, Filarowski A. 2013. H/D isotope effects in hydrogen bonded systems. Molecules 18, 4467–4476. (doi:10.3390/molecules18044467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buncel E, Jones JR.. 1991. Isotopes in the physical and biomedical sciences: isotopic applications in NMR studies. Amsterdam, The Netherlands: Elsevier Science Publishers. [Google Scholar]
- 36.Petrova M, Muhamadejev R, Vigante B, Duburs G, Liepinsh E. 2018. Data from: Intramolecular hydrogen bonds in 1,4-dihydropyridine derivatives Dryad Digital Repository. (http://dx.doi.org/10.5061/dryad.j3m73) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Petrova M, Muhamadejev R, Vigante B, Duburs G, Liepinsh E. 2018. Data from: Intramolecular hydrogen bonds in 1,4-dihydropyridine derivatives Dryad Digital Repository. (http://dx.doi.org/10.5061/dryad.j3m73) [DOI] [PMC free article] [PubMed]
Data Availability Statement
Supporting data are available at Dryad (http://dx.doi.org/10.5061/dryad.j3m73) [36].