

Abstract
Hemopexin (Hpx) binds heme with extraordinary affinity, and after haptoglobin may provide a second line of defense against the toxicity of extracellular hemoglobin (Hb). In this series of experiments, the hypothesis that Hpx protects neurons from Hb neurotoxicity was evaluated in murine primary cultures containing neurons and glial cells. Contrary to hypothesis, Hpx increased neuronal loss due to micromolar concentrations of Hb by 4-12-fold, as measured by LDH release assay; conversely, the neurotoxicity of hemin was completely prevented. The endogenous fluorescence of Hpx was quenched by Hb, consistent with transfer of Hb-bound heme to Hpx. This was associated with precipitation of globin chains, as detected by immunostaining and fluorescent Hb labeling. A portion of this precipitate attached firmly to cells and could not be removed by multiple washes. Concomitant treatment with haptoglobin (Hp) prevented globin precipitation and most of the increase in neuronal loss. Hpx weakly attenuated the increase in culture non-heme iron produced by Hb treatment, quantified by ferrozine assay. However, Hb-Hpx toxicity was iron-dependent, and was blocked by deferoxamine and ferrostatin-1. Upregulation of cell ferritin expression, a primary cell defense against Hb toxicity, was not observed on Western blots of culture lysates that had been concomitantly treated with Hpx. These results suggest that Hpx destabilizes Hb in the absence of haptoglobin, leading to globin precipitation and exacerbation of iron-dependent oxidative cell injury. Combined therapy with hemopexin plus haptoglobin may be preferable to hemopexin alone after CNS hemorrhage.
Keywords: heme, intracerebral hemorrhage, iron, stroke, subarachnoid hemorrhage
Graphical abstract
Hemopexin (Hpx) is a heme-binding protein that is considered the second line of defense against hemoglobin (Hb) toxicity, after depletion of haptoglobin (Hp). However, its effect on Hb neurotoxicity has not been defined. Hb dimers alone remain in solution and robustly induce ferritin, which sequesters iron and is protective. Hpx extracts heme moieties from Hb, leading to globin precipitation, heme delivery to low density lipoprotein receptor-related protein 1 (LRP1)-expressing neurons, blunting of the ferritin response, possible globin radical formation, and increased iron-dependent neuronal loss. Haptoglobin (Hp) prevents Hpx-mediated Hb degradation and is protective. These results suggest that combined replacement of Hpx and Hp may be preferable to Hpx monotherapy after hemorrhagic CNS insults.
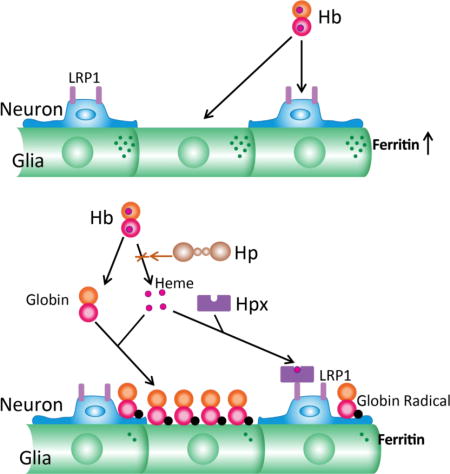
Introduction
Hemoglobin (Hb) is released in high concentrations after an intracerebral hemorrhage (ICH), and as a consequence of its heme groups is a potent oxidative neurotoxin. The primary defense mechanism against extracellular heme is scavenging, mediated by haptoglobin (Hp) and hemopexin (Hpx). These glycoproteins act in series to sequester Hb-bound or free heme, respectively, and transport it to the liver for breakdown and iron recycling (Schaer et al. 2013; Smith and McCulloh 2015). Hp binds Hb dimers with very high affinity and is well-suited to Hb clearance after mild intravascular hemolysis, but is readily depleted due to its relatively low concentration in the circulation and its endosomal degradation along with Hb. Hp binding sites are likely to be rapidly saturated in an intracerebral hematoma, as the very low CNS Hp concentration will provide negligible local supply (Galea et al. 2012). Hpx is widely regarded as the backup scavenger that is utilized after Hp depletion (Deuel et al. 2015). The potent pro-oxidant effect of heme is neutralized after Hpx binding, which likely contributes to its efficacy in ICH models (Chen et al. 2011; Leclerc et al. 2016). However, over the past decade other effects of Hpx have been identified that may also be relevant to hemorrhagic stroke, including dampening of the inflammatory response and facilitation of cellular heme export (Yang et al. 2010; Lin et al. 2012b)
Due to its extraordinary binding affinity (Kd < 1 pM, (Tolosano et al. 2010), Hpx directly extracts heme from Hb (Kassa et al. 2016), a reaction that may initiate the breakdown of extracellular Hb when Hp binding sites are saturated (Hrkal et al. 1974; Miller et al. 1996; Kassa et al. 2016). This transfer is favored when heme moieties are oxidized to their ferric form (methemoglobin), as is predictably observed over days after an ICH due to Hb autoxidation (Bradley 1993). Heme extraction is not random, but rather proceeds by sequential loss of heme moieties from Hb β chains followed by loss from Hb α chains (Hrkal et al. 1974). After heme moiety loss, Hb dimers or tetramers are markedly less soluble than either Hb or methemoglobin, and form aggregates that precipitate in vitro (Miller et al. 1996). Globin per se is tolerated under some experimental conditions (Yoo et al. 1999). However, hydrogen peroxide generated during Hb autoxidation may react with residual ferric heme moieties to produce highly reactive ferrylhemoglobin and globin radicals, which can initiate lipid, protein and nucleic acid oxidation (McArthur and Davies 1993).
Although hemopexin mitigates the pro-oxidant effect of Hb in the presence of serum Hp (Deuel et al. 2015), surprisingly little is known of the consequences of extracellular Hb degradation by Hpx on neuronal vulnerability to Hb. In the present series of experiments, this process was investigated in a defined model of Hb neurotoxicity.
Methods
Cell culture preparation
All animal use was approved by the Thomas Jefferson University Institutional Animal Care and Use Committee (Protocol #00013). Animals (2-6 month old males and females, 22-30 grams) were bred and reared in the Thomas Jefferson University animal facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Founding mice were originally purchased from Taconic Bioscience (RRID:IMSR_TAC:b6129). Breeding mice were housed with one or two companions in 75 JAG micro-vent mouse cages (Allentown, Inc., Allentown, NJ, USA), and were provided with food and water ad libitum. Visibly pregnant and lactating females were housed separately until weaning. Cultures containing both neurons and glial cells were prepared from cortices of fetal mice at gestational day 14-16 as previously described in detail (Rose et al. 1993; Rogers et al. 2003). Plating medium contained 5% equine serum and 5% fetal bovine serum (Hyclone, Logan, UT, USA), 2 mM glutamine, and MEM (Gibco 11430) or DMEM (Gibco 11960). Glial-only cultures were prepared from postnatal day 2-4 mice, also following a previously published method (Li et al. 2009b).
Two thirds of the medium of neuron-glia cultures was replaced on day 5 in vitro with feeding medium, which contained 10% equine serum and lacked fetal bovine serum. Feeding was repeated on day 9 and then daily beginning on day 11. Medium replacement of glial-only cultures began on day 5-6 and was repeated every four days thereafter.
Cortical cell cultures prepared via this method have been extensively characterized. Neurons are readily distinguished from glial cells due to the phase-bright appearance of their somata. Prior studies have demonstrated that these cells express neuronal markers, exhibit spontaneous action potentials, and reliably depolarize with an increase in membrane conductance in response to an excitatory stimulus (Choi et al. 1987; Peters et al. 1987; Christine and Choi 1990).
Hemoglobin, hemin, and hemopexin exposure
The study time-line is depicted in Fig. 1. All direct comparisons were made between cultures prepared under identical conditions and using the same serum lots. Researchers conducting experiments were blinded to plating order, which may produce minor alterations in neuron density of mixed cultures. The primary inclusion criterion was culture age greater than 11 days in vitro; cultures were excluded only if they had evidence of bacterial or fungal contamination, cell peeling or cell death prior to the experiment. All experiments were conducted on cultures at days 12-28 in vitro. On the day prior to the experiment, two thirds of the culture medium was replaced with serum-free medium, reducing serum content to 3.3%. All Hb exposures were conducted in serum-free MEM supplemented with glucose (MEM10, total glucose concentration 10 mM). Assignment to study groups was guided by a random number generator (www.random.org). The experimenter was blinded to experimental conditions during outcome assessments, which were prespecified and standard for this model.
Fig. 1.
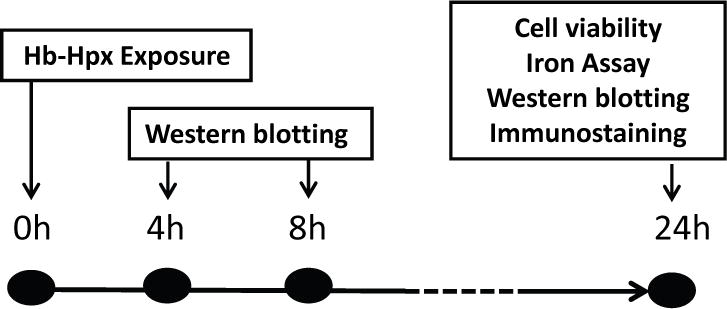
Study time-line, indicating time points (hours) after hemoglobin (Hb) ± hemopexin (Hpx) treatment at which indicated testing was conducted.
Injury Assessment
Neuronal death was quantified by LDH release assay, which provides an accurate and automated measure of neuronal death in this cell culture system (Koh and Choi 1988), following a previously-described method (Regan et al. 2008). This assay was initially developed to quantify neuronal death due to glutamate toxicity in these cultures, and was adapted to quantify hemoglobin neurotoxicity when it was observed that Hb also produced cell injury that was selective for neurons and spared glial cells (Regan and Panter 1993; Regan et al. 2000; Chen-Roetling and Regan 2006). The LDH activity detected in control cultures subjected to medium exchanges only was subtracted to quantify the signal specific to Hb neurotoxicity. All values in mixed neuron-glia cultures were normalized to those in sister cultures treated with 300 μM N-methyl-D-aspartate (NMDA), which produces 100% neuronal death without injuring glial cells. LDH values in toxicity experiments using glia-only cultures were normalized to those in cultures treated with 0.1% Triton-X 100, which lyses all cells.
Iron assay
Non-heme chlelatable iron was quantified on culture lysates using the method of Pountney et al. (Pountney et al. 1999; Chen-Roetling and Regan 2016). This assay measures free or loosely-bound iron that is available for chelation by ferrozine, resulting in a colored product that is detected by spectrophotometry (Abs 562 nm).
Immunoblotting and immunostaining
Cells were harvested after incubation with Hb alone or with Hpx for 4 or 8 hours, which are time points that precede cell lysis after exposure to 3 μM Hb in this model. Culture medium was completely aspirated, and cultures were washed with MEM10 using an Eppendorf Repeater pipette to remove unbound aggregates (>1000-fold dilution). Lysis buffer contained ice cold 210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EDTA, 0.1 % sodium dodecyl sulfate, and 0.1 % Triton X-100. For immunostaining, cultures were fixed in 100% methanol for 10 minutes. Standard immunoblotting and immunostaining methods were then followed (Chen-Roetling et al. 2011). Antibodies and dilutions are listed in Materials subsection. Validation data were provided by manufacturer and confirmed by Western blotting.
Fluorescent protein labeling
Hb and hemopexin solutions (2 mg/ml) were fluorescently labeled with FITC-EX or Alexa Fluor 568, using protein labeling kits (Molecular Probes Life Technologies, Carlbad, CA, RRID:SCR_013318). Unreacted dye was separated from the protein solution using Amicon Ultra centrifugal filter units with 10K membranes (EMD Millipore, Billerica, MA, RRID:SCR_008983).
Heme transfer assay
Heme transfer from Hb to hemopexin was monitored fluorometrically following the method of Miller et al. (Miller et al. 1996), which is based on the quenching of hemopexin fluorescence by heme binding. Hb and Hpx were diluted on 96 well plates in a bicarbonate-based buffer containing the following (mM): NaCl 117.9, KCl 5.3, MgSO4 0.8, NaH2PO4 1.0, CaCl2 1.8, Glucose 10. The plate was incubated at 37°C in a 5% CO2 incubator until the designated reading time. Fluorescence (Ex 285 nm, Em 345 nm) was quantified using a Molecular Devices Spectramax M2 plate reader.
Statistical analysis
All data were amenable to parametric analysis. Statistical significance was tested via one way ANOVA and the Bonferroni multiple comparisons test, using GraphPad Prism 4 software. Sample size was determined by power calculation, using a two tailed α= 0.05 and β = 0.20, and the expected standard deviation for the parameters to be tested. Each unit is defined as a single culture.
Materials
Antibodies were purchased from the following vendors, and used at indicated dilutions:
HO-1: Enzo Life Sciences, Farmingdale, NY, USA, ADI-SPA-896 (RRID:AB_10614948), 1:2000 Ferritin, Sigma-Aldrich, St. Louis, MO, USA, F6136, RRID:AB_259684, 1:250
Actin: Sigma-Aldrich, A2066, RRID: AB_476693, 1:1000
NeuN: Millipore Sigma MAB377X (RRID: AB_2149209) or MAB377 (RRID:AB_2298772), 1:25-1:100
LRP1: Santa Cruz Biotechnology sc-25469 (RRID: AB_2139163)
α-Hemoglobin: Santa Cruz Biotechnology sc-514378, RRID: AB_2716828, 1:2000
β-Hemoglobin, Santa Cruz Biotechnology sc-21757, RRID:AB_627713, 1:2000
Hemin was purchased from Frontier Scientific, Logan, UT, USA (RRID:SCR_000914)
Purified proteins were provided as gifts as follows:
Human plasma hemopexin, CSL Behring, King of Prussia, PA; additional human plasma hemopexin was purchased from Athens Research and Technology, Athens, GA (RRID:SCR_001079).
Human plasma haptoglobin, Bio Products Laboratory (BPL), Herfordshire, UK
Human hemoglobin A, Hemosol Inc, Etobicoke, Ontario, Canada
Preregistration statement
This study was not preregistered.
Results
Hemopexin increases neuronal vulnerability to Hb in the absence of haptoglobin
Cultures treated with 3-10 μM Hb sustained weak neuronal loss (Fig. 2) that was increased by concomitant treatment with 1 mg/ml Hpx. A similar effect was observed when Hpx purchased from Athens Research and Technology was used (data not shown). Injury was limited to phase-bright cells that assembled in clusters on the culture monolayer, which is the typical appearance of neurons in these primary cultures (Chen-Roetling and Regan 2016). The glial monolayer remained intact, without any appearance of cell injury. Consistent with an injury that was limited to neurons, no significant LDH release was detected in glial-only cultures subjected to the same treatments (Fig. 2D–F, H). A fine granular precipitate was observed under phase-contrast microscopy in cultures treated with Hb + Hpx which was not removed by medium exchanges or by washing viable or fixed cultures. In contrast to its deleterious effect on Hb toxicity, Hpx completely prevented neuronal injury in mixed cultures treated with hemin rather than Hb (Fig. 2G).
Fig. 2.
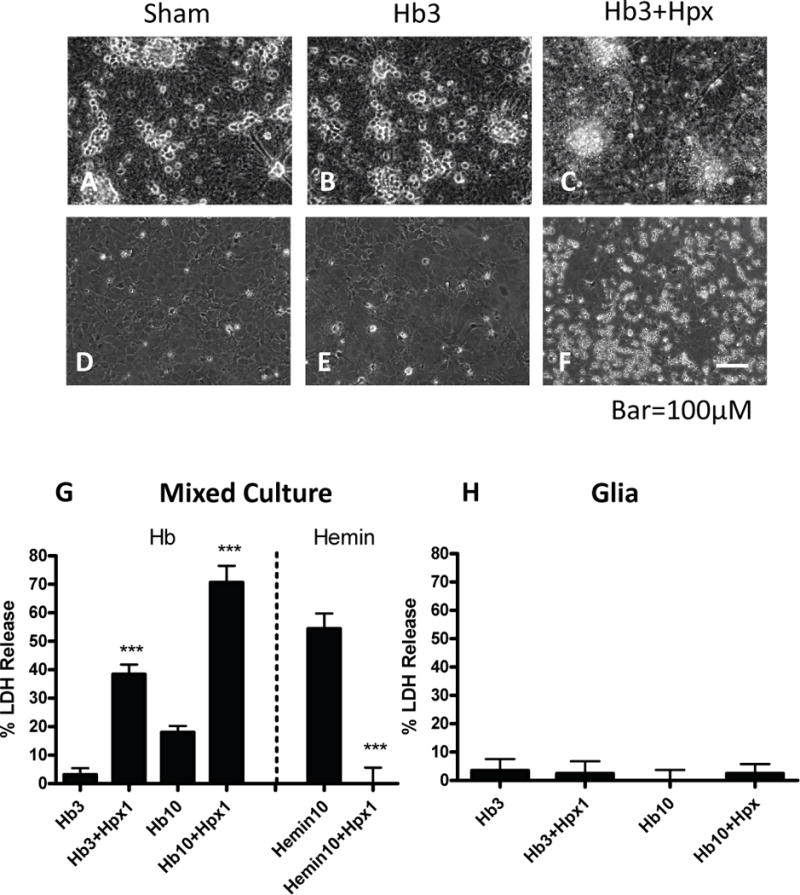
Effects of hemopexin on hemoglobin and hemin toxicity. (A-C) Phase contrast images of mixed neuron-glia cultures subjected to: (A) sham medium exchange only; phase bright neuron cell bodies cluster in groups and overlie a confluent glial monolayer; B) hemoglobin (Hb) 3 μM for 24 h; (C) Hb 3 μM plus hemopexin (Hpx) 1 mg/ml; neuronal degeneration and a fine precipitate are apparent, while the glial monolayer remains intact. (D-F) Glial-only cultures treated as in A-C; glial monolayer remains intact; precipitate is apparent in Hb+Hpx condition. (G) Bars represent mean neuronal loss (± S.E.M., n = 7 cultures for Hb 3 μM condition, 15 cultures/condition for Hb 10 μM, and 8 cultures/condition for hemin experiments) in mixed neuron-glia cultures as measured by LDH assay. (H) Percentage glial cell loss (± S.E.M., n = 12 cultures/condition) in glia-only cultures treated with Hb alone or with 1 mg/ml Hpx as in G. The low LDH values in sham cultures were subtracted from each mean value to yield the signal associated with neurotoxicity. ***p < 0.001 versus value in Hb or hemin only groups.
Hb+Hpx neurotoxicity is oxidative and iron-dependent
Hb+Hpx neurotoxicity was significantly reduced by antioxidants (ascorbate, Trolox) and by the ferric iron chelator deferoxamine (Fig. 3), in agreement with their effects on the toxicity of Hb alone (Regan and Panter 1993; Regan and Guo 1998; Zille et al. 2017). It was completely prevented by ferrostatin-1.
Fig. 3.
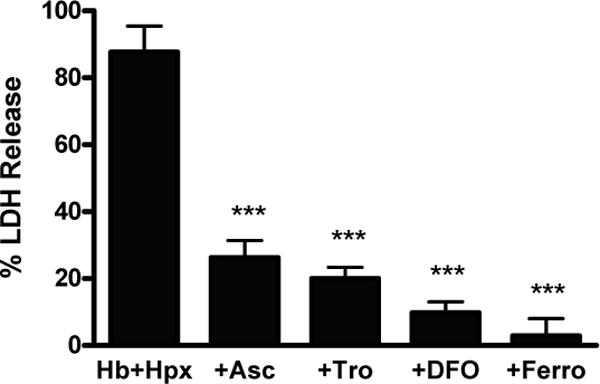
Hemoglobin+hemopexin toxicity is oxidative and iron-dependent. Percentage neuron loss in mixed neuron-glia cultures treated with Hb 3 μM+Hpx 1 mg/ml alone (n = 14 cultures) or with 100 μM ascorbate (Asc, n = 10 cultures), 100 μM Trolox (Tro, n = 10 cultures), 100 μM deferoxamine (DFO, n = 10 cultures), or 10 μM ferrostatin-1 (Ferro, n = 10 cultures). ***p < 0.001 versus Hb+Hpx condition.
Hemopexin extracts heme from hemoglobin
Prior work has demonstrated that Hpx removes heme moieties from Hb, resulting in formation of protein aggregates (Hrkal et al. 1974; Miller et al. 1996). It was hypothesized that a similar phenomenon was occurring when the Hb and Hpx preparations used in the present experiments were combined, accounting for the precipitate seen in cultures. Consistent with this hypothesis, Hpx fluorescence quenching in the presence of Hb, a marker of heme transfer (Miller et al. 1996), was observed within 10 minutes and continued over the subsequent hour (Fig. 4).
Fig. 4.
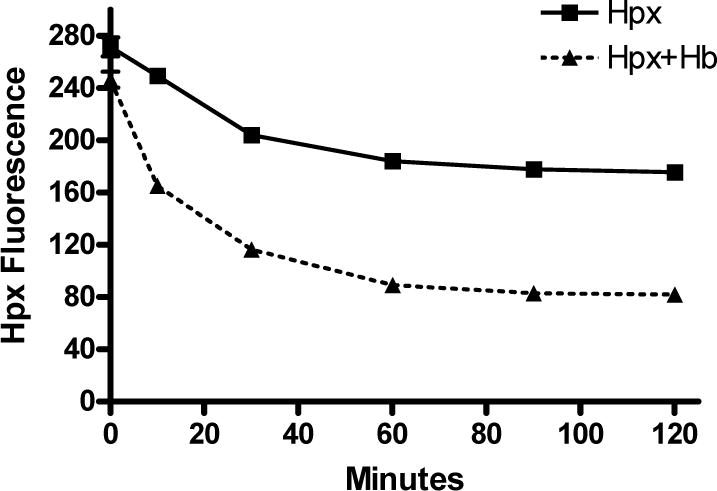
Hemopexin binds heme moieties of Hb. Fluorescence quenching of 1 μM Hpx solution by Hb (0.25 μM containing 1 μM heme) over two hours, consistent with transfer of heme to Hpx. Each point represents mean of 5 replicates.
Neurons express LRP1 and bind hemopexin
In agreement with prior reports (Bu et al. 1994; Hvidberg et al. 2005), neurons expressed the Hpx receptor LRP1 in this culture system (Fig. 5A–D). Consistent with neuronal LRP1 expression, Alexa Fluor 568-labeled Hpx colocalized with anti-NeuN when applied to fixed cells (Fig 5E–H). The labeled apoform, which also binds to LRP1 but with lower affinity than the Hpx-hemin complex (Hvidberg et al. 2005), was used in these experiments due to fluorescence quenching of the labeled Hpx by hemin binding. However, in living cultures treated overnight with Alexa Fluor 568-labeled Hpx plus equimolar hemin, uptake produced a strong fluorescent signal that was localized to phase bright cells with the typical appearance of neurons, suggesting restoration of fluorescence by viable cells (Fig. 5I–J).
Fig. 5.
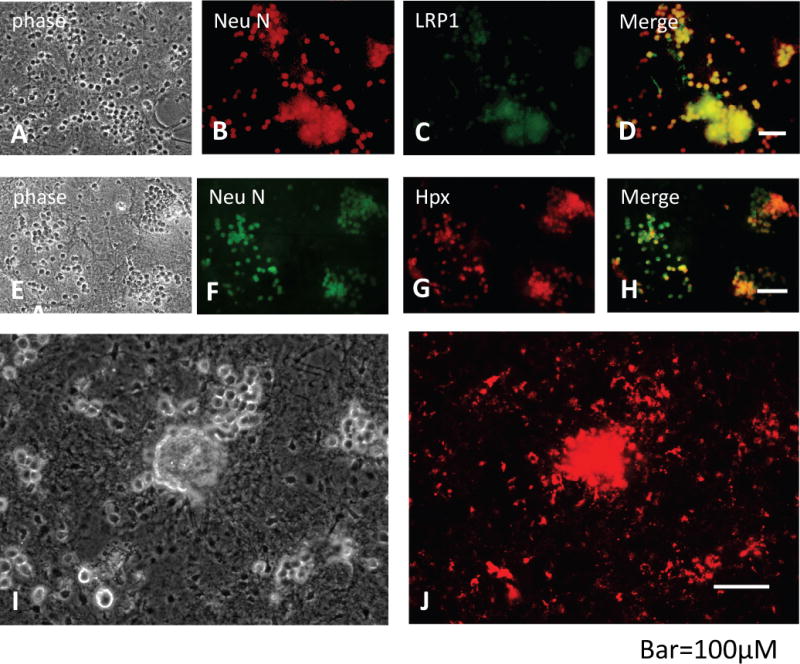
Neurons express LRP1 and bind hemopexin. (A-D) Phase contrast and fluorescence images of fixed cultures immunostained with anti-NeuN and anti-LRP1. (E-H) Fixed cultures stained with anti-NeuN and 80 μg/ml solution of Alexa Fluor 568-labeled Hpx for 4 h; the labeled Hpx apoform was used since hemin binding quenched fluorescence when applied to fixed cells. (I, J) Viable culture incubated with 1 μM Alexa Fluor 568-labeled Hpx-hemin solution for 24 h. Uptake by phase bright neuron somata is apparent.
Hemopexin without haptoglobin precipitates globin chains
Based on observations that heme extraction decreases Hb solubility (Jacob and Winterhalter 1970; Hrkal et al. 1974), it was hypothesized that the precipitate observed in cultures treated with Hb plus Hpx without haptoglobin consisted of Hb or globin aggregates rather than heme or a culture medium component. When cultures were treated with FITC-labeled Hb plus unlabeled Hpx, a strong fluorescent signal was observed in the precipitate (Fig. 6). Addition of Hp to Hb+Hpx kept Hb in solution, as no granular precipitate was observed with phase-contrast or fluorescence microscopy. The weak fluorescent signal in Hb+Hpx+Hp-treated cultures was localized to phase-bright neurons, consistent with prior observations of Hb uptake by rodent neurons (Lara et al. 2009; Chen-Roetling and Regan 2016). Ascorbate weakly increased Hb precipitation, while deferoxamine and ferrostatin-1 had no effect.
Fig. 6.
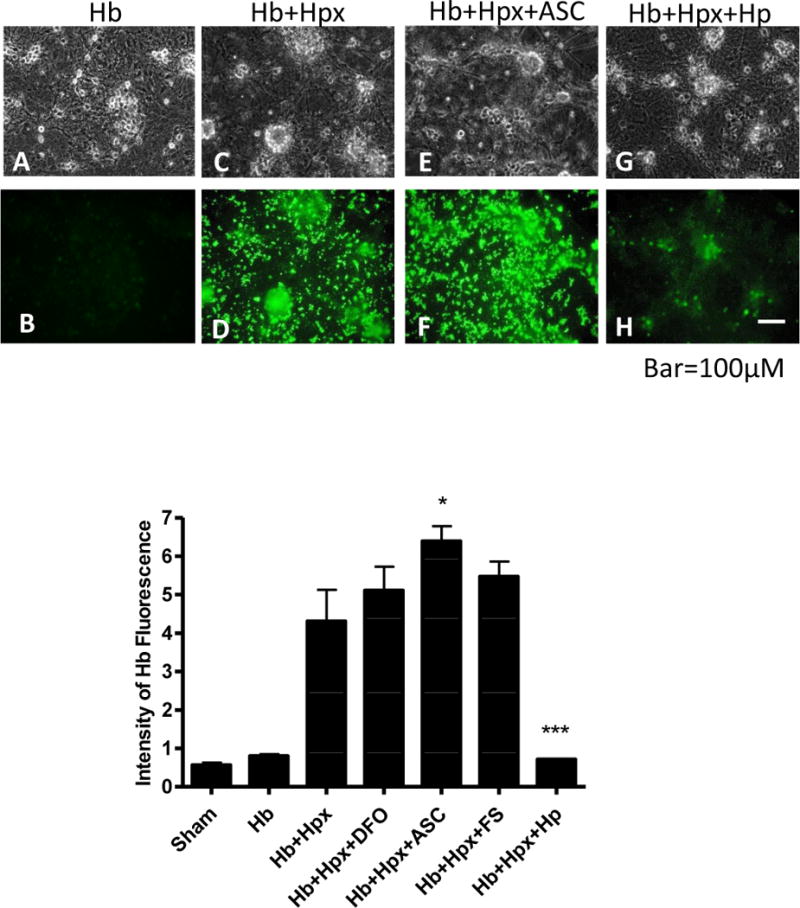
Hemoglobin precipitation by hemopexin is prevented by haptoglobin. Viable cultures were treated for 24 hours with FITC-conjugated Hb (3 μM) alone (A,B), with unlabeled Hpx 1 mg/ml (C,D), or with Hb+Hpx plus 100 μM ascorbate (Asc, E,F) or 1 mg/ml haptoglobin (Hp, G,H). Bars represent mean fluorescence intensity in cultures treated as above or with deferoxamine (DFO) 100 μM or ferrostatin-1 (FS) 10 μM. *p < 0.05, ***p < 0.001 versus Hb+Hpx condition, n = 6 cultures/condition for sham, Hb and Hb+Hpx, 4 cultures/condition for +DFO, +Asc and +FS, and 5 cultures/condition for +Hp.
In order to determine if Hpx increased the Hb protein content of cultures, immunoblots of culture lysates were immunostained with two anti-Hb monoclonal antibodies that were specific to α or β-chains (Fig. 7). Compared with the signal in cultures treated with Hb alone, Hpx increased the globin signal over twofold, with similar results observed from both antibodies. However, since precipitate was still visible on cells after multiple washes, this signal is likely due at least in part to globin chains tightly bound to cell membranes.
Fig. 7.
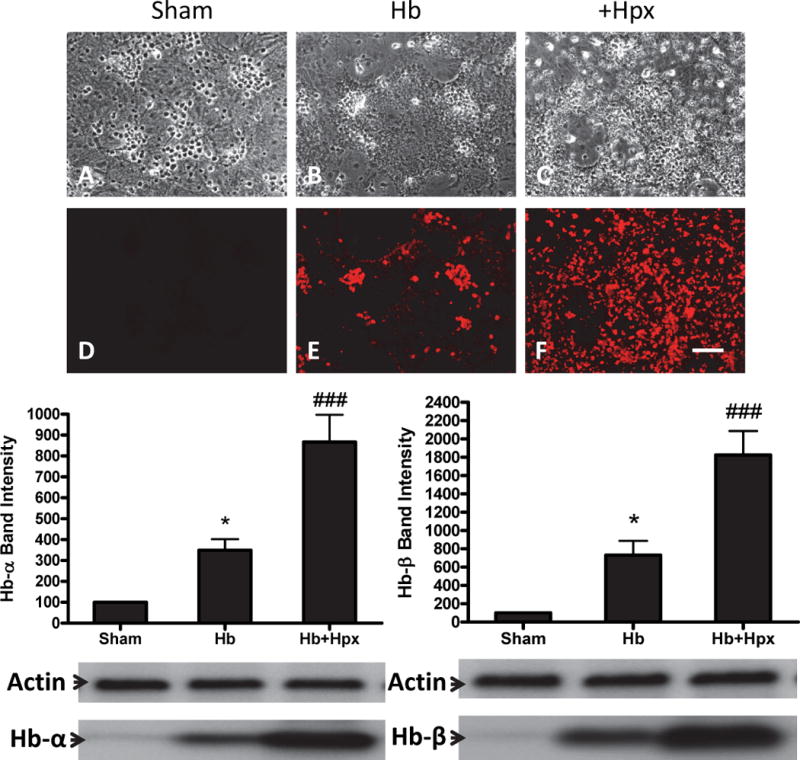
Hemopexin increases hemoglobin precipitation and cell hemoglobin uptake. (A-F) Cultures were fixed after 8h exposure to Hb 3 μM plus Hpx 1 mg/ml, then stained with antibody to α-Hb. Bars represent mean Western blot band intensities (± S.E.M., 13 cultures/condition) from cultures receiving the same treatment, stained with antibodies specific to human α-Hb and β-Hb chains. Gel lane order corresponds with bar order. *p < 0.05 versus sham, ###p < 0.001 versus Hb alone condition.
Haptoglobin attenuates Hb+Hpx neurotoxicity
Most of the increase in neuronal death produced by adding Hpx to Hb was prevented by an equal concentration of Hp (Fig. 8A). However, cell death in Hp-treated cultures remained at a level that was greater than that produced by Hb alone, as recently reported (Chen-Roetling and Regan 2016). Cell nonheme free or loosely-bound iron in Hb-treated cultures was weakly but significantly reduced by Hpx alone at 24 hours after exposure onset, and was further reduced by addition of Hp (Fig. 8b). This decrease in culture iron content by Hpx was not due to iron loss into the culture medium from lysing neurons, since similar results were observed when neuronal death was prevented by ferrostatin-1 (Fig. 8C).
Fig. 8.
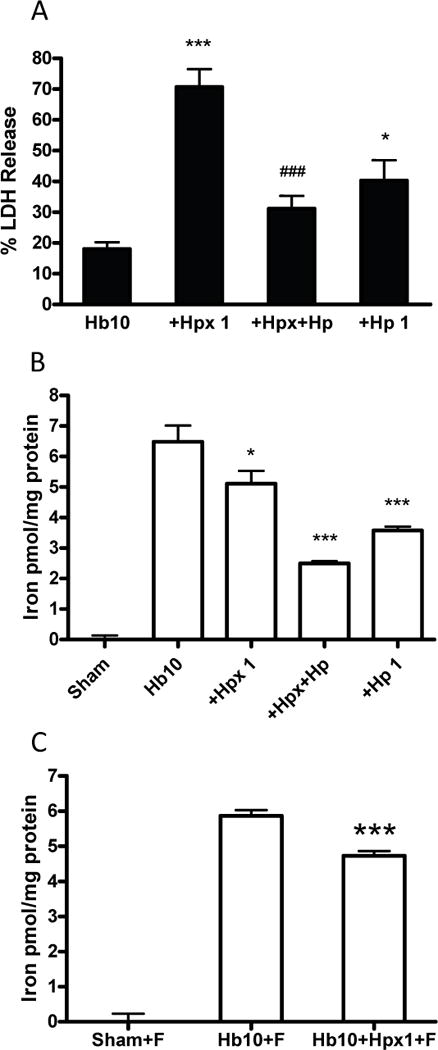
Haptoglobin attenuates Hb+Hpx neurotoxicity and nonheme chelatable iron increase. (A) Percentage neuron loss, measured by LDH release assay, in cultures treated for 24 h with Hb 10 μM alone (n = 15 cultures) or with Hpx 1 mg/ml (n = 15 cultures), Hpx 1 mg/ml+Hp 1 mg/ml (n = 8 cultures), or Hp 1 mg/ml without Hpx (n = 8 cultures); (B) nonheme iron content of cultures (n = 7-8) subjected to same treatment, as detected by ferrozine assay; (C) cultures (10/condition) treated with Hb alone or with Hpx as in B, but with addition of 10 μM ferrostatin-1 (F) to prevent cell lysis and neuron iron loss. *p < 0.05, ***p < 0.001 versus Hb alone or Hb + ferrostatin-1 condition, ###p < 0.001 versus Hb+Hpx condition.
Hemopexin reduces heme oxygenase-1 and ferritin induction
In order to further investigate the discordant result that Hpx weakly attenuated the increase in Hb-mediated iron content in these cultures but increased iron-dependent neuronal death, its effect on endogenous cellular defenses was investigated. Cells were harvested for this experiment at 4-8 hours after treatment with Hb ± Hpx, time points preceding LDH release, to prevent any confounding by neuronal death and protein loss. Cell heme is detoxified by the sequential activities of heme oxygenase-1 (HO-1) and ferritin (Balla et al. 1992; Teng et al. 2004; Regan et al. 2008). HO-1 induction was reduced by concomitant Hpx treatment, and ferritin upregulation was completely prevented (Fig. 9).
Fig. 9.
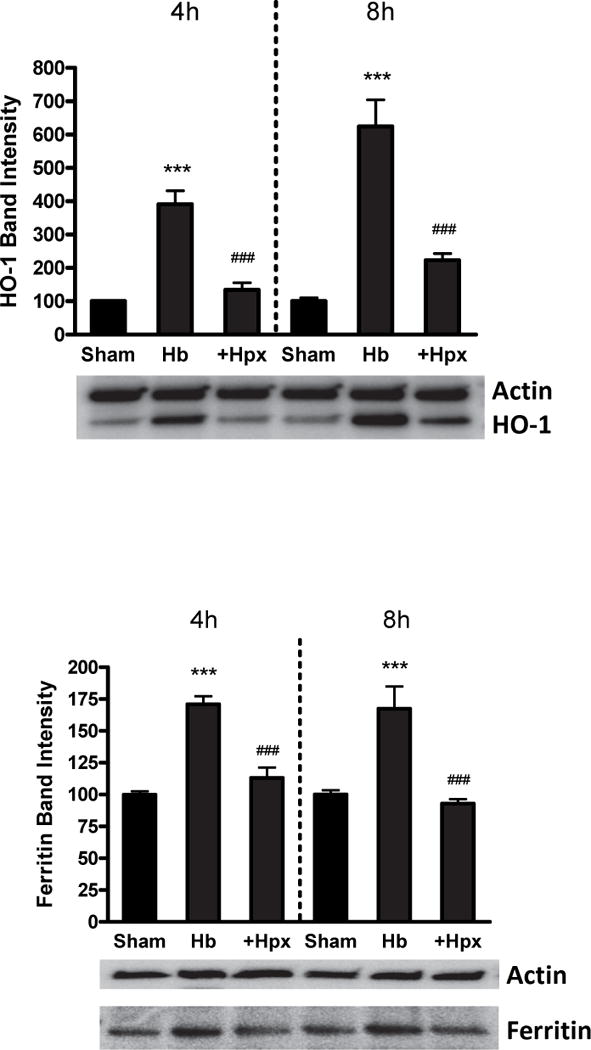
Hemopexin reduces heme oxygenase-1 (HO-1) and ferritin induction by hemoglobin. HO-1 and ferritin expression in cultures treated with Hb 3 μM alone or with 1 mg/ml Hpx for 4 or 8 hours. ***p < 0.001 versus sham, ###p < 0.001 versus Hb alone condition, n = 6 cultures/condition.
Discussion
This paper reports three novel observations. First, a physiologically-relevant concentration of Hpx unexpectedly increased neuronal loss after Hb exposure in Hp-free medium, challenging the concept of Hpx as a second line of defense against Hb toxicity. Second, in agreement with prior observations in cell-free models (Miller et al. 1996), Hpx destabilized Hb, resulting in precipitation of globin chains, their binding to cell membranes, and perhaps cell uptake. Third, despite a slight reduction in cell nonheme iron, the increase in neuronal injury produced by Hpx was iron-dependent, and was associated with loss of the protective ferritin response to Hb exposure.
These results contrast with the beneficial effects of Hpx and its LRP1 receptor that have been previously reported in multiple models of hemorrhagic stroke (Chen et al. 2011; Leclerc et al. 2016; Ma et al. 2016; Wang et al. 2017). The latter studies were conducted in mice expressing Hp, which in the present study prevented Hb precipitation by Hpx and attenuated neuronal loss. The effect of Hpx on hemorrhagic CNS injury when Hp is reduced or absent has not been specifically reported. Zhao et al. (Zhao et al. 2009) observed that Hp depletion by Hb pretreatment or Hp gene knockout worsened injury in a blood injection ICH model. Although Hpx levels were not reported, Hp KO mice have a compensatory increase in Hpx expression (Tolosano et al. 2002). A deleterious effect of Hpx on intravascular hemolysis-induced renal injury has been reported by Tolosano et al. (Tolosano et al. 2002). Compared with WT or Hp KO mice, Hpx-Hp double knockouts sustained less tubular Hb deposition, consistent with the present observation that Hpx induced Hb precipitation in the absence of Hp. When hemolytic stress was severe, renal injury and mortality were also reduced in double knockouts. The protective effect of Hp against extracellular Hb may therefore be due in part to prevention of heme removal from Hb by Hpx and the consequent reduction in Hb solubility (Miller et al. 1996). However, any benefit of Hp in this cell culture model is limited by neuronal expression of CD163, which targets Hb-Hp to neurons and away from glial cells that are better equipped for Hb breakdown (Chen-Roetling and Regan 2016).
The protective effect of Hpx after hemorrhagic stroke is likely mediated by regulation of heme trafficking rather than direct scavenging of the heme moieties of Hb after Hp depletion. The primary mechanism of hematoma resolution is erythrophagocytosis by microglia and macrophages. These cells abundantly express the heme export protein FLVCR1, consistent with their role in heme recycling (Quigley et al. 2004; Keel et al. 2008). After Hb breakdown, the export of excess heme is largely dependent on extracellular Hpx as acceptor protein. Yang et al. reported that macrophages exported 52% of a heme load within 2 hours when Hpx was present in the culture medium, compared with only 6% when Hpx was replaced by equimolar albumin (Yang et al. 2010). Consistent with a role for Hpx in heme export by microglia and macrophages, the heme content of an intracerebral hematoma decreased more rapidly in WT mice than in Hpx knockouts (Chen et al. 2011). Although some exported heme may be taken up by LRP1-expressing neurons, equimolar Hpx reduces the net uptake of hemin in this culture system by over 90% (Chen-Roetling et al. 2012), in agreement with its protective effect on hemin neurotoxicity (Li et al. 2009a). Sequestration of exported heme by Hpx has the added benefit of mitigating its inflammatory effect, which is mediated by TLR4 binding (Lin et al. 2012a; Lin et al. 2012b).
The nonheme iron content detectable by the ferrozine assay in cultures treated with Hb+Hpx was slightly but significantly less than that of cultures treated with Hb alone at 24 hours. Nevertheless, the cell injury produced by Hb+Hpx, like that of Hb alone (Zille et al. 2017), was iron-dependent and was robustly reduced by the Fe (III) chelator deferoxamine. The observations that Hpx increases the iron-dependent toxicity of Hb without increasing the cellular iron burden may be reconciled by consideration the effects of extracellular iron and ferritin on Hb+Hpx toxicity.
Extracelluar oxyhemoglobin spontaneously oxidizes to Fe(III)-methemoglobin, concomitantly producing hydrogen peroxide (H2O2) (Rifkind et al. 2014). Subsequent reaction of H2O2 with Fe(III)-methemoglobin yields Fe(IV)-ferrylhemoglobin, followed by transfer of unpaired electrons to globin chains to produce a variety of highly reactive protein radicals (Deterding et al. 2004). In the presence of Hpx these globin radicals would be localized to the membrane-bound or cellular globin precipitate, potentiating oxidative injury by proximity. Fe(IV)-ferrylhemoglobin also reacts with H2O2 to produce heme degradation products plus Fe(III) (Rifkind et al. 2014); the latter appears to be critical to globin radical generation, which is blocked by deferoxamine (McArthur and Davies 1993).
Ferritin has a high capacity for iron sequestration and detoxification, and is rapidly increased in these cultures by Hb treatment (Chen-Roetling and Regan 2016). In the present study, this upregulation was blocked by Hpx. Although ferritin expression is almost exclusively localized to glial cells, it provides robust protection to cultured neurons treated with Hb (Regan et al. 2008), which may be mediated by the combined effects of glial iron trapping and ferritin secretion (Greco et al. 2010; Hohnholt and Dringen 2013). The failure of Hpx-treated cultures to appropriately increase ferritin in response to Hb may contribute to their increased neuronal loss. Further investigation is needed to define the mechanism by which Hpx blocks ferrritin upregulation in this model, but more selective heme delivery likely plays a key role. Hb-bound heme is diffusely taken up by glial cells and neurons in these cultures (Chen-Roetling and Regan 2016), while Hpx-bound heme localizes to LRP1+ neurons, which have little capacity for ferritin synthesis in response to iron loading (Bishop and Robinson 2001; Healy et al. 2016). LRP1 expression has also been reported in glial cells in the intact CNS, although neuronal predominance has been observed in the mouse hippocampus and cortex (Xu et al. 2012; Auderset et al. 2016), consistent with these primary cell culture results. However, it is possible that the effect of Hpx on ferritin expression and Hb neurotoxicity may differ in brain regions with predominantly glial LRP1 expression.
The relevance of these in vitro observations to CNS hemorrhage and other hemolytic diseases remains to be determined. Although Hb degradation by Hpx and subsequent protein aggregate formation was first reported over four decades ago (Hrkal et al. 1974), it has not previously been considered a potential source of toxicity. The present results indicate that monitoring for Hb chain precipitation would be appropriate during therapeutic trials of Hpx, particularly in experimental models and disease states that are associated with Hp depletion. Combined therapy with Hpx plus Hp may be preferable to Hpx monotherapy.
Acknowledgments
This study was supported by NIH grants R21NS088986 and RO1NS095205. The authors thank CSL Behring, King of Prussia, PA and Bio Products Laboratory (BPL), Herfordshire, UK for generous supplies of hemopexin and haptoglobin, respectively.
Abbreviations
- Hb
hemoglobin
- Hp
haptoglobin
- Hpx
hemopexin
- ICH
intracerebral hemorrhage
- LDH
lactate dehydrogenase
- RRID
Research resource identifiers
Footnotes
PROF. RAYMOND F REGAN (Orcid ID : 0000-0003-2567-6632)
Conflict of interest disclosure
The authors report no conflicts of interest.
References
- Auderset L, Cullen CL, Young KM. Low Density Lipoprotein-Receptor Related Protein 1 Is Differentially Expressed by Neuronal and Glial Populations in the Developing and Mature Mouse Central Nervous System. PLoS One. 2016;11:e0155878. doi: 10.1371/journal.pone.0155878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: A cytoprotective strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- Bishop GM, Robinson SR. Quantitative analysis of cell death and ferritin expression in response to cortical iron: implications for hypoxia-ischemia and stroke. Brain Res. 2001;907:175–187. doi: 10.1016/s0006-8993(01)02303-4. [DOI] [PubMed] [Google Scholar]
- Bradley WG., Jr MR appearance of hemorrhage in the brain. Radiology. 1993;189:15–26. doi: 10.1148/radiology.189.1.8372185. [DOI] [PubMed] [Google Scholar]
- Bu G, Maksymovitch EA, Nerbonne JM, Schwartz AL. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem. 1994;269:18521–18528. [PubMed] [Google Scholar]
- Chen-Roetling J, Regan RF. Effect of heme oxygenase-1 on the vulnerability of astrocytes and neurons to hemoglobin. Biochem Biophys Res Commun. 2006;350:233–237. doi: 10.1016/j.bbrc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Regan RF. Haptoglobin increases the vulnerability of CD163-expressing neurons to hemoglobin. J Neurochem. 2016;139:586–595. doi: 10.1111/jnc.13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Chen L, Regan RF. Apotransferrin protects cortical neurons from hemoglobin toxicity. Neuropharmacology. 2011;60:423–431. doi: 10.1016/j.neuropharm.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Liu W, Regan RF. Hemopexin decreases hemin accumulation and catabolism by neural cells. Neurochem Int. 2012;60:488–494. doi: 10.1016/j.neuint.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhang X, Chen-Roetling J, Regan RF. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J Neurosurg. 2011;114:1159–1167. doi: 10.3171/2010.10.JNS10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christine CW, Choi DW. Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. J Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J Biol Chem. 2004;279:11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- Deuel JW, Vallelian F, Schaer CA, Puglia M, Buehler PW, Schaer DJ. Different target specificities of haptoglobin and hemopexin define a sequential protection system against vascular hemoglobin toxicity. Free Radic Biol Med. 2015;89:931–943. doi: 10.1016/j.freeradbiomed.2015.09.016. [DOI] [PubMed] [Google Scholar]
- Galea J, Cruickshank G, Teeling JL, Boche D, Garland P, Perry VH, Galea I. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem. 2012;121:785–792. doi: 10.1111/j.1471-4159.2012.07716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Seeholzer SH, Mak A, Spruce L, Ischiropoulos H. Quantitative mass spectrometry-based proteomics reveals the dynamic range of primary mouse astrocyte protein secretion. J Proteome Res. 2010;9:2764–2774. doi: 10.1021/pr100134n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy S, McMahon J, Owens P, FitzGerald U. Significant glial alterations in response to iron loading in a novel organotypic hippocampal slice culture model. Sci Rep. 2016;6:36410. doi: 10.1038/srep36410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohnholt MC, Dringen R. Uptake and metabolism of iron and iron oxide nanoparticles in brain astrocytes. Biochem Soc Trans. 2013;41:1588–1592. doi: 10.1042/BST20130114. [DOI] [PubMed] [Google Scholar]
- Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur J Biochem. 1974;43:73–78. doi: 10.1111/j.1432-1033.1974.tb03386.x. [DOI] [PubMed] [Google Scholar]
- Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106:2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- Jacob HS, Winterhalter KH. The role of hemoglobin heme loss in Heinz body formation: studies with a partially heme-deficient hemoglobin and with genetically unstable hemoglobins. J Clin Invest. 1970;49:2008–2016. doi: 10.1172/JCI106421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassa T, Jana S, Meng F, Alayash AI. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio. 2016;6:876–884. doi: 10.1002/2211-5463.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: Differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara FA, Kahn SA, da Fonseca AC, Bahia CP, Pinho JP, Graca-Souza AV, Houzel JC, de Oliveira PL, Moura-Neto V, Oliveira MF. On the fate of extracellular hemoglobin and heme in brain. J Cereb Blood Flow Metab. 2009;29:1109–1120. doi: 10.1038/jcbfm.2009.34. [DOI] [PubMed] [Google Scholar]
- Leclerc JL, Santiago-Moreno J, Dang A, Lampert AS, Cruz PE, Rosario AM, Golde TE, Dore S. Increased brain hemopexin levels improve outcomes after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16679170. 271678X16679170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RC, Saleem S, Zhen G, Cao W, Zhuang H, Lee J, Smith A, Altruda F, Tolosano E, Dore S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab. 2009a;29:953–964. doi: 10.1038/jcbfm.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen-Roetling J, Regan RF. Increasing expression of H- or L-ferritin protects cortical astrocytes from hemin toxicity. Free Radic Res. 2009b;43:613–621. doi: 10.1080/10715760902942808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation. 2012a;9:46. doi: 10.1186/1742-2094-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Sammy F, Yang H, Thundivalappil S, Hellman J, Tracey KJ, Warren HS. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J Immunol. 2012b;189:2017–2022. doi: 10.4049/jimmunol.1103623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Day JP, Phillips H, Slootsky B, Tolosano E, Dore S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J Neuroinflammation. 2016;13:26. doi: 10.1186/s12974-016-0490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur KM, Davies MJ. Detection and reactions of the globin radical in haemoglobin. Biochim Biophys Acta. 1993;1202:173–181. doi: 10.1016/0167-4838(93)90002-9. [DOI] [PubMed] [Google Scholar]
- Miller YI, Smith A, Morgan WT, Shaklai N. Role of hemopexin in protection of low-density lipoprotein against hemoglobin-induced oxidation. Biochemistry (Mosc) 1996;35:13112–13117. doi: 10.1021/bi960737u. [DOI] [PubMed] [Google Scholar]
- Peters S, Koh J, Choi DW. Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- Pountney DJ, Konijn AM, McKie AT, Peters TJ, Raja KB, Salisbury JR, Simpson RJ. Iron proteins of duodenal enterocytes isolated from mice with genetically and experimentally altered iron metabolism. Br J Haematol. 1999;105:1066–1073. doi: 10.1046/j.1365-2141.1999.01441.x. [DOI] [PubMed] [Google Scholar]
- Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118:757–766. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- Regan RF, Guo Y. Toxic effect of hemoglobin on spinal cord neurons in culture. J Neurotrauma. 1998;15:645–653. doi: 10.1089/neu.1998.15.645. [DOI] [PubMed] [Google Scholar]
- Regan RF, Guo YP, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Chen M, Li Z, Zhang X, Benvenisti-Zarom L, Chen-Roetling J. Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol Dis. 2008;31:242–249. doi: 10.1016/j.nbd.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkind JM, Mohanty JG, Nagababu E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front Physiol. 2014;5:500. doi: 10.3389/fphys.2014.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Rad Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine neocortical cell culture. In: Tyson CA, Frazier JM, editors. Methods in Toxicology; Part A: In Vitro Biological Systems. Vol. 1. Academic Press; San Diego: 1993. pp. 46–60. [Google Scholar]
- Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–1284. doi: 10.1182/blood-2012-11-451229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, McCulloh RJ. Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol. 2015;6:187. doi: 10.3389/fphys.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng ZP, Chen J, Chau LY, Galunic N, Regan RF. Adenoviral transfer of the heme oxygenase-1 gene protects cortical astrocytes from heme-mediated oxidative injury. Neurobiol Dis. 2004;17:179–187. doi: 10.1016/j.nbd.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal. 2010;12:305–320. doi: 10.1089/ars.2009.2787. [DOI] [PubMed] [Google Scholar]
- Tolosano E, Fagoonee S, Hirsch E, Berger FG, Baumann H, Silengo L, Altruda F. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood. 2002;100:4201–4208. doi: 10.1182/blood-2002-04-1270. [DOI] [PubMed] [Google Scholar]
- Wang G, Manaenko A, Shao A, Ou Y, Yang P, Budbazar E, Nowrangi D, Zhang JH, Tang J. Low-density lipoprotein receptor-related protein-1 facilitates heme scavenging after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. 2017;37:1299–1310. doi: 10.1177/0271678X16654494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Green CC, Fromholt SE, Borchelt DR. Reduction of low-density lipoprotein receptor-related protein (LRP1) in hippocampal neurons does not proportionately reduce, or otherwise alter, amyloid deposition in APPswe/PS1dE9 transgenic mice. Alzheimers Res Ther. 2012;4:12. doi: 10.1186/alzrt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Philips JD, Doty RT, Giraudi P, Ostrow JD, Tiribelli C, Smith A, Abkowitz JL. Kinetics and specificity of feline leukemia virus subgroup C receptor (FLVCR) export function and its dependence on hemopexin. J Biol Chem. 2010;285:28874–28882. doi: 10.1074/jbc.M110.119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo YM, Kim KM, Kim SS, Han JA, Lea HZ, Kim YM. Hemoglobin toxicity in experimental bacterial peritonitis is due to production of reactive oxygen species. Clin Diagn Lab Immunol. 1999;6:938–945. doi: 10.1128/cdli.6.6.938-945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, Aronowski J. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–1043. doi: 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]