

Abstract
Determining the magnitude of local immune response during mucosal exposure to viral pathogens is critical to understanding the mechanism of viral pathogenesis. We previously showed that vaginal inoculation of lymphocytic choriomeningitis virus (LCMV) fails to induce a robust innate immune response in the lower female reproductive tract (FRT), allowing high titer viral replication and a delay in T cell-mediated viral control. Despite this immunological delay, LCMV replication remained confined mainly to the FRT and the draining iliac lymph node. Here, we show that rectal infection with LCMV triggers type I/III interferon responses, followed by innate immune activation and lymphocyte recruitment to the colon. In contrast to vaginal exposure, innate immunity controls LCMV replication in the colon, but virus rapidly disseminates systemically. Virus-induced inflammation promotes the recruitment of LCMV target cells to the colon followed by splenic viral dissemination by infected B cells, and to a lesser extent by CD8 T cells. These findings demonstrate major immunological differences between vaginal and rectal exposure to the same viral pathogen, highlighting unique risks associated with each of these common routes of sexual viral transmission.
Keywords: rectal viral infection, LCMV, circulating lymphocytes, systemic viral dissemination, innate-mediated viral control
Introduction
Successful prevention of viral dissemination upon mucosal exposure of pathogens requires an improved fundamental understanding of how protective immunity is elicited and how pathogen dissemination occurs after crossing mucosal barriers 1. While viral cell-to-cell transmission has been extensively studied in tissue culture, in vivo studies that enhance our understanding of how viral pathogens are disseminated following mucosal infections are scarce 2. For example, sexual HIV transmission probability per exposure event is much greater across the rectal versus vaginal mucosa 3, 4, but the exact reason for this difference is unknown. These mucosal barriers have to discriminate between harmful pathogens versus commensals, as well as food and sperm antigens, and thus must continuously balance tolerance and immunity 5. After breaching the mucosal barrier, the early events of host response can play a key role in determining the outcome of an infection 6, and differences in tolerance mechanisms at various mucosal sites can influence immunity to invading pathogens. While it is appreciated that the rectum and vaginal anatomy are different, we lack a basic understanding of the immunological characteristics that contribute to the variance observed in the rate of viral transmission and dissemination after rectal versus vaginal exposure to pathogens.
To address these questions and to enhance our understanding of immunological events that contribute to the outcome of mucosal viral infections, we have established a new model of rectal infection using a widely-used model pathogen, lymphocytic choriomeningitis virus (LCMV). LCMV is an enveloped single-stranded RNA virus of the Arenaviridae family, with mice being its natural host 7. LCMV-infected animals shed the virus in their feces, urine, saliva, breast milk, and semen 8; thus mucosal transmission of LCMV likely occurs in nature, despite the more commonly used systemic infections performed in laboratory settings using this model pathogen.
We recently showed that compared to the immunity elicited after systemic intraperitoneal or transcervical infection, intravaginal (i.vag.) infection with LCMV in WT mice elicits a dampened and delayed anti-viral immune response, including dampened induction of type I and III interferons (IFNs) in the lower female reproductive tract (LFRT). This also leads to delayed activation of the protective CD8 T cells and enhanced replication and prolonged viral persistence in the vaginal mucosa. However, notwithstanding this dampened immunity, viral replication remained localized in the FRT and the draining iliac lymph node (iLN), without significant dissemination to the spleen 9.
We used our new intrarectal (i.rec.) model of LCMV infection in mice to determine if this dampened immunity and localization of the infection is a feature of all mucosal barriers or unique to the LFRT. Surprisingly, unlike LCMV i.vag. infection, we find induction of innate immunity after i.rec. infection, which is sufficient to control but not inhibit colonic LCMV replication. LCMV i.rec. infection results in inflammation-induced recruitment of leukocytes to the colon and rapid dissemination of virus to the spleen. We further show an important role for infected B cells, and to a lesser extend CD8 T cells, in the systemic dissemination of LCMV following i.rec. infection. These findings highlight important immunological differences between vaginal versus rectal exposure to the same viral pathogen. While dampened induction of innate immunity in the LFRT allows for greater local viral replication, the lack of inflammation-induced recruitment of leukocytes to the site of infection likely prevents enhanced circulation of infected target cells. After i.rec. infection, strong induction of IFNs limits local LCMV replication; however, the IFNs and inflammatory signals also recruit circulating target cells to the colon. Once infected, the migrating lymphocytes disseminate LCMV to the spleen early during infection and fuel productive systemic viral replication. Our results indicate that viral dissemination patterns can be dictated by the initial elicited innate immunity at the mucosal barrier, which we show to be very different after vaginal versus rectal exposure to the same viral pathogen.
Results
Rectally inoculated LCMV replicates in the colon and rapidly disseminates to the spleen
To determine if LCMV infects and replicates in the colon, we established an i.rec. infection model with this pathogen. We fasted WT mice for 16 hours to limit gut content, and after bowel movement, animals were rectally inoculated with 1 × 106 PFU of LCMV, without causing tissue abrasion (Figure 1a). At different times post-infection (p.i.), using an established quantitative RT-PCR (qRT-PCR) assay 9, we compared viral loads in the colon, the main colon-draining lymph nodes (LNs) including iLN and mesenteric (m)LN 10, 11 as well as the inguinal (ing)LN and the spleen. We found that LCMV robustly replicates in the colon and rapidly disseminates to the iLN and other lymph nodes, coinciding with rapid systemic spread to the spleen (Figure 1b). Viral replication peaks at day 5 p.i., with highest titers observed in the iLN, and gradually declines over about 2 weeks. We also confirmed that overnight fasting of the mice does not alter the magnitude of systemic viral dissemination at the peak of infection (Supplementary Figure S1).
Figure 1.
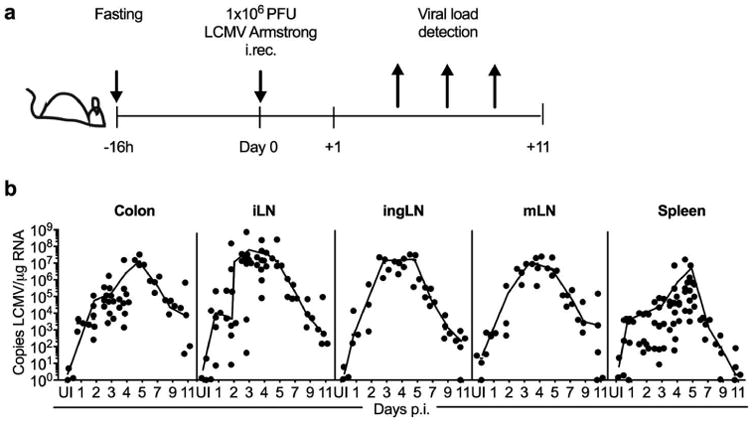
Rectally inoculated LCMV replicates in the colon and rapidly disseminates to the spleen. (a) Experimental outline used throughout the study. (b) C57BL/6N (WT) male mice were fasted overnight and then i.rec. infected with 1×106 PFU of LCMV, and viral copies in total RNA from indicated tissues and times post-infection were determined by qRT-PCR. Data are pooled from three (ingLN, mLN, spleen), five (colon) or six (iLN) independent experiments. n = 3–16 per time point, and each dot represents an individual animal. UI, uninfected.
Systemic viral dissemination has also been observed after rectal SIV 12, 13 and ZIKV 14 infection in nonhuman primate (NHP) models, suggesting that rapid dissemination may be a general characteristic of viral infections that occur via the rectal mucosa. Since we did not observe a similar systemic dissemination after i.vag. LCMV infection 9, we asked if the observed differences in viral dissemination may be the result of alterations in the induction of protective innate or adaptive immunity after each route of infection. Therefore, we opted to use our i.rec. LCMV mouse model to investigate the immunological differences between this route of infection with what we had previously observed after i.vag. infection and to identify the mechanism(s) that contribute to systemic viral dissemination after rectal LCMV infection.
CD8 T cells are activated and mediate viral clearance after LCMV rectal infection
Viral clearance after acute systemic LCMV infection is dependent on CD8 T cells 15, 16. Similarly, after i.vag. LCMV infection, CD8 T cells are also required to restrict the virus to the FRT and iLN, as systemic viral dissemination does occur in CD8−/− animals 9. To determine if LCMV control is also dependent on CD8 T cells after i.rec. infection, we infected WT, CD8−/−, MHC-II−/− and muMT−/− mice and quantitated the number of viral copies in the spleen and colon at day 15 p.i.. WT, MHC-II−/− and muMT−/− mice efficiently cleared LCMV in the spleen and the colon, while CD8−/− mice maintained peak viral loads even at day 15 p.i. (Figure 2a). This data suggests that similar to systemic and i.vag. LCMV infection, CD8 T cells are required to control rectally infected LCMV, independent of CD4 T cells or B cells.
Figure 2.
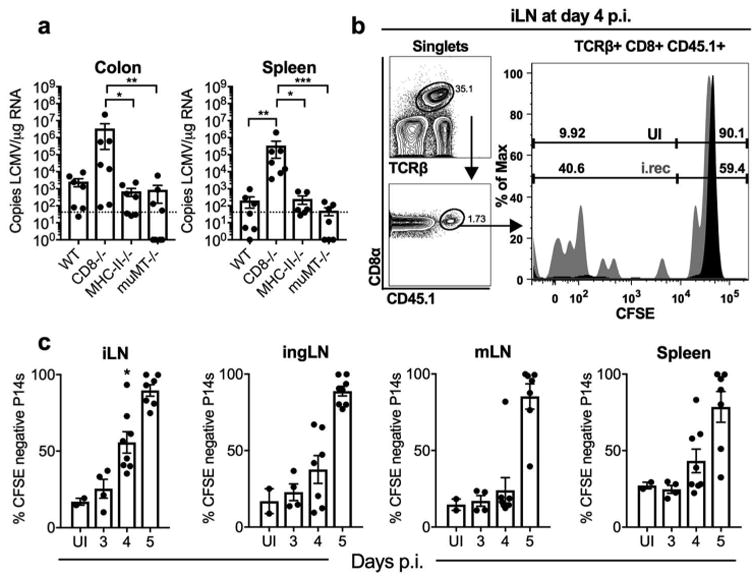
CD8 T cells are activated and mediate viral clearance after LCMV rectal infection. (a) WT, CD8-/-, MHC-II-/- and muMT-/- male and female mice were i.rec. infected and at day 15 p.i., LCMV copies in total RNA from spleen and colon were determined using qRT-PCR. (b-c) Naive CD45.1+ P14 CD8+ T cells (P14 cells) were isolated and labeled with CFSE, and 500,000 cells were adoptively transferred into male CD45.2+ WT recipient mice 1 day before i.rec. LCMV infection. Based on the representative gating strategy (b), the frequencies of CFSE-negative P14 cells in the iLN, ingLN, mLN and spleens were determined at indicated times post infection and statistical significance was determined among tissues at day 4, with * showing significant difference between iLN and mLN (c). Data are pooled from two (a, c) independent experiments. n = 7 per group (a) or 4–7 per infection time point (c). Error bars represent mean ± SEM. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01. UI, uninfected.
To eliminate the possibility that i.rec. LCMV infection results in systemic spread due to inadequate CD8 T cell mediated immunity, we determined the kinetics and location of antigen-specific CD8 T cell priming. We adoptively transferred congenically marked (CD45.1+) and CFSE-labeled P14 CD8 T cells (P14 cells), that express a transgenic TCR recognizing the gp33 epitope of LCMV 17, into WT mice prior to i.rec. infection. We measured the rate of CFSE dilution of P14 cells in various lymph nodes and the spleen. While the mLN is generally considered to be the major draining lymph node of the intestine, the significantly faster kinetics of P14 priming in the iLN at day 4 p.i. (Figure 2b-c) suggests that this LN may be the major site of CD8 T cell priming after i.rec. viral infection in mice. Together these data eliminate the possibility that lack of antigen-specific CD8 T cell activation is responsible for viral systemic dissemination.
Interferons and interferon-stimulated genes are induced in the colon after i.rec. LCMV Infection
Despite high viral titers after i.vag. LCMV infection, type I/III IFNs are not significantly induced in the vaginal mucosa, and recruitment of immune cells is highly delayed; yet, systemic viral dissemination is not observed 9. To determine if absence of Type I/III IFN induction is perhaps responsible for the rapid systemic LCMV dissemination after i.rec. infection, we measured the induction of IFNs and various IFN stimulated genes (ISGs) in the colons of i.rec. infected mice. Surprisingly, type I and III IFNs and the inflammatory cytokine IL-6 are significantly induced on days 1 and 2 p.i. compared to uninfected or mock infected animals. This is followed by gradual activation of ISGs such as IRF7, RIG-I, and MDA5 (Figure 3), which likely help to further amplify cytoplasmic innate sensing of viral RNA and the IFN response 18, 19. Furthermore, expression of the lymphocyte attractant ligands CXCL-9 and CXCL-10, which promote lymphocyte trafficking to the inflamed tissue 20, is also significantly enhanced after LCMV i.rec. infection.
Figure 3.
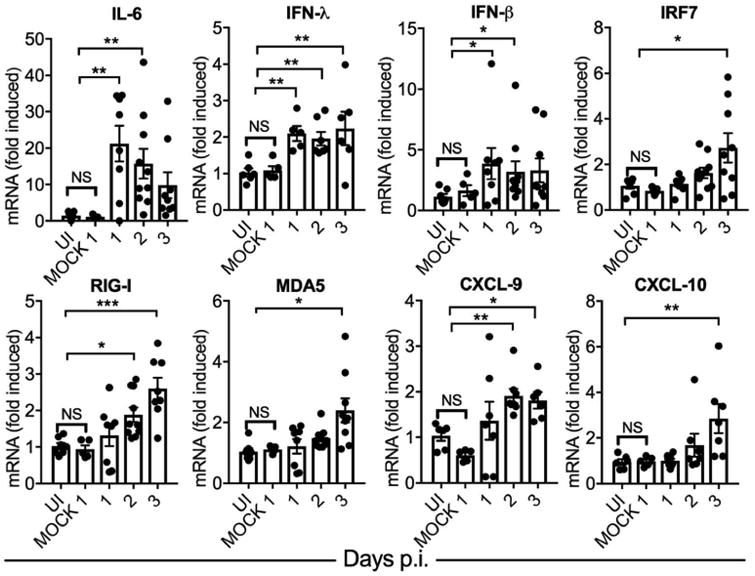
Interferons and interferon-stimulated genes are induced in the colon after i.rec. LCMV Infection. Male WT mice were uninfected, i.rec. mock infected with PBS, or infected with LCMV. Levels of each indicated mRNA at the indicated time points were determined from total colonic homogenates using qRT-PCR, normalized to GAPDH, and expressed as fold-change over uninfected controls. See Table S1 for primer sequences. Data are pooled from two independent experiments. n = 6–8 per time point. Error bars represent mean ± SEM. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01, ***P < 0.001. UI, uninfected.
Rectal infection with LCMV results in recruitment and activation of immune cells to the colonic lamina propria and enhances systemic lymphocyte migration
Consistent with induction of IFNs, cytokines, and chemokines, we observed an increase in the numbers (Figure 4a-b) and activation (Figure 4a,c) of inflammatory monocytes as well as dendritic cells in the colonic lamina propria (LP) at 2-3 days p.i. We also determined lymphocyte cellularity in the colonic LP, iLN, and spleen of i.rec. LCMV infected animals at days 1–3 p.i. After infection, a gradual increase in the number of B and T cells was observed in the LP; notably CD19+ B cell numbers doubled by day 2 p.i. (Figure 4d). This pattern of rapid recruitment of innate and adaptive cells to the site of infection highly contrasted the lack of leukocyte recruitment to the vaginal mucosa after LCMV i.vag. infection 9. A dramatic increase in the number of all lymphocytes was observed in the iLN at day 3 p.i. (Figure 4e). In the spleen, an increase in B cell numbers was detected as early as day 1 p.i., while splenic T cells, in particular CD8 T cells, decreased in number during this time frame (Figure 4f), likely due to their inflammation-induced migration to other infected tissues. Together, this data demonstrates enhanced migration of leukocytes and their rapid recruitment to the LP after i.rec. LCMV infection.
Figure 4.
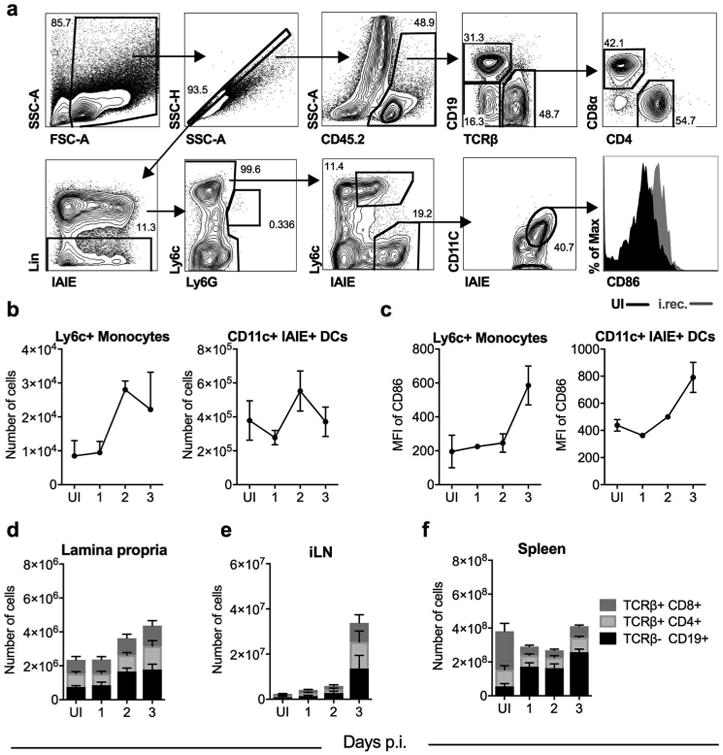
Rectal infection with LCMV results in recruitment and activation of immune cells to the colonic lamina propria and enhances systemic lymphocyte migration. Male WT mice were i.rec. infected with LCMV, and at indicated time points, the frequency and activation of indicated immune cell populations in the lamina propria, iLN and spleen was determined by flow cytometry. (a) Representative gating strategy depicting a LP sample at day 3 p.i.. Lin: TCRβ/CD19/NK1.1/CD326 (EpCam). (b) Number of Ly6c+ monocytes and dendritic cells (DCs) in the LP. (c) Median fluorescence intensity (MFI) of CD86 on DCs and Ly6c+ monocytes. Absolute counts of various lymphocytes in the lamina propria of the colon (d), iLN (e), and spleen (f). B cells: TCRβ- CD19+; CD8 T: TCRβ+ CD8+; CD4 T: TCRβ+ CD4+. Data represented from one of three independent experiments (b, c), or were pooled from two independent experiments (d-f). n = 3 (b, c) or 6 (d-f) per time point. Error bars represent mean ± SEM. UI, uninfected.
Systemic dissemination of LCMV is mediated by productively infected immune cells
The rapid increase in lymphocyte numbers, in particular B cells, in colonic LP and spleen led us to hypothesize that infected B cells are the main vehicle for splenic dissemination of LCMV after rectal infection. To determine if B cells are preferentially infected by LCMV, we sorted CD45+CD19+ B cells, CD45+TCRβ+ T cells, CD45+CD19-TCRβ- non-lymphocytes, and CD45- non-immune cells from the spleen of LCMV i.rec.-infected animals at day 5 p.i., and determined viral copies per μg of RNA for each sorted population. Based on this calculation, the various splenic cell types carried similar amounts of viral RNA per μg of total RNA (Figure 5a), suggesting that LCMV infects and equally replicates in all of these cell types. However, when we normalized viral load data to total cell numbers, thus accounting for the frequency of infected cell types, lymphocytes, notably B cells, carried the majority of the virus in the spleen (Figure 5b). We did not detect significant viral copies in the plasma of infected mice at this time point, further suggesting splenic viral dissemination likely occurs via circulating lymphocytes rather than cell-free via blood circulation.
Figure 5.

Systemic dissemination of LCMV is mediated by productively infected immune cells. (a,b) WT male mice were infected i.rec. with LCMV. At day 5 p.i. indicated cell populations were sorted from spleens and copies of LCMV were determined in each population by qRT-PCR. Viral loads were expressed as (a) copies of LCMV per μg RNA or (b) copies per organ to account for the abundance of each population. (c) Schematic design of experiments depicted in d-f. WT male and female mice were infected i.rec. with 1.5 × 106 PFU of recombinant LCMV expressing GFP and sacrificed at days 2 or 5 p.i.. (d) Cell populations positive for LCMV GFP at indicated time points and tissues were determined by flow cytometry, and presented as frequency of total cells. (e) Gating strategy for the sorting of LCMV GFP positive immune cells from blood, lymph nodes and spleens at day 5 p.i.. (f) Copies of LCMV were determined by qRT-PCR from total splenic RNA of recipient mice at day 5 after adoptive transfer of 8,000 LCMV GFP+ cells. Viral loads were expressed as copies per μg RNA. Data are pooled from two (a, b, d (blood and LP at day 2), f) or three (d (iLN, spleen at day 2; iLN, spleen, LP, blood at day 5)) independent experiments. n = 4–8 (a-b), 8–13 per population and time point (d), 7 per transferred population (f). Error bars represent mean ± SEM. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01, ***P < 0.001. UI, uninfected.
The tropism of LCMV includes many cell types, including lymphocytes and non-lymphocytes 7, 21. To determine whether migrating lymphocytes become infected by LCMV in the LP after rectal infection, and if they have the capacity to disseminate the virus systemically to the spleen, we i.rec.-infected WT mice with 1.5 × 106 PFU of a recombinant LCMV GFP virus (LCMV-GFP) 22. At days 2 and 5 p.i., we determined the fraction of GFP+ CD45+ B cells (TCRβ- CD19+), CD8 T cells (TCRβ+ CD8+), CD4 T cells (TCRβ+ CD4+), and non-lymphocyte (TCRβ- CD19-) immune cells in the LP, blood, spleen, and iLN (Figure 5c). We detected infected immune cells in all tissues at day 2 p.i. and a general increase in the frequency of these cells at day 5 p.i. (Figure 5d). Infection of immune cells after rectal LCMV transmission occurred similarly in male and female mice (Supplementary Figure S2). CD19+ B cells were the most abundant LCMV GFP+ lymphocyte population in LP, blood, spleen and iLN while infected CD8+ T cells were also readily detected in the spleen and the iLN. In contrast, infected GFP+ CD4 T cells were scarcer and were found at lower frequency at day 2 p.i. than the other cell types in all the examined organs (Figure 5d).
Next, we proceeded to determine whether GFP+ cells are productively infected with LCMV and if they can facilitate infection upon adoptive transfer into uninfected animals. We collected cells from blood, lymph nodes, and spleen of LCMV-GFP infected mice at day 5 p.i., and sorted TCRβ-CD19+, TCRβ+CD8+, TCRβ+CD4+ and TCRβ-CD19- among the CD45+ and GFP+ immune cells (Figure 5c). The ratio between the sorted populations correlated with the infection pattern previously observed in the individual tissues (Figure 5d), with the majority of GFP+ sorted cells being B cells and non-lymphocytes (Figure 5e). We transferred 8,000 GFP+ cells of each population into groups of uninfected WT mice, and then measured LCMV viral loads in their spleens at day 4 post adoptive transfer of infected cells. Productive infection was achieved in most mice except for those that received TCRβ+CD4+ cells (Figure 5f). These data suggest that lymphocytes, in particular B cells and CD8 T cells, as well as myeloid cells can be productively infected by LCMV, and have the potential to systemically spread the infection.
Viral dissemination to the spleen is predominately mediated by B cells and to a lesser extent by CD8 T cells
To identify the exact lymphocytes and exclude the role of circulating CCR2-dependent monocytes in systemic viral dissemination of LCMV, we i.rec.-infected WT, RAG1-/-, muMT-/-, CD8-/-, MHC-II-/- and CCR2-/- mice. We measured viral loads in various tissues at day 5 p.i., which is the peak of viremia in WT mice and found no significant differences in viral copies in the colon (Figure 6a) or iLN (Figure 6b) among the various KO mice. However, viral titers were significantly lower in the spleens of RAG1-/- and muMT-/- mice, and a trend toward lower viral dissemination was notable in CD8-/- mice (Figure 6c). This data is consistent with lymphocytes carrying the majority of the viral load in the spleen at this time point (Figure 5b), as well as CD8 T cells being involved in viral clearance starting at around day 5 post infection (Figures 1-2). Thus, at day 5 post infection, the RAG1-/- and CD8-/- mice must balance lower viral dissemination by lymphocytes, with the lack of protective CD8 T cells that are needed to control and clear the virus. We therefore next performed these experiments at early time points, at days 1-3 p.i., when lymphocyte-mediated viral dissemination is not yet counteracted by the protective role of CD8 T cells. We used mice lacking B, CD8 T, and CD4 T cells and compared the kinetics of viral dissemination to the spleen after LCMV i.rec. infection in each group. We detected fewer than 100 viral copies in the muMT-/- mice during the first 2 days after i.rec. infection, which also resulted in highly significant differences in viral titers between WT and muMT-/- male and female mice at day 3 p.i. (Figure 6d and Supplementary Figure S3). Interestingly, a significant decrease in viral dissemination was also observed in CD8-/- animals only at days 1 and 2 p.i., and no significant difference was observed between WT and MHCII-/- animals at any time point (Figure 6c,d). This data is consistent with high frequency of LCMV-GFP+ B cells and CD8 T cells during the first few days after infection (Figure 5d), and the efficient ability of GFP+ B cells and CD8 T cells to spread infection in uninfected animals upon adoptive transfer (Figure 5f). Together, these data demonstrate the role of B cells, and to a lesser extent CD8 T cells, in systemic dissemination of LCMV after i.rec. infection.
Figure 6.
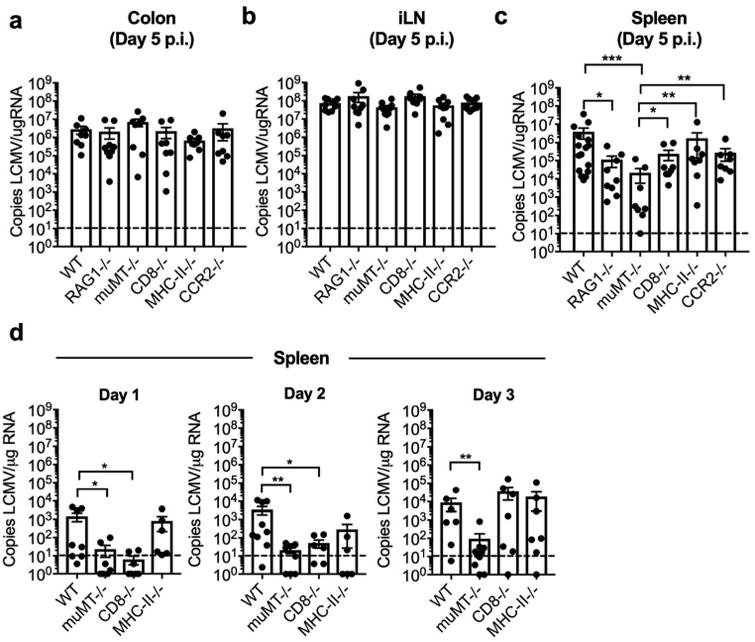
Viral dissemination to the spleen is predominately mediated by B cells and to a lesser extent by CD8 T cells. C57BL/6N (WT), RAG-/-, muMT-/-, CD8-/-, MHC-II-/-, and CCR2-/- male and female mice were infected i.rec. with LCMV. (a-c) Copies of LCMV per μg RNA were determined by qRT-PCR for specified tissues at day 5 p.i. or at days 1 to 3 p.i. (d). Data are pooled from three independent experiments. n = 8-12 per group (a-c), or 6-10 per group (d). Error bars represent mean ± SEM. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01, ***P < 0.001. UI, uninfected.
Discussion
Upon breaching the mucosal barrier, viral pathogens are sensed by the host, which results in the induction of type I and III interferons (IFNs) by the immune and non-immune cells, respectively 23, 24. This IFN induction is responsible for controlling local viral replication and can also help eliminate viral dissemination by recruiting and activating protective immune cells to the site of infection 25. At the same time, such inflammation-induced recruitment of lymphocytes, as has also been described in HIV/SIV studies 13, 26-28 and coxsackievirus infection 29, can increase the pool of available target cells to support viral infection and replication. Once infected, these lymphocytes can also re-enter circulation and disseminate the virus to other distal sites 2.
Our data confirms that LCMV can replicate in a variety of immune and non-immune cells, with B cells being the main disseminator of LCMV following rectal inoculation. We were able to detect infected B cells in the colonic LP, blood, spleen, and iLN of WT animals as early as day 2 post rectal infection, while in the absence of B cells, LCMV copies were significantly reduced in the spleen in the first 5 days post infection. Interestingly, depletion of B cells significantly prevents systemic dissemination of intraperitoneally administered type B coxsackievirus, a ssRNA virus from the group of enteric viruses that productively infect B lymphocytes 29. Furthermore, B cells are implicated in the early dissemination of anthrax (Bacillus anthracis) from the lung after inhalation of this bacterium 30. In addition, following intranasal administration, B cells are able to transport virus-like particles from the lung to the spleen in a B cell receptor-dependent manner 31. B cells were shown to also facilitate infection without active replication. For example, even though HIV-1 cannot replicate in B cells, it can bind to the CD21 receptor expressed on mature B cells, achieving cell-to-cell spread facilitated by B cells 32. Together, these findings highlight B cells as an important vehicle of both viral and bacterial dissemination. Surprisingly however, very little is known about the exact characteristic of B cells capable of pathogen dissemination. In regards to viral dissemination after rectal infection, it is known that along with IgA-producing resident B cells, both plasma cells and naïve IgM+ IgD+ B cells exist in the healthy colon and their numbers significantly increase in the large intestine under inflammatory conditions 33. Whether these B cells are capable of systemic viral dissemination is an important area for future investigation.
Sex-based differences in innate and adaptive immunity can contribute to the variances observed among males and females in severity of autoimmune diseases and cancers as well as altered response to infectious diseases and vaccines 34-36. Of particular interest, in a rectal model of HSV-2 infection in mice, BALB/c male and female mice showed the same disease progression, whereas the female C57BL/6 mice were very resistant to HSV-2 infection and showed minimal signs of neurological disease 37. In contrast, in a nonhuman primate model of rectal SHIV-1 infection, the females progressed faster to disease than their male counterparts, which was linked to an earlier and more robust inflammatory immune response in the rectum of the females, as well as increased expansion of Proteobacteria in their rectal mucosa 38. Interestingly however, we did not observe significant differences in LCMV infection and dissemination between male and female mice. It is tempting to speculate that involvement of plasmacytoid (p)DCs in disease pathogenesis of these various models of rectal viral infections contributes to the sex-based differences that have been reported, as it is well documented that pDCs in females produce more IFNα, which ultimately results in stronger immune activation in females 39-41. While pDCs are protective against systemic HSV infection in mice 42, persistent stimulation of pDCs and excessive production of IFNα during HIV/SIV infection further enhances viral pathogenesis 35; thus potentially explaining the differences seen in disease outcome after rectal infection of females using these pathogens. It is also possible that the lack of sex-based differences in our LCMV rectal infection model in C57BL/6 mice is due to the absence of notable pDC recruitment to the LP after infection (data not shown), while monocyte and cDC recruitment and activation was readily observed. Further studies are needed to determine the exact mechanism that contributes to different disease outcomes in male versus female mice after rectal viral infections.
Using the same viral pathogen inoculated via two different routes of infection, our previous and current study uncovers major immunological differences between vaginal and rectal mucosa, two common routes of all sexually transmitted pathogens, that result in different patterns of viral dissemination. While dampened immunity in the vaginal mucosa results in higher viral replication, the absence of rapid lymphocyte recruitment to the site of infection eliminates lymphocyte-mediated viral dissemination 9. On the other hand, the rapid induction of IFNs and inflammation after rectal infection with the same viral pathogen results in faster control of colonic viral replication, but also results in recruitment of target lymphocytes capable of disseminating the virus to distal sites. Thus, better understating of how pathogens are sensed and innate immunity elicited at the mucosal barriers is fundamental to understanding the pattern of dissemination and in vivo pathogenesis, particularly for pathogens that target migrating and circulating immune cells.
Materials and Methods
Mice
The following mouse strains were used: C57BL/6NJ (WT) (JAX); Rag1tm1Mom (RAG-/-) 43; Tg(TcrLCMV)327Sdz (P14 mice) 17 mice crossed to CD45.1; H2dlAb1-Ea (MHC-II-/-, lacking CD4 T cells) 44; CD8atm1Mak (CD8-/-) (JAX) 45; CCR2tm1Ifc (CCR2-/-) 46; Igh-6tm1Cgn (muMT-/-, lacking B cells) 47. The mice were housed and bred in the Gladstone animal facility. Male and female mice (8–25-weeks-old) were used throughout this study, except where indicated. Age-matched animals were assigned blindly to various treatment groups. All animal experiments were conducted in accordance with guidelines set by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Adoptive transfer of CFSE labeled P14 CD8 T cells
For priming experiments, LCMV-specific P14 chimeras were generated by adoptive transfer of ∼5 × 105 naive CD45.1+ P14 CD8 T cells (P14 cells) on a RAG−/− background into CD45.2+ congenic WT recipient mice. P14 CD8 T cells from donor mice were collected with the use of EasySep™ Mouse CD8 T Cell Isolation Kit (StemCell Technologies) based on the manufacturers protocol. Isolated P14 cells were labeled with 0.25 μM CFSE (Molecular Probes) at 37°C for 10 min, and 5 × 105 cells in 200 μl of FACS buffer (PBS + 1% FBS) were transferred into each recipient mouse via retroorbital injection, 1 day before infection.
Viral infections
Two batches of LCMV Armstrong strain were used in the experiments, with similar results, as well as a recombinant LCMV Armstrong construct with tri-segmented genome stably expressing a GFP reporter gene (LCMV-GFP) 22. LCMV was propagated in BHK-21 cells [C-13] (CCL-10; ATCC) as described 48. Viral titers of new batches were established by a modified FACS-based TCID50 assay on Vero cells (CCL-81; ATCC) as described 49. Briefly, 10-fold dilutions of viral stocks were cultured with Vero cells. After 3 days of incubation, cells were fixed with 0.8% PFA and stained with a rat monoclonal antibody (VL4) against LCMV-nucleoprotein (LCMV-NP; Bio X Cell) conjugated to FITC in 0.1% Triton X-100 containing FACS buffer for 45 min at 4°C. Infected (LCMV-NP+) versus uninfected (LCMV-NP-) wells at each dilution were counted to establish TCID50 values and a derivative PFU read-out using the Poisson distribution. For i.rec. infections, mice were first fasted 16 h prior to infection as variable luminal content might significantly alter infection rate and severity. Later mice were anesthetized using isoflurane and 30 μl of viral suspension (or PBS for mock infections) was inoculated into the rectum via the rectal opening twice in 10-min intervals for a total infectivity load of 1 × 106 PFU of LCMV Armstrong or 1.5 × 106 PFU of LCMV-GFP using a p200 micropipette and without causing tissue abrasion. Animals remained sedated for at least 15 minutes post final viral inoculation.
Isolation of immune cells, flow cytometry and cell sorting
LN and spleen tissues were collected into sterile cold FACS buffer and processed into single-cell suspensions for further application. The large intestines were harvested between the rectal opening and the caecum (in text referred to as colon), and the lumen was rinsed with sterile PBS, opened longitudinally and chopped into small pieces. Single-cell suspensions were obtained from the gut tissue as described 50. In brief, the epithelial lining was removed from tissue pieces during two rounds of a 20-min incubation in Ca+ and Mg+-free PBS with 5 mM EDTA and 1 mM DTT (Sigma-Aldrich) under constant rotation (40×g) at 37°C. The supernatant containing intra-epithelial lymphocytes was collected separately on ice, and the remaining lamina propria was digested in 1 mg/ml of collagenase type IV (Worthington Biochemical Corporation) and 75 μg/ml DNase I (Roche) in RPMI medium for 30 min under constant rotation (40×g) at 37°C. After digestion, single-cell suspensions were generated with the GentleMACS Dissociator (Miltenyi Biotec), according to the manufacturer's protocol, and passed through a 70μm cell strainer. Cell counts of collected single-cell suspensions were obtained using an Accuri C6 cytometer (BD), and cell numbers were normalized to 1 million cells before antibody surface staining. In order to determine the number of individual cell populations in each tissue, we first determined the percent of each population out of all cells (excluding debris and doublets) and then multiplied the fraction by the total number of live cells (based on FSC/SSC gating) obtained from the Accuri counts of each tissue. Fc receptors were blocked with anti-CD16/CD32 Ab. Cells were incubated with a mixture of fluorescence-conjugated anti-mouse antibodies for 30 min at 4°C. Stained cells were washed once and examined with a BD LSR II flow cytometer or sorted with the BD FACSAria II cell sorter, both utilizing the FACSDiva software (BD). The gating strategies are represented in the main figures. PE-Cy7 CD8α (SK1), Percp-Cy5.5 CD45.1 (A20), AF700 IAIE (MHC-II; M5/114.15.2), PE-Cy7 Ly6c (HK1.4), PB TCRβ (H57-597), APC-Cy7 CD11c (N418) and biotin conjugated CD326 (G8.8.) antibodies were from BioLegend. PE-CF594 Ly6G (1A8), BV605 CD86 (GL-1), BV650 CD19 (SJ25C1), BV650 NK.1.1 (PK136), BV605 CD4 (RM4-5), APC-Cy7 TCRβ (H57-597), and biotin-conjugated TCRβ (H57-597) antibodies were from BD. Biotin-conjugated CD19 (1D3), PE CD8α (YTS169.4), AF488 CD45.2 (AL1-4A2) and AF700 CD4 (GK.15) antibodies were from the UCSF Monoclonal Antibody Core.
Transfer of LCMV-GFP positive immune cells
WT mice were infected i.rec. with 1.5 × 106 PFU LCMV-GFP as described under the section Viral infections. At day 5 p.i. blood, lymph nodes and spleens from infected mice were harvested and stained with surface antibodies as described under the section Isolation of immune cells, flow cytometry and cell sorting. Individual LCMV-GFP+ cell populations were sorted with the BD FACSAria II cell sorter into 5 ml tubes containing PBS supplemented with 20% FBS and cooled to 4°C. 8,000 cells of each population were transferred into each recipient mouse via retroorbital injection. At day 4 post adoptive transfer of infected cells, splenic viral loads of the recipient mice were determined using qRT-PCR as described below.
RNA isolation and quantitative real-time PCR
Pieces of the large intestine were collected in TRIzol tubes filled with 3 mm zirconium beads, and homogenized in three 10-s pulses with a Beadbug microtube homogenizer (Benchmark Scientific). Fractions of the LN and spleen cell suspensions were collected in TRIzol-containing Eppendorf tubes without beads. Sorted cell samples were collected into Eppendorf tubes in FACS buffer and cell pellets after centrifugation were re-suspended in TRIzol. RNA from the TRIzol samples was isolated according to the manufacturer's instructions (Invitrogen). 1–5 μg of RNA was reverse transcribed into cDNA with a Maxima First Strand cDNA Synthesis kit (Thermo Fisher Scientific). Real-time PCR was performed using 2× SensiFAST probe Hi-Rox mix (Bioline) with gene-specific primers (Table S1) and acquired on an ABIPrism 7900 sequence detector with the ΔΔCt method from SDS 2.4 software (Applied Biosystems). The relative expression of genes was calculated with the formula 2−ΔCt, where ΔCt = Ct target gene – Ct endogenous control gene, and GAPDH was used as the endogenous control housekeeping gene. Viral copies per μg of RNA were calculated from a standard curve obtained using serial 10-fold dilutions of LCMV plasmid51.
Statistical analysis
Data analysis and visualization were performed with Prism 6.0 (GraphPad Software). Significance among multiple groups was determined with Kruskal-Wallis test. No experimental data were excluded from the statistical analysis. A P-value ≤0.05 was considered statistically significant.
Supplementary Material
Supplementary figure S1. Fasting before i.rec. infection does not affect viremia at the peak of infection. C57BL/6N (WT) male mice were i.rec. infected with 1×106 PFU of LCMV without prior fasting, and viral copies in total RNA from indicated tissues were determined by qRT-PCR at day 5 post i.rec. LCMV infection. Each dot represents an individual animal.
Supplementary figure S2. Infection of immune cells after rectal LCMV transmission occurs similarly in male and female mice. WT male and female mice were infected i.rec. with 1.5 × 106 PFU of recombinant LCMV expressing GFP. At day 5 p.i., cell populations positive for LCMV GFP in the spleen were determined by flow cytometry, and are presented as frequency of total cells. Data are pooled from two independent experiments. n = 4-5 per group.
Supplementary figure S3. Male and female (WT and muMT-/-) mice have comparable viral titers in the spleen at day 3 post rectal LCMV infection. C57BL/6N (WT) and muMT-/- male and female mice were infected i.rec. with LCMV. Copies of LCMV per μg RNA were determined by qRT-PCR for the spleen at day 3 p.i. Data are pooled from two independent experiments. n = 5-6 per group. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Table S1. Primer sequences for qRT-PCR analyses
Acknowledgments
We thank J. C. de la Torre (Scripps) for LCMV-GFP; C. Allen (UCSF) for muMT mice; E. Woodruff, J. Luong, and I. Lew for maintaining animal colonies and technical assistance. We are grateful to W. C. Greene (Gladstone), J. G. Cyster (UCSF), N. R. Roan (UCSF), M. Ott (Gladstone) and K. Fontaine (Gladstone) for critical reading of the manuscript, and to G. Howard (Gladstone) for editorial assistance. This publication was made possible with help from the University of California San Francisco-Gladstone Institute of Virology & Immunology Center for AIDS Research (CFAR), an NIH-funded program (P30 AI027763), NIH S10 RR028962, and the James B. Pendelton Charitable Trust, all supporting the Gladstone Flow Core and cell sorter. The Gladstone Institutes received support for its animal care facility from a National Center for Research Resources grant (RR18928). This work was supported by grants from the National Institutes of Health (DP2 AI112244), a University of California Hellman Award, a Center for AIDS Research (CFAR) Pilot Award (P30 AI027763) and CNIHR (P30 AI027767) to S. Sanjabi.
Abbrevations
- dLN
draining LN
- PFU
plaque-forming unit
- FRT
female reproductive tract
- iLN
iliac LN
- ingLN
inguinal LN
- KO
knock-out
- I/III IFNs
type I and III interferons
- ISG
IFN-stimulated gene
- i.vag
intravaginal
- i.rec
intrarectal
- LCMV
lymphocytic choriomeningitis virus
- LCMV-NP
LCMV-nucleoprotein
- LFRT
lower female reproductive tract
- medLN
mediastinal LN
- NHP
nonhuman primates
- ssRNA
single stranded RNA
- qRT-PCR
quantitative RT-PCR
- WT
wild-type
Footnotes
Author Contributions: Conceptualization, S. Sanjabi and M. Trapecar; Methodology, M. Trapecar, B. L. Cohn, S. Khan and S. Sanjabi; Formal analysis, M. Trapecar; investigation, M. Trapecar, S. Khan., B. L. Cohn, F. Wu; Resources, S. Sanjabi; Writing (original draft), S. Sanjabi and M. Trapecar; Writing (review and editing), S. Sanjabi, M. Trapecar, S. Khan; Visualization, M. Trapecar and S. Sanjabi; Supervision, S. Sanjabi and M. Trapecar; and Funding acquisition, S. Sanjabi.
Disclosure: The authors declare no competing financial interests or other conflicts of interest.
References
- 1.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nature Reviews Immunology. 2012;12(8):592–605. doi: 10.1038/nri3251. [DOI] [PubMed] [Google Scholar]
- 2.Sewald X, Motamedi N, Mothes W. Viruses exploit the tissue physiology of the host to spread in vivo. Curr Opin Cell Biol. 2016;41:81–90. doi: 10.1016/j.ceb.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw GM, Hunter E. HIV transmission. Cold Spring Harb Perspect Med. 2012;2(11) doi: 10.1101/cshperspect.a006965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel P, Borkowf CB, Brooks JT, Lasry A, Lansky A, Mermin J. Estimating per-act HIV transmission risk: a systematic review. AIDS. 2014;28(10):1509–1519. doi: 10.1097/QAD.0000000000000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sansonetti PJ, Medzhitov R. Learning tolerance while fighting ignorance. Cell. 2009;138(3):416–420. doi: 10.1016/j.cell.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16(4):343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou X, Ramachandran S, Mann M, Popkin DL. Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future. Viruses. 2012;4(11):2650–2669. doi: 10.3390/v4112650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton LL, Mets MB, Beauchamp CL. Lymphocytic choriomeningitis virus: Emerging fetal teratogen. Am J Obstet Gynecol. 2002;187(6):1715–1716. doi: 10.1067/mob.2002.126297. [DOI] [PubMed] [Google Scholar]
- 9.Khan S, Woodruff EM, Trapecar M, Fontaine KA, Ezaki A, Borbet TC, et al. Dampened antiviral immunity to intravaginal exposure to RNA viral pathogens allows enhanced viral replication. J Exp Med. 2016;213(13):2913–2929. doi: 10.1084/jem.20161289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14(10):667–685. doi: 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- 11.Houston SA, Cerovic V, Thomson C, Brewer J, Mowat AM, Milling S. The lymph nodes draining the small intestine and colon are anatomically separate and immunologically distinct. Mucosal Immunol. 2016;9(2):468–478. doi: 10.1038/mi.2015.77. [DOI] [PubMed] [Google Scholar]
- 12.Ribeiro Dos Santos P, Rancez M, Pretet JL, Michel-Salzat A, Messent V, Bogdanova A, et al. Rapid dissemination of SIV follows multisite entry after rectal inoculation. PLoS One. 2011;6(5):e19493. doi: 10.1371/journal.pone.0019493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu W, Ma F, Churbanov A, Wan Y, Li Y, Kang G, et al. Virus-host mucosal interactions during early SIV rectal transmission. Virology. 2014;464-465:406–414. doi: 10.1016/j.virol.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haddow AD, Nalca A, Rossi FD, Miller LJ, Wiley MR, Perez-Sautu U, et al. High Infection Rates for Adult Macaques after Intravaginal or Intrarectal Inoculation with Zika Virus. Emerg Infect Dis. 2017;23(8) doi: 10.3201/eid2308.170036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8(2):167–175. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68(12):8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346(6285):629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 19.Clingan JM, Ostrow K, Hosiawa KA, Chen ZJ, Matloubian M. Differential roles for RIG-I-like receptors and nucleic acid-sensing TLR pathways in controlling a chronic viral infection. J Immunol. 2012;188(9):4432–4440. doi: 10.4049/jimmunol.1103656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 21.King CC, de Fries R, Kolhekar SR, Ahmed R. In vivo selection of lymphocyte-tropic and macrophage-tropic variants of lymphocytic choriomeningitis virus during persistent infection. J Virol. 1990;64(11):5611–5616. doi: 10.1128/jvi.64.11.5611-5616.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emonet SF, Garidou L, McGavern DB, de la Torre JC. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc Natl Acad Sci U S A. 2009;106(9):3473–3478. doi: 10.1073/pnas.0900088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paludan SR. Innate Antiviral Defenses Independent of Inducible IFNalpha/beta Production. Trends in immunology. 2016 doi: 10.1016/j.it.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Pott J, Stockinger S. Type I and III Interferon in the Gut: Tight Balance between Host Protection and Immunopathology. Front Immunol. 2017;8:258. doi: 10.3389/fimmu.2017.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207(10):2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haase AT. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu Rev Med. 2011;62:127–139. doi: 10.1146/annurev-med-080709-124959. [DOI] [PubMed] [Google Scholar]
- 27.Deruaz M, Murooka TT, Ji S, Gavin MA, Vrbanac VD, Lieberman J, et al. Chemoattractant-mediated leukocyte trafficking enables HIV dissemination from the genital mucosa. JCI Insight. 2017;2(7):e88533. doi: 10.1172/jci.insight.88533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, et al. HIV-infected T cells are migratory vehicles for viral dissemination. Nature. 2012;490(7419):283–287. doi: 10.1038/nature11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard J, Whitton JL. The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol. 1999;155(4):1205–1215. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rayamajhi M, Delgado C, Condon TV, Riches DW, Lenz LL. Lung B cells promote early pathogen dissemination and hasten death from inhalation anthrax. Mucosal Immunol. 2012;5(4):444–454. doi: 10.1038/mi.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bessa J, Zabel F, Link A, Jegerlehner A, Hinton HJ, Schmitz N, et al. Low-affinity B cells transport viral particles from the lung to the spleen to initiate antibody responses. Proc Natl Acad Sci U S A. 2012;109(50):20566–20571. doi: 10.1073/pnas.1206970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malaspina A, Moir S, Nickle DC, Donoghue ET, Ogwaro KM, Ehler LA, et al. Human immunodeficiency virus type 1 bound to B cells: relationship to virus replicating in CD4+ T cells and circulating in plasma. J Virol. 2002;76(17):8855–8863. doi: 10.1128/JVI.76.17.8855-8863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimomura Y, Ogawa A, Kawada M, Sugimoto K, Mizoguchi E, Shi HN, et al. A unique B2 B cell subset in the intestine. J Exp Med. 2008;205(6):1343–1355. doi: 10.1084/jem.20071572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16(10):626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 35.Griesbeck M, Scully E, Altfeld M. Sex and gender differences in HIV-1 infection. Clin Sci (Lond) 2016;130(16):1435–1451. doi: 10.1042/CS20160112. [DOI] [PubMed] [Google Scholar]
- 36.Jaillon S, Berthenet K, Garlanda C. Sexual Dimorphism in Innate Immunity. Clin Rev Allergy Immunol. 2017 doi: 10.1007/s12016-017-8648-x. [DOI] [PubMed] [Google Scholar]
- 37.Olson K, Macias P, Hutton S, Ernst WA, Fujii G, Adler-Moore JP. Liposomal gD ectodomain (gD1-306) vaccine protects against HSV2 genital or rectal infection of female and male mice. Vaccine. 2009;28(2):548–560. doi: 10.1016/j.vaccine.2009.09.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren W, Ma Y, Yang L, Gettie A, Salas J, Russell K, et al. Fast disease progression in simian HIV-infected female macaque is accompanied by a robust local inflammatory innate immune and microbial response. AIDS. 2015;29(10):F1–8. doi: 10.1097/QAD.0000000000000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berghofer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 Ligands Induce Higher IFN-Production in Females. The Journal of Immunology. 2006;177(4):2088–2096. doi: 10.4049/jimmunol.177.4.2088. [DOI] [PubMed] [Google Scholar]
- 40.Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. 2009;15(8):955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-alpha Production in Women. J Immunol. 2015;195(11):5327–5336. doi: 10.4049/jimmunol.1501684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swiecki M, Wang Y, Gilfillan S, Colonna M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. 2013;9(10):e1003728. doi: 10.1371/journal.ppat.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 44.Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, et al. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96(18):10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, et al. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65(3):443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 46.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. The Journal of clinical investigation. 1997;100(10):2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350(6317):423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 48.Welsh RM, Seedhom MO. Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr Protoc Microbiol. 2008;Chapter 15 doi: 10.1002/9780471729259.mc15a01s8. Unit 15A 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korns Johnson D, Homann D. Accelerated and improved quantification of lymphocytic choriomeningitis virus (LCMV) titers by flow cytometry. PLoS One. 2012;7(5):e37337. doi: 10.1371/journal.pone.0037337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reissig S, Hackenbruch C, Hovelmeyer N. Isolation of T cells from the gut. Methods Mol Biol. 2014;1193:21–25. doi: 10.1007/978-1-4939-1212-4_3. [DOI] [PubMed] [Google Scholar]
- 51.McCausland MM, Crotty S. Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J Virol Methods. 2008;147(1):167–176. doi: 10.1016/j.jviromet.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure S1. Fasting before i.rec. infection does not affect viremia at the peak of infection. C57BL/6N (WT) male mice were i.rec. infected with 1×106 PFU of LCMV without prior fasting, and viral copies in total RNA from indicated tissues were determined by qRT-PCR at day 5 post i.rec. LCMV infection. Each dot represents an individual animal.
Supplementary figure S2. Infection of immune cells after rectal LCMV transmission occurs similarly in male and female mice. WT male and female mice were infected i.rec. with 1.5 × 106 PFU of recombinant LCMV expressing GFP. At day 5 p.i., cell populations positive for LCMV GFP in the spleen were determined by flow cytometry, and are presented as frequency of total cells. Data are pooled from two independent experiments. n = 4-5 per group.
Supplementary figure S3. Male and female (WT and muMT-/-) mice have comparable viral titers in the spleen at day 3 post rectal LCMV infection. C57BL/6N (WT) and muMT-/- male and female mice were infected i.rec. with LCMV. Copies of LCMV per μg RNA were determined by qRT-PCR for the spleen at day 3 p.i. Data are pooled from two independent experiments. n = 5-6 per group. Significance was determined with the Kruskal-Wallis test; *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Table S1. Primer sequences for qRT-PCR analyses