

Abstract
KRAS is the most common oncogenic driver in lung adenocarcinoma (LUAC). We previously reported that STK11/LKB1 (KL) or TP53 (KP) co-mutations define distinct subgroups of KRAS-mutant LUAC. Here, we examine the efficacy of PD-1 inhibitors in these subgroups. Objective response rates to PD-1 blockade differed significantly among KL (7.4%), KP (35.7%), and K-only (28.6%) subgroups (P<0.001) in the SU2C cohort (174 patients) with KRAS-mutant LUAC and in patients treated with nivolumab in the CheckMate-057 phase 3 trial (0% vs 57.1% vs 18.2%, P=0.047). In the SU2C cohort, KL LUAC exhibited shorter progression-free (P<0.001) and overall survival (P=0.0015) compared to KRASMUT;STK11/LKB1WT LUAC. Among 924 LUAC, STK11/LKB1 alterations were the only marker significantly associated with PD-L1 negativity in TMBIntermediate/High LUAC. The impact of STK11/LKB1 alterations on clinical outcomes with PD-1/PD-L1 inhibitors extended to PD-L1-positive NSCLC. In Kras-mutant murine LUAC models, Stk11/Lkb1 loss promoted PD-1/PD-L1 inhibitor resistance, suggesting a causal role. Our results identify STK11/LKB1 alterations as a major driver of primary resistance to PD-1 blockade in KRAS-mutant LUAC.
Keywords: STK11/LKB1, KRAS, PD-1 blockade, lung adenocarcinoma, non-small cell lung cancer
Introduction
Despite improvements in overall survival and clinical responses of unprecedented duration with the use of therapeutic monoclonal antibodies that target programmed cell death −1 (PD-1) or programmed cell death −1 ligand (PD-L1), the majority of patients with non-small cell lung cancer (NSCLC) fail to respond to PD-1/PD-L1 axis inhibitors (1–8). The landscape of primary resistance to PD-1 blockade in NSCLC is largely unknown, with no single factor capable of accurately segregating responders from non-responders. Expression of PD-L1 on the membrane of tumor and immune cells is associated with enhanced objective response rates to PD-1/PD-L1 inhibition, but is neither sensitive nor specific (1–3, 7, 9–12). A higher burden of non-synonymous somatic mutations (tumor mutation burden; TMB) further correlates with increased likelihood of clinical benefit and is undergoing evaluation as a predictive biomarker in many tumor types (4, 13–15).
KRAS mutations are the most prevalent oncogenic driver in NSCLC, accounting for ~25% of LUAC (16, 17). We previously reported that co-occurring genomic alterations in the STK11/LKB1 (KL) and TP53 (KP) tumor suppressor genes define subgroups of KRAS-mutant LUAC with distinct biology, therapeutic vulnerabilities and immune profiles (18). STK11/LKB1 encodes a serine threonine kinase with an established role in the regulation of cellular metabolism/energy homeostasis, growth and polarity through phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) and 12 AMPK-related kinases (19). Inactivation of STK11 (or its protein product, LKB1) by mutational or non-mutational mechanisms is associated with an inert or “cold” tumor immune microenvironment, with reduced density of infiltrating cytotoxic CD8+ T lymphocytes in both human tumors and genetically engineered murine models (18, 20, 21). Based on these findings, we hypothesized that STK11/LKB1 genomic alterations may predict for lack of clinical benefit from PD-1/PD-L1 blockade in KRAS-mutant LUAC and conducted a study to address this hypothesis and examine the inter-relationship between individual genetic alterations, TMB and PD-L1 expression.
Results
Patient characteristics
174 patients that met the pre-specified eligibility criteria were included in the SU2C dataset [MDACC (N=62), MSKCC (N=56), DFCI/MGH (N=56)] (Table 1). The overall cohort was representative of the general population of patients with KRAS-mutant LUAC with median patient age of 66 years (range 42-87), high percentage of current/former smokers (88.5%) and typical frequencies of distinct KRAS-mutant alleles (Figure S1A and S1B) (16–18, 22). Across the entire cohort, 31 % of tumors were classified as KL, 32% were KP and 37% K-only (Figure S1C). The majority of patients received PD-1 inhibitor monotherapy (165/174, 95%) and the remainder received combination with CTLA-4 blockade (9/174, 5%) (Figure S1A). Demographic and clinical characteristics were generally well balanced between the co-mutation defined subgroups (Figure S1A and Figure S1D). STK11/LKB1 mutations were in their overwhelming majority predicted to be deleterious (Figure S2).
Table 1.
Clinical cohorts included in the study.
| Cohort | Foundation Medicine (FM) | Stand Up to Cancer (SU2C) | Checkmate-057 (CM-057) |
MDACC (PD-L1≥1%) |
||
|---|---|---|---|---|---|---|
| N | MDACC | MSKCC | DFCI/MGH | |||
| 924 (346 KRASMUT) | 174 | 44 | 66 | |||
| 62 | 56 | 56 | ||||
| Nivolumab | NA | 146 | 24 | 16 | ||
| Pembrolizumab | NA | 19 | NA | 40 | ||
| Atezolizumab | NA | 0 | NA | 5 | ||
| a-PD(L)-1/a-CTLA-4 | NA | 9 | NA | 3 | ||
| Docetaxel | NA | NA | 20 | NA | ||
| Other | NA | NA | NA | 2* | ||
One patient with STK11/LKB1 mutant tumor was treated with Nivolumab and NKTR-214 (CD122-based agonist) and one patient with STK11/LKB1 wild-type tumor was treated with pembrolizumab and OX40 agonist.
Co-mutations in STK11/LKB1 are associated with inferior clinical outcome with PD-1 blockade in multiple independent cohorts of KRAS-mutant LUAC
The objective response rates to PD-1 inhibition in KL, KP and K-only groups were significantly different (P<0.001, Fisher’s exact test) (Figure 1A and Figure 1C). KL tumors were mostly resistant to PD-1 axis blockade (ORR 7.4% overall), with consistently low response rates seen in each of the three independent datasets (MDACC: 9.1%, MSKCC: 9.1%, DFCI/MGH: 4.8%). In contrast, KP LUAC were more sensitive to PD-1 inhibitors (ORR 35.7% overall). K-only tumors with no identifiable mutations in either STK11/LKB1 or TP53 had an intermediate response rate (28.6%). Assessment of additional co-occurring genetic alterations in the few KL tumors that responded to PD-1 blockade did not identify any obvious unifying molecular features (Figure S3).
Figure 1. STK11/LKB1 co-mutations are associated with inferior objective response rate with PD-1 blockade in KRAS-mutant LUAC.
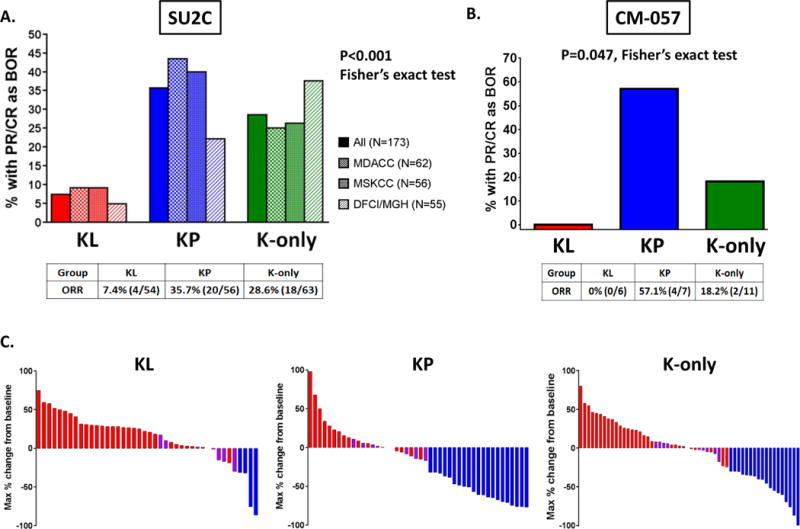
A. Objective response rate (RECISTv1.1) to PD-1 axis blockade in the KL, KP and K-only subgroups in the overall SU2C population (N=173 response-evaluable patients) and in each of the three independent cohorts (MDACC, MSKCC, DFCI/MGH). A two-tailed Fisher’s exact test (computed from a 2×3 contingency table) was used to assess the significance of the association between group membership and best overall response (PR/CR vs SD/PD). B. Objective response rate to nivolumab in the KL, KP and K-only subgroups in the CheckMate-057 international randomized phase III clinical trial (N=24). A two-tailed Fisher’s exact test (computed from a 2×3 contingency table) was used to assess the significance of the association between group membership and best overall response (PR/CR vs SD/PD). C. Waterfall plots illustrating individual patient-level maximal % change in tumor burden from baseline in response to PD-1/PD-L1 inhibition in the SU2C cohort. Only data from response-evaluable patients with measurable disease are graphed.
In order to replicate these findings in the context of a randomized clinical trial, we further analyzed the impact of STK11/LKB1 and TP53 genetic alterations on clinical outcomes in 44 patients with KRAS-mutant NSCLC (96% LUAC) with available WES data that were randomly assigned to treatment with nivolumab (N=24) or docetaxel (N=20) in the CheckMate-057 randomized phase 3 clinical trial (NCT01673867). In agreement with data from the SU2C cohort, ORR differed significantly between the KL, KP and K-only subgroups in the nivolumab arm of CM-057 (P=0.047), with KL tumors being refractory (ORR: 0%, 0/6) and KP more sensitive (ORR: 57.1%, 4/7) to nivolumab (Figure 1B). Although ORR did not differ significantly between the three subgroups in the docetaxel arm (P=0.65), it is relevant to note that the ORR in the KL subgroup was 0% (0/3) [ORR was 0% (0/6) and 18.2% (2/11) in KP and K only subgroups, respectively]. Given the relatively small numbers within subgroups, it cannot be determined whether STK11/LKB1 mutation is prognostic or predictive of treatment outcomes in the CM-057 dataset.
Progression-free survival differed between the three groups in the SU2C cohort (P=0.0018), with significantly shorter PFS for patients with KL compared to either KP (HR 1.77, 95% CI 1.16-2.69; P=0.0072) or K-only tumors (HR 1.98, 95% CI 1.33-2.94; P<0.001) in pair-wise comparisons (Figure 2A, left panel). In contrast, patients with KP and K-only tumors had similar PFS. Because STK11/LKB1 abrogation likely determines immunotherapy resistance in this context, we further compared PFS in patients with STK11/LKB1 wild-type and mutant tumors by merging the KP and K-only cohorts. PFS was significantly shorter in KL tumors compared to KRAS-mutant LUAC with wild-type STK11/LKB1 (HR 1.87, 95% CI 1.32 to 2.66; P<0.001) (Figure 2A, right panel). The CM-057 study had limited power to detect PFS or OS differences due to the small size of subgroup cohorts and no significant differences were seen in PFS or OS in either arm (Figure S4 and Figure S5).
Figure 2. STK11/LKB1 genetic alterations are associated with shorter progression-free and overall survival with PD-1 blockade among KRAS-mutant LUAC in the SU2C cohort.
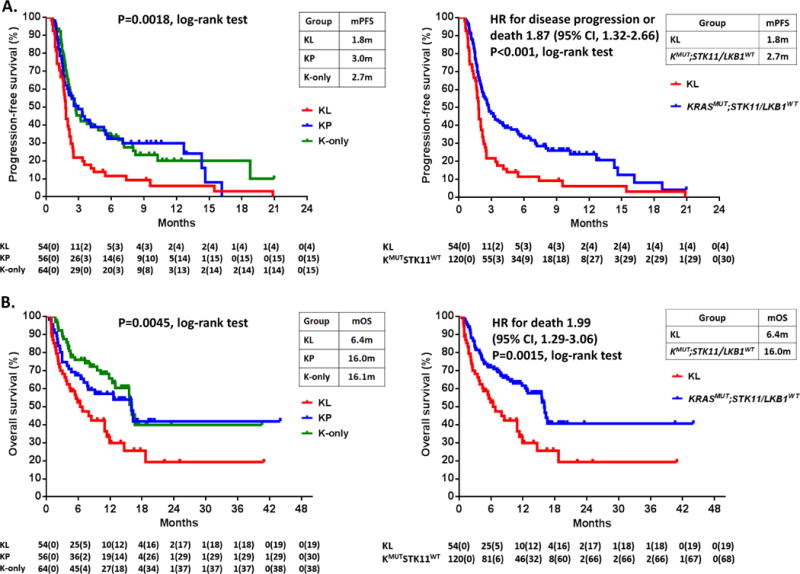
A. Kaplan-Meier estimates of progression-free survival with PD-1 blockade in the KL, KP, K-only subgroups (left panel) and in the two-group comparison between KRASMUT;STK11/LKB1MUT (KL) and KRASMUT;STK11/LKB1WT LUAC (encompassing KP and K-only tumors) (right panel). Tick marks represent data censored at the last time the patient was known to be alive and without disease progression (date of last radiological assessment). B. Kaplan-Meier estimates of overall survival with PD-1 inhibitors in the KL, KP, K-only subgroups (left panel) and in the two group comparison between KRASMUT;STK11MUT (KL) and KRASMUT;STK11/LKB1WT tumors (right panel). Tick marks represent data censored at the last time the patient was known to be alive.
Overall survival also varied significantly between the three groups in the SU2C cohort (P=0.0045) (Figure 2B, left panel). Median overall survival was 6.4 months in KL compared with 16.0 months in KP and 16.1 months in K-only LUACs. In the two group comparison, overall survival was significantly shorter in STK11/LKB1-mutant compared to wild-type tumors (HR 1.99, 95% CI 1.29 to 3.06; P=0.0015) (Figure 2B, right panel). KRAS subgroup remained a significant independent predictor of OS on multivariate analysis (P=0.00055). Notably, STK11/LKB1 mutation or deficiency were not associated with worse OS in the TCGA cohort, arguing against a purely prognostic role for STK11/LKB1 inactivation in this setting of predominantly early stage, surgically resected tumors (Figure S6), in agreement with previous studies in metastatic tumors (23–25).
Because non-mutational mechanisms can also account for STK11/LKB1 inactivation in LUAC (19), we further assessed expression of LKB1 (the protein product of the STK11 gene) by IHC in a subset of tumors for which archival tissue was available (26). KRASMUT;STK11/LKB1MUT (KL) tumors expressed low to undetectable levels of LKB1 whereas KRASMUT;STK11/LKB1WT tumors displayed variable levels of LKB1 expression, with 17.6% having a LKB1 H-score of zero (Figure 3A). Patients bearing STK11/LKB1-deficient tumors (STK11/LKB1MUTor STK11/LKB1WT and LKB1 H-score zero) exhibited significantly shorter PFS (HR 1.80, 95%CI 1.15-2.82; P=0.0094) (Figure 3B, left panel) and OS (HR 2.03, 95% CI 1.13-3.65; P=0.016) (Figure 3B, right panel) compared to those harboring STK11/LKB1-proficient tumors (STK11/LKB1WT and LKB1 H-score>0).
Figure 3. LKB1 expression by IHC can identify LKB1-deficient LUAC in the absence of STK11/LKB1 alterations.
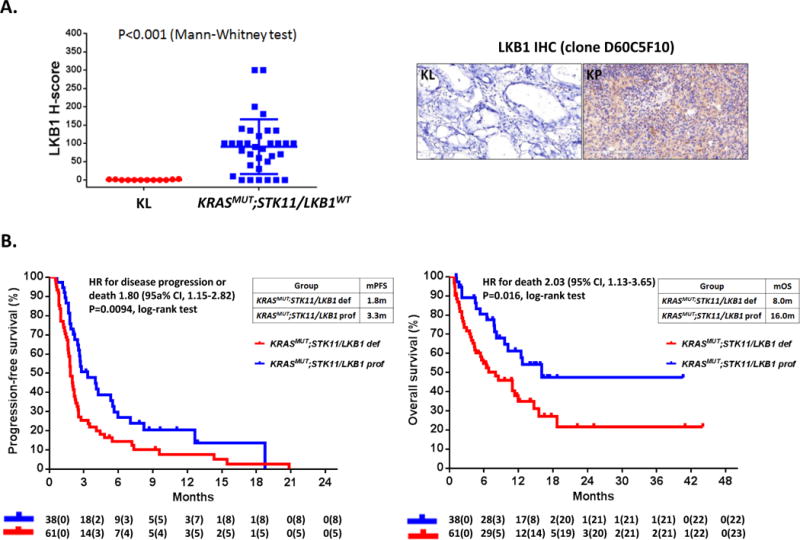
A. LKB1 IHC expression (H-score) in KRASMUT;STK11/LKB1MUT (KL) and KRASMUT;STK11/LKB1WT LUAC. Quantitative IHC using a commercially available LKB1 rabbit monoclonal antibody (clone D60C5F10, Cell Signaling Technology) is technically robust and can identify LKB1-deficient tumors with intact STK11/LKB1 genomic locus (26). (Left panel) KL LUAC (N=12) exhibit absent or minimal cytoplasmic LKB1 staining, whereas KRASMUT;STK11/LKB1WT LUAC (N=34) display variable LKB1 H-score. LUAC were therefore considered LKB1-proficient if they had intact STK11/LKB1 locus and expressed LKB1 by IHC at any level (LKB1 H-score > 0) and LKB1-deficient if they were STK11/LKB1-altered and/or exhibited LKB1 H-score = 0. Representative images of KL and KP LUAC immuno-stained for LKB1 are included (right panel). Staining was performed as previously described (26). B. Kaplan-Meier estimates of progression-free survival (left panel) and overall survival (right panel) with PD-1 blockade in LKB1-deficient (STK11/LKB1-mutant and/or LKB1 H-score = 0; N=61) and LKB1-proficient (STK11/LKB1-wild-type and LKB1 H-score > 0; N=38) KRAS-mutant LUAC.
STK11/LKB1 mutations are significantly enriched among TMB intermediate/high, PD-L1 – negative tumors
In a parallel, unbiased analysis, we sought to identify candidate genomic drivers of absent PD-L1 expression (as an indicator of a “cold” or non-T-cell inflamed immune microenvironment) in LUAC using the large FM dataset (Figure S7). We focused on TMB intermediate and high (TMBI/H) tumors and excluded TMB low (TMBL) LUAC since low TMB has been associated with impaired response to PD-1 axis inhibitors in retrospective studies, likely due to poor tumor immunogenicity (4, 13). We then compared the prevalence of individual genomic alterations in PD-L1 negative (PD-L1Neg; TMBI/H) vs high positive (PD-L1HP; TMBI/H) tumors (Figure 4A). This analysis identified STK11/LKB1 as the only significantly enriched gene in the PD-L1 negative group (adjusted P<0.001). Further interrogation of the PD-L1/TMB landscape indicated that STK11/LKB1 alterations are most prominently enriched in TMBI; PD-L1Neg samples and LUAC bearing STK11/LKB1 alterations are less likely to be either PD-L1HP or TMBL (Figure 4B). Furthermore, STK11/LKB1 was significantly enriched (P<0.001) for PD-L1Neg; TMBI/H negative status even when the analysis was restricted to KRAS-mutant samples. Thus, we conclude that STK11/LKB1 is associated with higher likelihood of absent PD-L1 expression among TMBI/H tumors irrespective of KRAS status.
Figure 4. STK11/LKB1 genomic alterations are enriched in LUAC with intermediate or high TMB that are negative for PD-L1 expression.
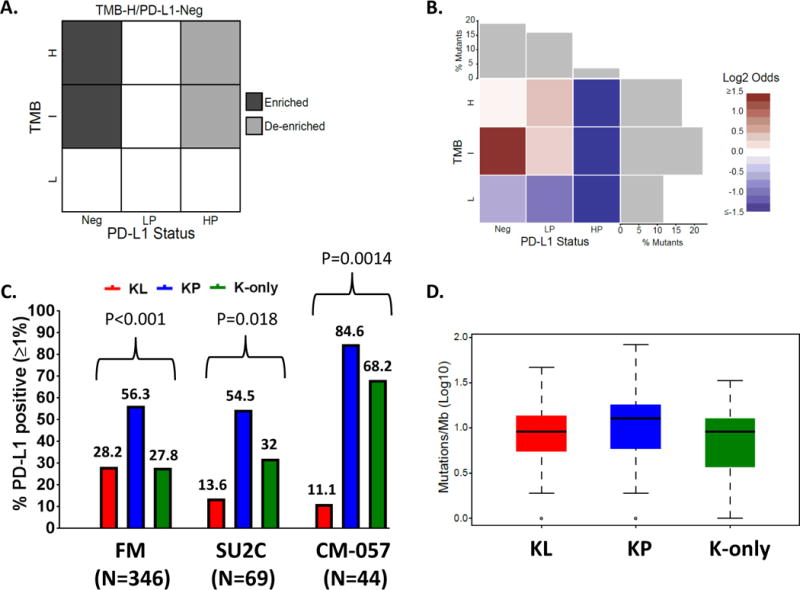
(A) PD-L1/TMB landscape matrix illustrating the enrichment analysis strategy in 924 LUAC samples with available CGP and PD-L1 expression (FM cohort). Enrichment of individual genomic alterations in PD-L1Neg; TMBI/H vs PD-L1HP; TMBI/H tumors was assessed using a one-sided Fisher’s exact test. (B) Heatmap of log-odds values reflecting the prevalence of STK11/LKB1 alterations in different cells of the PD-L1/TMB matrix. Alterations in STK11/LKB1 primarily cluster in TMBI;PD-L1Neg LUAC. (C) PD-L1 expression in the KL, KP and K-only subgroups in the FM (N=346), SU2C (N=69) and CM-057 (N=44) cohorts. A two-tailed Fisher’s exact test (computed from a 2×3 contingency table) was used to assess the significance of the association between group membership and PD-L1 expression status [PD-L1 positive (≥1%) or negative (0%)]. (D) TMB (Log10) in the KL, KP and K-only subgroups among 346 KRAS-mutant LUAC in the FM cohort.
We further analyzed PD-L1 expression and TMB in the KL/KP/K-only subgroups and their KRAS wild-type counterparts (Figure 4C and Figure S8). PD-L1 expression varied significantly between the KRAS sub-groups (Figure 4C, P < 0.001), with KL least likely to be PD-L1HP (P < 0.001) (Figure S8). Among KRAS wild-type tumors STK11/LKB1 alterations were also associated with lower likelihood of high PD-L1 expression (Figure S8). KP LUAC exhibited the highest rate of PD-L1 positivity (56.3% PD-L1-positive, 31.3% PD-L1HP) followed by the TP53-altered, KRAS-wild-type group (32.3% PD-L1-positive, 11% PD-L1HP) (Figure S8). In contrast, median TMBs across samples with KRAS, STK11/LKB1or TP53 alterations were comparable, ranging between 8.1 and 11.7 mutations/Mb (Figure 4D and Figure S8).
Consistent with the observed association between STK11/LKB1 genomic alterations and low PD-L1 expression in the FM cohort, significant difference in the rate of PD-L1 positivity (PD-L1≥1%) was also noted between KL, KP and K-only tumors in the SU2C and CheckMate-057 cohorts (Figure 4C and Figure S9), with KL exhibiting the lowest frequency of PD-L1-positive (13.6% in the SU2C and 11.1% in the CM-057 cohort) and PD-L1HP tumors (0% in both cohorts) (Figure 4C and Figure S9).
We further directly interrogated the composition of the tumor immune microenvironment in surgically resected LUAC specimens (PROSPECT cohort) with available whole exome sequencing and automated quantitative IHC-based immune profiling (27, 28). In agreement with lower tumor cell PD-L1 expression, STK11/LKB1-mutated tumors exhibited lower densities of infiltrating CD3+ (P=0.0019) and CD8+ (P=0.0072) T-lymphocytes but not FOXP3+ cells (P=0.7648) (Figure S10A). Furthermore, in the TCGA dataset STK11/LKB1-mutated LUAC (or STK11/LKB1-deficient LUAC as determined using a previously validated gene expression signature (29)) exhibited lower T-cell signature scores (30) (Figure S10B) and expressed lower PD-L1 (CD274) mRNA levels (Figure S10C). Thus, we provide compelling evidence from multiple independent cohorts that STK11/LKB1 genomic alterations are associated with, and may actually promote, a non-T-cell-inflamed immune microenvironment with lack of tumor cell PD-L1 expression, despite an intermediate or high TMB.
STK11/LKB1 genomic alterations are associated with primary resistance to PD-1 axis inhibitors in PD-L1 positive NSCLC
In view of the strong association between STK11/LKB1 genomic alterations and lack of PD-L1 expression on tumor cells, we also sought to examine the impact of STK11/LKB1 mutations on clinical responses to PD-1 axis blockade in PD-L1 positive (≥1%) non-squamous NSCLC. For this analysis, we identified a distinct cohort of 66 non-squamous NSCLC patients (irrespective of KRAS status) treated with PD-1/PD-L1 inhibitors at MDACC, with available STK11/LKB1 genomic profiling and PD-L1 expression (assessed using the FDA-approved 22C3 pharmDx assay). Within this PD-L1-positive group, STK11/LKB1-mutated tumors exhibited significantly lower ORR to PD-1/PD-L1 blockade compared to NSCLC with intact STK11/LKB1 (ORR 0% vs 34.5%, P=0.026, Fisher’s exact test) (Figure 5A), despite inclusion of PD-L1 high-expressing tumors in the STK11/LKB1-mutant group (Figure 5B). Importantly, STK11/LKB1 mutations were associated with dramatically shorter PFS (HR 4.76, 95%CI 2.0-11.1, P=0.00012, log rank test) and OS (HR 14.3, 95% CI 3.4-50.0, P<0.0001, log-rank test) with PD-1 axis blockade (Figure 5C and Figure 5D). The effect of STK11/LKB1 genomic alterations on PFS and OS with PD-1/PD-L1 blockade did not differ significantly across PD-L1 high (PD-L1≥50%) and low (PD-L1<50%) expressing groups (interaction test; P=0.48 for PFS and P=0.59 for OS) (Figure S11). We therefore conclude that STK11/LKB1 genomic alterations impact response to PD-1/PD-L1 blockade at least partially independently of PD-L1 status and that their effect likely extends to the entire population of non-squamous NSCLC regardless of KRAS status. Extension of the effect of STK11/LKB1 inactivation to the broader population of non-squamous NSCLC is further supported by data from a separate cohort of TMBI/H non-squamous NSCLC patients (without available PD-L1 expression) treated with anti-PD-1/PD-L1 therapy, whereby STK11/LKB1 alterations (regardless of KRAS status) were associated with significantly shorter time on drug (HR 2.91, 95% CI 1.22-6.92; P=0.0156) (Figure S12).
Figure 5. STK11/LKB1 mutations are a genomic determinant of poor clinical outcome with PD-1 axis blockade in PD-L1 positive non-squamous NSCLC, regardless of KRAS status.
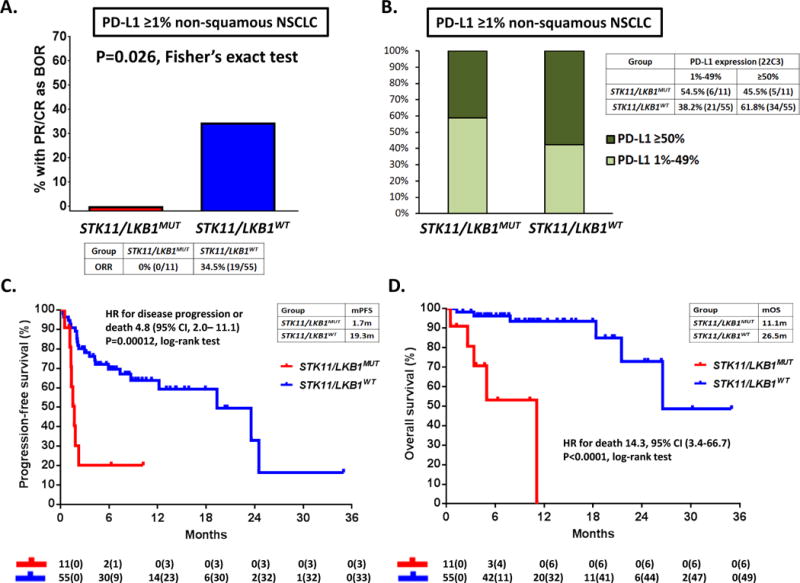
A. Objective response rate (RECISTv1.1) to PD-1/PD-L1 inhibitors in STK11/LKB1-mutant and wild-type patients with PD-L1-positive non-squamous NSCLC (≥1%) from MDACC (N=66). PD-L1 expression was assessed using the FDA-approved 22C3 pharmDx assay (Dako). A two-tailed Fisher’s exact test (computed from a 2×2 contingency table) was used to assess the significance of the association between group membership (STK11/LKB1-mutant versus STK11/LKB1 -wild-type) and best overall response (PR/CR vs SD/PD). B. Fractions of PD-L1 low-positive (1%-49%) and PD-L1 high-positive (≥50%) tumors in the STK11/LKB1-mutant and wild-type groups. C. Kaplan-Meier estimates of progression-free survival with PD-1/PD-L1 blockade in STK11/LKB1-mutant and wild-type groups. Tick marks represent data censored at the last time the patient was known to be alive and without disease progression (date of last radiological assessment). D. Kaplan-Meier estimates of overall survival with PD-1 inhibitors in the STK11/LKB1-mutant and wild-type groups. Tick marks represent data censored at the last time the patient was known to be alive.
TP53 co-mutations may impact response to PD-1 inhibitors in PD-L1 negative KRAS-mutant LUAC
As part of an exploratory analysis we interrogated the impact of KRAS co-mutations on clinical benefit from PD-1 blockade in PD-L1-negative tumors. Among PD-L1-negative KRAS-mutant LUAC in the SU2C cohort (n=46), DCR differed significantly between the subgroups (P=0.034) and was highest (70%) in KP tumors (Figure S13). The difference in ORR also favored the KP subgroup (30%), but did not reach statistical significance (P=0.11, Figure S13). This result further supports the notion that the predictive utility of co-mutations may extend beyond that of PD-L1 expression.
Stk11/Lkb1 ablation induces de novo resistance to PD-1 blockade in a syngeneic murine model of KRAS-mutant LUAC
In order to establish whether primary resistance to immunotherapy is causally linked with STK11/LKB1 inactivation we generated Stk11/Lkb1 proficient/deficient isogenic derivatives of the LKR13 Kras-mutant murine LUAC cell line (previously established from a spontaneously arising LUAC in the KrasLA1/+ model) using CRISPR/Cas9-mediated bi-allelic disruption of the Stk11/Lkb1 locus. Upon confirmation of Stk11/Lkb1 knockout (by immunoblotting for the LKB1 protein) (Figure S14) isogenic cell lines were implanted into the right flank of syngeneic recipient mice and cohorts of tumor-bearing mice were randomized to treatment with anti-PD-L1 mAb or IgG control. Treatment with anti-PD-L1 monoclonal antibody potently suppressed LKR13-derived tumors but growth of Stk11/Lkb1-deficient LKR13KO continued unabated (Figure 6A). In agreement with findings in human STK11/LKB1 deficient tumors, lower numbers of CD3+CD8+ and CD3+CD8+/PD1+ T-lymphocytes were present in Stk11/Lkb1-deficient LKR13KO tumors compared to their Stk11/Lkb1-proficient counterparts, whereas numbers of CD45+ and CD3+CD4+ cells were not significantly different (Figure S15). Furthermore, we did not observe enrichment of tumor associated neutrophils in the microenvironment of Stk11/Lkb1-deficient tumors in this model (Figure S15), contrary to a previous report (20). Similar results were obtained using a second syngeneic tumor model based on the LKR10 Kras-mutant murine LUAC cell line in response to treatment with anti-PD-1 mAb or isotype control (Figure 6B). Thus, Stk11/Lkb1 loss directly promotes primary resistance to PD-1/PD-L1 blockade and fosters establishment of a non-T-cell-inflamed tumor immune microenvironment in immune competent murine models of KRAS-mutant LUAC.
Figure 6. Stk11 ablation directly promotes primary resistance to PD-L1/PD-1 blockade in immune-competent murine models of Kras-mutant LUAC.
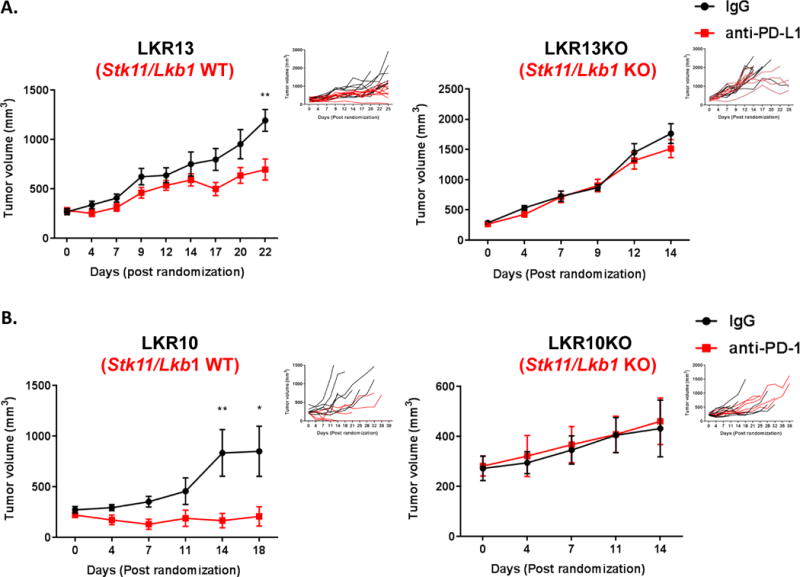
Stk11-proficient/deficient isogenic derivatives of the LKR13 (A) and LKR10 (B) cell lines were used in preclinical experiments. Changes in mean (main panels) and individual (insert panels, “spider plots”) subcutaneous tumor volume following treatment with (A) anti-PD-L1 (mIgG1-D265AFc clone 80) or IgG control antibody (LKR13/LKR13KO isogenic pair) and (B) anti-PD-1 monoclonal antibody (clone RMPI-14; BioXCell) or isotype control antibody (clone 2A3; BioXCell) (LKR10/LKR10KO isogenic pair) are graphed. Error bars represent standard error of the mean. Mean tumor volume plots are depicted from the time of randomization to the time that the first mouse in any of the two treatment arms was sacrificed. Spider plots indicate individual tumor volume trajectories for the entire duration of the in vivo experiment (25 days for the LKR13/LKR13KO and 39 days for the LKR10/LKR10KO model). Note that PD-1/PD-L1 blockade blunts the in vivo growth of Stk11/Lkb1-proficient Kras-mutant LUAC, whereas Stk11/Lkb1 knockout renders tumors recalcitrant to PD-1/PD-L1 inhibition. The Mann-Whitney U test was used to compare mean tumor volumes between IgG control and anti-PD-L1/anti-PD-1 treated mice in each syngeneic model. Asterisks denote statistical significance at the P≤0.05 (*) and P≤0.01 (**) level.
Discussion
In this study we identify genomic alterations in STK11/LKB1 as a tumor cell-intrinsic determinant of primary resistance to PD-1 axis blockade in three independent retrospective cohorts of KRAS-mutant LUAC, a fourth cohort of PD-L1-positive NSCLC regardless of KRAS status, as well as in patients with KRAS-mutant NSCLC treated with nivolumab in the pivotal CheckMate-057 randomized phase 3 clinical trial. Somatic mutations in STK11/LKB1 are prevalent in LUAC (16.7% in the large FM cohort), particularly among KRAS-mutant tumors (25.4% in the combined FM/SU2C cohort) and foster establishment of a non-T-cell-inflamed tumor immune microenvironment with frequently undetectable tumor cell PD-L1 expression (18, 20, 21). Furthermore, we show that genetic ablation of Stk11/Lkb1 directly promotes resistance to anti-PD-1/anti-PD-L1 therapy in two immune competent Kras-mutant LUAC murine models. Therefore, STK11/LKB1 inactivation represents a major driver of immune escape and innate resistance to PD-1 blockade in KRAS-mutant LUAC.
Our work demonstrates that alterations in STK11/LKB1 are associated with lack of PD-L1 expression in tumor cells across multiple independent cohorts, despite the presence of intermediate or high TMB. This finding is consistent with lower densities of infiltrating CD8+ cytotoxic T lymphocytes in both human and murine STK11/LKB1 deficient tumors. However, the negative impact of STK11/LKB1 genomic alterations on clinical response to PD-1 axis inhibitors also extends to PD-L1-positive NSCLC, indicating that it is at least partially independent of PD-L1 expression. In addition, in an exploratory analysis among PD-L1-negative KRAS-mutant LUAC, KP tumors exhibited more favorable response to PD-L1 blockade, with PR/SD achieved in 7/10 patients. Therefore, analysis of STK11/LKB1 and TP53 co-mutations may help refine response prediction algorithms in both PD-L1 positive and negative tumors as well as in cases where tumor biopsy for assessment of PD-L1 expression is unavailable or impractical but profiling of ctDNA (“liquid biopsy”) has been obtained.
It is important to note that non-mutational mechanisms may also account for STK11/LKB1 inactivation in a subset of LUAC (19). Quantitative IHC for LKB1 (the protein product of the STK11 gene) can capture STK11/LKB1-deficient tumors in the absence of STK11/LKB1 genomic alterations (26). Therefore, evaluation of LKB1 expression by IHC may further enhance the predictive utility of a composite biomarker panel encompassing PD-L1 expression, TMB and STK11/LKB1 genomic alterations.
Although our study primarily examined clinical response to PD-1 axis inhibitors in KRAS-mutant tumors, we anticipate that the effect of STK11/LKB1 inactivation extends to the entire LUAC population, regardless of KRAS status. This hypothesis is supported by: a) poor ORR and shorter PFS and OS with PD-1/PD-L1 blockade in STK11/LKB1-mutant tumors in a cohort of PD-L1-positive NSCLC encompassing both KRAS-mutant and wild-type tumors; b) shorter time on PD-1 inhibitor in a separate cohort of patients with STK11/LKB1-altered TMBI/H tumors ; c) evidence of a “cold” immune microenvironment in STK11/LKB1-altered LUAC irrespective of KRAS status; and was further proposed in a recent separate study (31). However, application to the wider population of non-squamous NSCLC will require further validation in larger datasets.
The mechanistic basis of T-cell exclusion in STK11/LKB1-deficient tumors is under active investigation and did not constitute a focus of the current study. However, based on established and emergent STK11/LKB1 functions a number of possibilities are proposed including altered cytokine/chemokine milieu (20), metabolic restriction of effector T cells (32) or impaired antigenicity, possibly as a result of STK11/LKB1-dependent changes in the epigenetic landscape of tumor cells (33). In a set of elegant in vivo experiments, inducible expression of c-Myc in KrasG12D-driven murine lung adenomas triggered rapid expulsion of CD3+ T-lymphocytes (as well as B cells and NK cells) from the tumor micro-environment via induction of IL-23 and CCL9 (34). STK11/LKB1 loss has been reported to promote transcriptional up-regulation of C-MYC via the MZF1 transcription factor (35). In a colorectal cancer murine isograft model T-cell exclusion and suppression of TH1 cell differentiation were mediated by TGFβ signaling (36), that has also been shown to be subject to modulation by STK11/LKB1 (37). Furthermore, loss of PTEN (phosphatase and tensin homologue), that – similar to STK11/LKB1 alterations – results in mTOR pathway activation, has been associated with impaired CD8+ T-cell recruitment in melanoma (38). In this tumor type, prior seminal work highlighted active WNT/β-catenin signaling as a key molecular driver of the non-T-cell-inflamed phenotype, via ATF3-mediated suppression of CCL4 production and impaired recruitment of CD103+ dendritic cells to the tumor immune microenvironment (30); interestingly, STK11/LKB1 deficiency has previously been associated with WNT pathway activation (39). Finally, it was recently demonstrated that tumor cell-derived prostaglandin E2 (PGE2) can also impair recruitment of conventional type 1 dendritic cells (cDC1) to the tumor microenvironment both directly, through down-regulation of chemokine receptor expression in cDC1, and indirectly, via attenuation of NK cell viability and function (40). It was previously reported that expression of cyclooxygenase-2 (COX-2) – that catalyzes the conversion of arachidonic acid to prostaglandins – is enhanced in STK11/LKB1 deficient NSCLC cells via activated CRTC1 (CREB-regulated transcription co-activator 1) (41). Thus, multiple and potentially non-overlapping mechanisms may underpin establishment and maintenance of a “cold” tumor immune microenvironment in STK11/LKB1-deficient NSCLC and further work is required to elucidate nodal downstream effectors and signaling cascades.
Delineation of pathways and mechanisms of immune escape downstream of STK11/LKB1 inactivation is also critical in order to inform rational combination therapeutic approaches aimed at invigorating anti-tumor immunity. Several strategies to convert non-T-cell-inflamed into T-cell-inflamed tumors have been proposed and are undergoing pre-clinical and clinical evaluation, including activation of innate immune recognition with STING agonists, TLR agonists, ionizing radiation or expression of LIGHT in tumor cells (42–45). Such approaches, as well as efforts that tackle specific STK11/LKB1 loss-dependent immunosuppressive cascades will require prospective evaluation in patients with STK11/LKB1-deficient NSCLC.
In contrast, evidence of pre-existing CD8+ T-cell infiltrate and adaptive immune resistance in the majority of KP (and possibly K-only STK11/LKB1-proficient) tumors supports simultaneous targeting of multiple immune inhibitory pathways in this subgroup (43).
Taken together, our data reveal a novel, frequent driver of de novo resistance to PD-1 blockade in KRAS-mutant LUAC and potentially the entire LUAC population. More broadly, somatic genomic alterations in individual genes may modulate the efficacy of PD-1/PD-L1 inhibitors in given tumor types and across tumor types. Although the fully integrated set of determinants of response to these agents is not yet completely defined, our results suggest that the development of tailored immunotherapy approaches for NSCLC may be facilitated by genomic profiling to allow simultaneous characterization of specific somatic alterations including KRAS, STK11/LKB1, and TP53, in addition to TMB and PD-L1 expression.
Methods
Patients
Patients with stage IV KRAS-mutant LUAC who received at least one cycle of PD-1 inhibitor therapy or combined PD-1/PD-L1 and CTLA-4 blockade, were alive for ≥ 14 days thereafter and had available molecular profiling of KRAS, STK11/LKB1 and TP53 were identified by retrospective electronic medical record review. Three independent cohorts were studied from members of the Stand Up To Cancer/American Cancer Society Lung Cancer Translational Research Dream Team: MD Anderson Cancer Center (MDACC), Memorial Sloan Kettering Cancer Center (MSKCC), and a combined cohort from Dana-Farber Cancer Institute and Massachusetts General Hospital (DFCI/MGH), cumulatively forming the SU2C cohort. A fourth cohort of 66 patients with PD-L1 positive (≥1%) non-squamous NSCLC (regardless of KRAS status) from MDACC with available tumor molecular profiling was assessed to determine the impact of STK11/LKB1 genomic alterations on clinical outcomes with anti-PD-1/PD-L1 therapy specifically in PD-L1 positive tumors. The study was conducted in accordance with the ethical standards outlined in the Declaration of Helsinki and its subsequent amendments. All participating patients at each institution provided written informed consent for the collection of clinical, demographic, and molecular data as well as the use of tissue for immunohistochemical and molecular studies, which proceeded in accordance with IRB-approved protocols at each of the participating institutions.
A fifth independent cohort of 44 patients with KRAS-mutant NSCLC (24 treated with nivolumab and 20 treated with docetaxel) with available STK11/LKB1 and TP53 mutational status and tumor cell PD-L1 expression from the CheckMate-057 (CM-057) international phase 3 randomized controlled trial (RCT) (NCT01673867) was also analyzed (46).
Finally, a separate large cohort of 924 unselected patients with LUAC who submitted samples to FM (Foundation Medicine, Cambridge, MA) for hybrid capture-based comprehensive genomic profiling (CGP) were included in an integrated analysis of TMB, PD-L1 expression, and genomic alterations of individual cancer-related genes. Duration of therapy (“time on drug”) was known for a subset of patients with CGP that received PD-1/PD-L1 inhibitors. Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817).
Study assessments
Tumor response was assessed by dedicated thoracic radiologists (MDACC, MSKCC, DFCI) or the study investigators (MGH) using Response Evaluation Criteria in Solid Tumors, version 1.1 (RECISTv1.1). Objective response rate (ORR) was defined as the percentage of patients achieving a confirmed or unconfirmed complete or partial response. Attribution of stable disease as best overall response to therapy required a minimum interval of ≥ 30 days between the first day of the first cycle of treatment (C1D1) and radiological evaluation. Patients who died before radiological re-assessment were deemed to have progressive disease. Progression-free survival (PFS) was defined as the time from C1D1 to the date of disease progression or death from any cause. Overall survival (OS) was defined as the time from C1D1 to the date of death from any cause. Efficacy endpoints for patients included in CM-057 were evaluated as previously described (46).
In the SU2C cohort, tumor cell PD-L1 expression was assessed in the most recent pre-immunotherapy formalin-fixed paraffin-embedded tumor biopsy tissue at each institution using the PD-L1 E1L3N® XP® rabbit monoclonal antibody (Cell Signaling Technology), and quantified as the percent of tumor cell membranes exhibiting specific staining of any intensity. PD-L1 expression in CM-057 samples was assessed using the validated 28-8 pharmDx assay (Dako, Carpinteria, CA) (3). PD-L1 staining of tumor samples submitted to FM was performed using the VENTANA PD-L1 (SP142) assay. Tumors were characterized as PD-L1 negative (PD-L1 < 1%), low positive (1% ≤ PD-L1 < 50%), or high positive (PD-L1 ≥ 50%). Assessment of PD-L1 expression in the separate cohort from MDACC (N=66) was based on the FDA-approved 22C3 pharmDx assay (Dako, Carpinteria, CA).
Molecular profiling platforms and study group definitions
CGP of tumor and/or circulating cell free tumor DNA utilized CLIA-certified assays available in each of the participating institutions (47–52). Samples from CM-057 underwent whole exome sequencing according to previously described methodology (4). Samples submitted to FM were processed at a CLIA-certified laboratory as previously described (53). TMB was measured by FM as previously described (54). Raw TMB values were measured in units of mutations per Mb and characterized as low (TMB<6), intermediate (6 ≤TMB<20), or high (TMB≥20).
KRAS-mutant LUAC bearing non-synonymous somatic mutations in STK11/LKB1 and/or mono-or bi-allelic loss of the STK11/LKB1 locus were denoted as KL. KRAS-mutant LUAC harboring non-synonymous somatic mutations in TP53 and/or mono- or bi-allelic loss of the TP53 locus were classified as KP. KRAS-mutant tumors with intact STK11 and TP53 were referred to as K-only (these tumors include a multitude of additional genetic alterations in addition to mutant KRAS). Triple-mutant tumors (KRAS;TP53;STK11 : KPL) were classified as KL(18). In CM-057 tumors bearing non-synonymous somatic mutations in STK11 were denoted as KL. In the FM cohort, a KRAS-mutant LUAC sample was considered altered in STK11 (KL) or TP53 (KP), if there was detection of a known non-synonymous somatic mutation, any truncating alteration, or bi-allelic loss.
Preclinical studies
2 × 106 LKR10/LKR10KO or LKR13/LKR13KO Kras-mutant murine LUAC cells were injected subcutaneously in the right flank of syngeneic recipient male mice (129Sv genetic background). Mice bearing tumors ≥ 200mm3 were randomly assigned to intraperitoneal treatment with : a) six doses of 200μg anti-PD-1 (clone RMPI-14; BioXCell) or isotype control antibody (clone 2A3; BioXCell) administered twice weekly (N=5-8 mice per group) (LKR10/LKR10KO isogenic system) or b) six doses of 200μg anti-PD-L1 (mIgG1-D265AFc clone 80) or IgG control antibody administered twice weekly (N=8-9 mice per group) (LKR13/LKR13KO isogenic system). Tumor caliper measurements were obtained twice weekly. Mice were sacrificed when tumor volume reached 1500mm3 (LKR10/LKR10KO) or 2000mm3 (LKR13/LKR13KO) or when moribund. Single cell suspension was established from excised tumors using a commercially available Tumor Dissociation Kit (Miltenyi) and the gentleMACS™ Dissociator (Miltenyi) and cell suspensions were prepared corresponding to 40mg of gross tumor per 100uL 1X PBS/0.05mM EDTA. 100μL of cell suspension per sample was stained with an antibody cocktail including CD3-FITC (clone 17A2), CD4-PerCP55 (clone RM4-5), CD8-PECy7 (clone 53-6.7), CD45-AF700 (clone 30-F11), CD11b-FITC (clone M1/70), Ly-6G-PerCP/Cy5.5 (clone 1A8) and Zombie dye according to the manufacturer’s protocol (Biolegend). Cells were analyzed using the BD FACSCanto™ multi-color flow cytometer and BD FACSDIVA™ software. The animal study was approved by the MD Anderson Institutional Animal Care and Use Committee (IACUC). The LKR13 and LKR10 murine cell lines were generously provided by Dr Tyler Jacks in 2005. All cell lines tested negative for Mycoplasma in March 2017 using the MycoAlert™ Mycoplasma Detection Kit (Lonza, LT-07-118). Cells were used in in vivo experiments within 10 passages from thawing.
Statistical analysis
The significance of the association between the KL, KP, and K-only subgroup allocations and objective response to PD-1 axis blockade was assessed using Fisher’s exact test. The Kaplan-Meier method was used to estimate PFS and OS. For the analysis of PFS, data for patients who were alive and had no evidence of disease progression at the time of the PFS data lockout (31st of December 2016) or who were lost to follow up were censored at the time of the last radiological tumor assessment. For the analysis of OS, data for patients who were alive or lost to follow up at the time of the OS data lockout (25th of April 2017) were censored at the time of the last documented patient contact. Analysis of both PFS and OS in the separate cohort of PD-L1-positive patients from MDACC was based on a January 15, 2018 data lockout. Differences between groups in PFS and OS were assessed based on the log-rank test. Bonferroni-adjusted p values were employed to account for multiple comparisons. Hazard ratios and the corresponding 95% confidence intervals were computed using the Cox-proportional hazards model. Wald test was applied for testing the hazard ratio of 1. A p value of ≤0.05 was considered statistically significant for all comparisons, unless stated otherwise.
Analysis of clinical endpoints in CM-057 was conducted as previously described and corresponds to a February 18, 2016 database lock (46).
For TMB and PD-L1 analysis, statistics were calculated using R version 3.3.2 (2016-10-31). The enrichment analysis for LUAC in the PD-L1/TMB landscape was limited to genes altered in >1% of samples (100 genes).
Supplementary Material
Statement of significance.
This work identifies STK11/LKB1 alterations as the most prevalent genomic driver of primary resistance to PD-1 axis inhibitors in KRAS-mutant lung adenocarcinoma. Genomic profiling may enhance the predictive utility of PD-L1 expression and TMB and facilitate establishment of personalized combination immunotherapy approaches for genomically defined LUAC subsets.
Acknowledgments
We thank the patients and their families who participated in this study. The research was supported by a Stand Up To Cancer - American Cancer Society Lung Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT17-15). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C; a Sabin Family Foundation Fellowship (FS); a Khalifa Bin Zayed Al Nahyan Foundation Scholar Award (FS); the Jane Ford-Petrin Fund; the Gil and Dody Weaver Foundation; Bill and Katie Weaver Charitable Trust; RP160652 Cancer Prevention Research Institute of Texas Multi-Investigator Research Award (JVH); 1R01 CA205150 (JVH); a V Foundation Translational Award (JVH); generous philanthropic contributions to the University of Texas MD Anderson Lung Cancer Moonshots Program; Lung SPORE grant 5 P50 CA070907; MD Anderson Cancer Center Support Grant P30 CA016672; MSKCC Core Grant P30 CA008748; 1R01 CA203636 (NM). We would like to thank Dr Patrick Hwu for critical review of the manuscript and Emily Roarty for editorial assistance.
Financial support:
Research supported by a Stand Up to Cancer – American Cancer Society Lung Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT17-15). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C; a Sabin Family Foundation Fellowship (FS); a Khalifa Bin Zayed Al Nahyan Foundation Scholar Award (FS); the Jane Ford-Petrin Fund; The Gil and Dody Weaver Foundation; Bill and Katie Weaver Charitable Trust; RP160652 Cancer Prevention Research Institute of Texas Multi-Investigator Research Award (JVH); 1R01 CA205150 (JVH); a V Foundation Translational Award (JVH); generous philanthropic contributions to the University of Texas MD Anderson Lung Cancer Moonshots Program; Lung SPORE grant 5 P50 CA070907; MD Anderson Cancer Center Support Grant P30 CA016672; MSKCC Core Grant P30 CA008748; 1R01 CA203636 (NM).
FS reports personal fees from BMS, outside the submitted work. MEG reports other (employment) from Foundation Medicine, Inc. DMG reports other (employment and stock ownership) from Bristol-Myers Squibb. MDH reports personal fees from Genentech, grants and personal fees from BMS, personal fees from Merck, personal fees from AstraZeneca, personal fees from Novartis, personal fees from Janssen, during the conduct of the study. MMA reports personal fees from AbbVie, personal fees from Ariad, personal fees from AstraZeneca, personal fees from Clovis, personal fees from BMS, personal fees from Merck, personal fees from Genentech, personal fees from Foundation Medicine, personal fees from Nektar Therapeutics, outside the submitted work. JFG reports grants from Stand Up To Cancer, during the conduct of the study; personal fees from BMS, personal fees from Merck, personal fees from Genentech/Roche, personal fees from Novartis, personal fees from LOXO Oncology, personal fees from Theravance, personal fees from Clovis, personal fees from Pfizer, personal fees from Ariad, outside the submitted work. ABS reports other (employment and stock ownership) from Foundation Medicine, Inc. RJH reports other (employment and stock ownership) from Foundation Medicine, Inc. SET reports other (employment and stock ownership) from Foundation Medicine, Inc. LG reports other (employment and stock ownership) from Foundation Medicine, Inc. SMA reports other (employment and stock ownership) from Foundation Medicine, Inc. JAE reports other (employment and stock ownership) from Foundation Medicine, Inc. GS reports other (employment and stock ownership) from Foundation Medicine, Inc. JSR reports other (employment and stock ownership) from Foundation Medicine, Inc. DF reports other (employment and stock ownership) from Foundation Medicine, Inc. PMS reports other (employment and stock ownership) from Bristol-Myers Squibb. HC reports other (employment and stock ownership) from Bristol-Myers Squibb. AS reports other (employment and stock ownership) from Bristol-Myers Squibb.SS reports other (employment and stock ownership) from Bristol-Myers Squibb. SK reports other (employment and stock ownership) from Bristol-Myers Squibb. JS reports other (employment and stock ownership) from Bristol-Myers Squibb. PV reports other (employment and stock ownership) from Bristol-Myers Squibb. RE reports other (employment and stock ownership) from Bristol-Myers Squibb. NS reports other from Novartis. SHIO reports personal fees from Foundation Medicine, Inc, outside the submitted work. NP reports personal fees from AstraZeneca, personal fees from Boehringer Ingelheim, personal fees from BMS, personal fees from Eli Lilly, personal fees from MSD, personal fees from Novartis, personal fees from Pfizer, personal fees from Roche, personal fees from Foundation Medicine, personal fees from Novellus Dx, personal fees from Guardant, personal fees from Takeda, outside the submitted work. MN reports other from BMS, other from Toshiba Medical Systems, other from Bayer; grants from Merck investigator studies program, grants from Toshiba Medical Systems, grants from NIH, outside the submitted work. WLD reports other (employment) from Juno Therapeutics Inc. LMS reports personal fees from Genentech, outside the submitted work. MMK reports personal fees from Roche, outside the submitted work. JZ reports personal fees from BMS, outside the submitted work. KKW reports grants from the NCI during the conduct of the study; grants and personal fees from AstraZeneca, grants and personal fees from Janssen, grants from Gilead, grants from Novartis, grants and personal fees from Eli Lilly, outside the submitted work. VAP reports other from Merck, other from Astra Zeneca, other from BMS, outside the submitted work. IIW reports personal fees from Genentech/Roche, personal fees from BMS, personal fees from Boehringer Ingelheim, personal fees from Medscape, personal fees from AstraZeneca/MedImmune, personal fees from Pfizer, personal fees from Eli Lilly, personal fees from Ariad, personal fees from HTG Molecular, personal fees from Asuragen, personal fees from Merck, outside the submitted work; grant support from Genentech, grant support from Oncoplex, grant support from HTG Molecular, grant support from DepArray, grant support from Merck, grant support from NCCN/BMS, grant support from MedImmune, grant support from Adaptive, grant support from AdaptImmune, grant support from EMD Serono, grant support from Pfizer, outside the submitted work. VAM reports other (employment and stock ownership) from Foundation Medicine, Inc. GMF reports other (employment and stock ownership) from Foundation Medicine, Inc. JDW reports personal fees from Merck, personal fees from BMS, personal fees from Genentech, personal fees from MedImmune, outside the submitted work; grant support from Merck, grant support from BMS, grant support from Genentech, grant support from MedImmune, during the conduct of the study. ATS reports personal fees from Pfizer, personal fees from Novartis, personal fees from Genentech, personal fees from Roche, personal fees from Ignyta, personal fees from Blueprint Medicine, personal fees from Daiichi-Sankyo, personal fees from Ariad, personal fees from Taiho Pharmaceuticals, personal fees from EMD Serono, personal fees from Foundation Medicine, Inc, outside this work. PAJ reports personal fees from AstraZeneca, personal fees from Boehringer Ingelheim, personal fees from Pfizer, personal fees from Merrimack Pharmaceuticals, personal fees from Roche, personal fees from Chugai, personal fees from ACEA Biosciences, personal fees from Ignyta, personal fees from LOXO Oncology, personal fees from ARIAD pharmaceuticals; other (Stock Ownership) from Gatekeeper Pharmaceuticals; other (postmarketing royalties) from LabCorp; grant support from Astellas Pharmaceuticals, grant support from AstraZeneca, grant support from Daiichi Sankyo, grant support from PUMA. PJS reports other (employment and stock ownership) from Foundation Medicine, Inc. CMR reports personal fees from BMS, Araxes, personal fees from AstraZeneca, personal fees from Celgene, personal fees from Harpoon, personal fees from G1 Therapeutics, personal fees from Chugai, outside the submitted work. WJG reports other (employment and stock ownership) from Bristol-Myers Squibb. LAA reports other (employment and stock ownership) from Foundation Medicine Inc. JVH reports grants from the NCI during the conduct of the study; personal fees from AstraZeneca, personal fees from Novartis, personal fees from Genentech, personal fees from Boehringer Ingelheim, personal fees from Eli Lilly, personal fees from Spectrum, personal fees from Guardant, outside the submitted work.
Footnotes
Declaration of Interests
All other authors declare no competing financial interests.
References
- 1.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England Journal of Medicine. 2015;372(21):2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 2.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 3.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. The New England Journal of Medicine. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. The New England Journal of Medicine. 2017;376(25):2415–26. doi: 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. The Lancet Oncology. 2016;17(11):1497–508. doi: 10.1016/S1470-2045(16)30498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–65. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 8.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. The New England Journal of Medicine. 2015;373(2):123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature Reviews Cancer. 2016;16(5):275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. The New England Journal of medicine. 2016;375(19):1823–33. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 13.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England Journal of Medicine. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discovery. 2015;5(8):860–77. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature Reviews Cancer. 2009;9(8):563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Research. 2016;76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. 2017;28(1):75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu HA, Sima CS, Shen R, Kass S, Gainor J, Shaw A, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol. 2015;10(3):431–7. doi: 10.1097/JTO.0000000000000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24(2):334–40. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonanno L, De Paoli A, Zulato E, Esposito G, Calabrese F, Favaretto A, et al. LKB1 Expression Correlates with Increased Survival in Patients with Advanced Non-Small Cell Lung Cancer Treated with Chemotherapy and Bevacizumab. Clinical Cancer Research. 2017;23(13):3316–24. doi: 10.1158/1078-0432.CCR-16-2410. [DOI] [PubMed] [Google Scholar]
- 25.Facchinetti F, Bluthgen MV, Tergemina-Clain G, Faivre L, Pignon JP, Planchard D, et al. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer. 2017;112:62–8. doi: 10.1016/j.lungcan.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. 2017;546(7656):168–72. doi: 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28(1):75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(6):1577–86. doi: 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufman JM, Yamada T, Park K, Timmers CD, Amann JM, Carbone DP. A Transcriptional Signature Identifies LKB1 Functional Status as a Novel Determinant of MEK Sensitivity in Lung Adenocarcinoma. Cancer Research. 2017;77(1):153–63. doi: 10.1158/0008-5472.CAN-16-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 31.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand (PD-L)-Ligand 1 Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. Journal of Clinical Oncology: official journal of the American Society of Clinical Oncology. 2018 doi: 10.1200/JCO.2017.75.3384. JCO2017753384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539(7629):390–5. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell. 2017;171(6):1301–15 e14. doi: 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai LH, Wu JY, Cheng YW, Chen CY, Sheu GT, Wu TC, et al. The MZF1/c-MYC axis mediates lung adenocarcinoma progression caused by wild-type lkb1 loss. Oncogene. 2015;34(13):1641–9. doi: 10.1038/onc.2014.118. [DOI] [PubMed] [Google Scholar]
- 36.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–43. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 37.Moren A, Raja E, Heldin CH, Moustakas A. Negative regulation of TGFbeta signaling by the kinase LKB1 and the scaffolding protein LIP1. The Journal of Biological Chemistry. 2011;286(1):341–53. doi: 10.1074/jbc.M110.190660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discovery. 2016;6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu W, Monahan KB, Pfefferle AD, Shimamura T, Sorrentino J, Chan KT, et al. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell. 2012;21(6):751–64. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172(5):1022–37 e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao C, Gao R, Zhang M, Amelio AL, Fallahi M, Chen Z, et al. Role of LKB1-CRTC1 on glycosylated COX-2 and response to COX-2 inhibition in lung cancer. Journal of the National Cancer Institute. 2015;107(1):358. doi: 10.1093/jnci/dju358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gajewski TF. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Seminars in Oncology. 2015;42(4):663–71. doi: 10.1053/j.seminoncol.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunology. 2013;14(10):1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;30(3):500. doi: 10.1016/j.ccell.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Reports. 2015;11(7):1018–30. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M, et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057) Journal of Clinical Oncology: official journal of the American Society of Clinical Oncology. 2017:JCO2017743062. doi: 10.1200/JCO.2017.74.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. Journal of Clinical Oncology: official journal of the American Society of Clinical Oncology. 2015;33(25):2753–62. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discovery. 2012;2(1):82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Medicine. 2017;23(6):703–13. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature Medicine. 2014;20(12):1479–84. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 51.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PloS One. 2015;10(10):e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lih CJ, Harrington RD, Sims DJ, Harper KN, Bouk CH, Datta V, et al. Analytical Validation of the Next-Generation Sequencing Assay for a Nationwide Signal-Finding Clinical Trial: Molecular Analysis for Therapy Choice Clinical Trial. The Journal of Molecular Diagnostics: JMD. 2017;19(2):313–27. doi: 10.1016/j.jmoldx.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31(11):1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine. 2017;9(1):34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.