

Abstract
The levels of various circulating blood proteins can change in response to cancer therapy. Monitoring therapy-induced secretomes (TIS) may have use as biomarkers for establishing optimal biological effect (such as dosing) or identifying sources of toxicity and drug resistance. While TIS can derive from tumor cells directly, non-tumor ‘host’ treatment responses can also impact systemic secretory programs. For targeted inhibitors of the tumor microenvironment, including antiangiogenic and immune-checkpoint therapies, host TIS could explain unexpected collateral ‘side-effects’ of treatment. Here we describe a comparative transcriptomic and proteomic analysis of host TIS in tissues and plasma from cancer-free mice treated with antibody and receptor tyrosine kinase inhibitors (RTKIs) of the VEGF, cMet/ALK, and PD-1 pathways. We found that all cancer therapies elicit TIS independent of tumor growth, with systemic secretory gene change intensity higher in RTKIs compared to antibodies. Our results show that host TIS signatures differ between drug target, drug class, and dose. Notably, protein and gene host TIS signatures were not always predictive for each other, suggesting limitations to transcriptomic-only approaches to clinical biomarker development for circulating proteins. Together, these are the first studies to assess and compare ‘off-target’ host secretory effects of VEGF and PD-1 pathway inhibition that occur independent of tumor stage or tumor response to therapy. Testing treatment impact on normal tissues to establish host-mediated TIS signatures (or ‘therasomes’) may be important for identifying disease agnostic biomarkers to predict benefits (or limitations) of drug combinatory approaches.
Keywords: secretomes, angiogenesis, immune-checkpoint, VEGF, PD-L1
Introduction
All cancer therapies cause unintended ‘side-effects’ that can have a negative impact on patients. These effects can include toxicities that reduce quality of life, increase risk for secondary adverse events (which often lead to treatment breaks or cessation), or even contribute to patient morbidity(1). One side-effect of nearly every cancer treatment is the induction of a broad array of cytokines, chemokines, and growth factors that can be detected in patients’ blood(2). Therapy-induced secretomes (TIS) have been proposed as potential surrogate biomarkers to gauge treatment effect, and have been investigated as possible drivers of drug resistance or promoters of cancer progression(3–5). While secretory products induced by traditional cytotoxics (such as radiation and chemotherapy) may derive from direct tissue damage and homeostatic repair/defense mechanisms (i.e., inflammation and wound healing), it is often underappreciated that molecular targeted treatments – including those aimed at the tumor microenvironment (TME) – can also induce systemic TIS(6). Monitoring circulating proteins induced by targeted agents may have use as surrogate markers for biological or anti-tumor drug effects(7–9).
Exactly how broadly TME inhibitors, including widely-used agents targeting angiogenesis and immune-checkpoints, can influence secretory programs remain unclear(2,5). TIS programs may derive from tumor cell populations, and thus may provide information about cancer stage and response, but may also derive from non-tumor ‘host’ cell populations(4,10). For example, a diverse array of secretory proteins can be induced in patients’ blood by inhibitors of the VEGF pathway that include receptor tyrosine kinase inhibitors (RTKIs), or antibodies that disrupt VEGF:VEGFR signaling(11,12). Clinically, these circulating protein changes vary among disease-stages, drug types, treatment timing, and doses(7,13–15). However, some circulating secretory profiles (including VEGF, sVEGFR2, PlGF, and several others) have been consistent amongst several VEGF RTKIs in multiple cancer settings(11). One possible explanation for this is that these changes are independent of disease and represent host secretory programs activated directly or indirectly by treatment. For example, we have shown that sunitinib, a VEGF RTKI, can activate host TIS in cancer-free mice that were dose dependent, included numerous ‘off-target’ molecules, and coincided with optimal dosing(7). But a comprehensive analysis of ‘host-only’ TIS has not been evaluated for targeted TME inhibitors. This is of immediate importance as PD-1 pathway inhibitors (which block T-cell inhibition) are increasingly administered in multiple diseases and stages (including metastatic and perioperative settings), and are currently being tested both as monotherapies and in combination with other agents, including with VEGF-targeted agents(16). Determining underlying host TIS signatures (or ‘therasomes’) may be useful as biomarkers of response, indicate toxicity independent of disease, or help predict optimal combination strategies.
In this study, we undertook a comparative transcriptomic and proteomic analyses of host secretory changes in cancer-free mice treated with several TME inhibitors and traditional cytotoxics (i.e. radiation and chemotherapy). We found that all targeted therapies can induce secretory gene expression changes, but found these changes were more pronounced for RTKI treatments. VEGF and PD-L1 pathway inhibitors shared several secretory gene and pathway activations, suggesting possible overlapping host response mechanisms. However, proteomic and genomic TIS signatures did not always align, indicating potential limitations to gene-only approaches for blood-based protein biomarker discovery. Together, this study is the first to broadly compare the tumor-independent ‘collateral’ effects of TME-targeted inhibitors that include host TIS in mice.
Methods and Materials
Animal studies
Animal studies were performed in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and according of the Canadian Council on Animal Care. Protocols used were approved by the Institutional Animal Care and Use Committee at Roswell Park Comprehensive Cancer Center (Protocol: 1227M – for JMLE) or the Sunnybrook Health Sciences Center Animal Care Committee (for RSK). 8-10 week old Balb/c or SCID mice were treated for 7 to 58 days, depending on the study. See Supplemental Table S3 and Supplemental Methods and Materials for drug information and details on tissue collection protocols.
Whole genome expression analysis
Expression profiling was performed from the Genomics and Bioinformatics Shared Resources at Roswell Park Comprehensive Cancer Center (RPCCC). Differentially expressed genes were identified using the Limma program(17). Secretome identification included analysis using differentially expressed genes with gene products located in the extracellular region (GO:00005576) identified using the Gene Ontology Databases(18,19). For volcano plots, differentially expressed genes with p<0.05 and fold change ≥ 1.5 or ≤ -1.5 are shown. For heat maps, differentially expressed genes were hierarchically clustered. Correlation matrix used Pearson Correlation Coefficient (PCC) for comparison of differentially expressed genes between each sample. Gene set enrichment analysis for canonical pathway gene sets, as well as gene ontology for biological processes, was performed. Immune deconvolution was performed using the ImmuCC (20) signature matrix through Cibersort (21,22). For quantification and analysis, undetectable cell-associated gene signatures were excluded. For instance, from the 25 immune cell types, no values were identified for memory B cells, T cells (CD8 activated), M0 macrophages, memory CD4+ T cells, and activated NK and DC cells. See Supplemental Methods and Materials for extended information. Complete microarray data were deposited in Gene Expression Omnibus repository (GEO, Accession numbers GSE112466, GSE112467, and GSE112468).
ELISA analysis
Mouse plasma protein concentration was measured for PlGF2 (dilution factor: 1), VEGF (dilution factor: 2), SDF1α (dilution factor: 1), G-CSF (dilution factor: 1), HGF (dilution factor: 1), TGFβ1 (dilution factor: 60), OPN (dilution factor: 100), sVEGFR2 (dilution factor: 20), CSF (dilution factor: 2), and Leptin (dilution factor: 10). All ELISA kits were purchased from R&D Systems (catalogue numbers: MP200, MMV00, MCX120, MCS00, DY2207, MB100B, MOST00, MVR200, MCK00, MOB00, respectively) and used according to manufacturer’s instructions. Protein levels after treatment were normalized to corresponding vehicle controls, and fold changes in log2 scale were graphed.
Statistical analysis
Results were subjected to statistical analysis using the GraphPad Prism software package v.7.03 (Graphpad software Inc., San Diego, CA) and R v.3.4.1 through RStudio v.1.0.143 (Integrated Development for R; RStudio, Inc., Boston, MA URL http://www.rstudio.com/). Results are expressed as mean ± standard deviation (SD). Comparisons between two groups were made with Student’s two-tailed unpaired t-test, whereas one-way ANOVA was used for comparison of more than two groups. A minimum significance level of 0.05 was used for all analyses.
See Supplementary Methods and Materials for information on drugs, doses, bioinformatics analysis, and sample collection.
Results
Tumor-independent global gene expression changes after targeted therapy
To investigate the impact of targeted therapies in the absence of cancer, Balb/c mice were treated for 7-days. Tissue and plasma were extracted for gene and circulating protein analysis, respectively (Figure 1A). We have previously optimized 7-day schedules in cancer-free mice to show that treatment can i) induce broad off-target protein changes in tissue and plasma in response to select VEGF RTKIs such as sunitinib(7), and ii) trigger host-mediated metastasis-promoting mechanisms when administered prior to i.v. implantation of cancer cells(23,24) (see (5) for comprehensive review). In this study, we performed whole genome expression analysis on lung tissues from mice treated with i) RTKIs targeting VEGFRs (sunitinib and axitinib) or cMET/ALK (crizotinib), or ii) antibodies targeting mouse VEGFR2 (DC101, or ‘αVEGFR2’), PD-1 (RMP1-141, or ‘αPD-1’), or PD-L1 (10F.9G2, or ‘αPD-L1’). Volcano plots show the number of differentially expressed genes between drug types (Figure 1B), with RTKIs found to induce greater than 350 gene expression changes compared to antibodies (less than 210) (Figure 1C). Hierarchical clustering of differentially expressed genes revealed 4 distinct clusters. These included upregulated genes found only in RTKI-treated mice (clusters i and iii), only in antibody-treated mice (cluster iv), or in all treated mice (cluster ii) (Figure S1A). Based on these clusters, we next performed a gene ontology search for biological processes, and found that RTKIs alter processes related to migration while antibodies predominantly alter processes related to immune functions (Figure S1B). Next, we performed gene set enrichment analysis and found that canonical pathways enriched by each drug treatment (when compared to each corresponding vehicle-treated control) were similar, ranging from 70 to 177 enriched gene sets (using axitinib and αVEGFR2, respectively, as examples) (Table S1). We found that significantly enriched gene-sets were shared between VEGF and PD-L1 inhibitors, with a small portion of gene-sets found to be up- or down-regulated in unison (Figure 1D). Together, these results show that targeted therapies can yield similar host-mediated gene effects based on their mechanism of action (e.g. RTKI vs. antibody), but that there were relatively few shared gene expression changes between the pathways targeted (e.g. VEGF vs. PD-1).
Figure 1. Tumor-independent global gene expression changes after targeted therapy.

(A) Schematic of experimental design showing tumor-free mice treated for 7 days with different cancer therapies with lung tissue and plasma collected for gene expression and protein analysis, respectively. (B-D) Whole genome expression analysis after treatment with RTKIs (Sunitinib; Axitinib; or Crizotinib) or Abs (αVEGFR2, αPD-L1, or αPD-1) (Balb/c mice; n=3). (B) Volcano blots show differentially expressed genes (red dots; left/downregulated, right/upregulated; see Figure S1A for heat map). (C) Bar graph of up- (red) and down- (blue) regulated genes in RTKI- and Ab-treated animals compared to vehicle-treated controls. (D) Gene set enrichment analysis for canonical pathways (1329 gene sets, Molecular Signatures Database). Common gene sets (with significant NOM-p-value) between VEGF pathway inhibitors (Sunitinib, Axitinib, and αVEGFR2) and a PD-1 pathway inhibitor (αPD-L1) are shown. Weight Rank Scores (-log(NOM p-value)*NES) were calculated for all gene sets. See Table S1 for full list of enriched gene sets, and Figure S1B for Gene ontology analysis for biological processes. Abs used: αVEGFR2 (DC101), αPD-L1 (10F.9G2), and αPD-1 (RMP1-14). RTKI; Receptor Tyrosine Kinase Inhibitor; Ab, Antibody; NES, Normalized Enrichment Score; NOM p-value, Nominal p-value.
Treatment-induced secretory gene expression changes are tumor-independent and differ between drug class and target
We next examined lung tissue from treated cancer-free mice for gene expression changes related to host secretory proteins. To do this, gene ontology enrichment analysis was performed for gene products outside and unattached to the cell (extracellular region, GO:0005576)(4). Hierarchical clustering of only differentially expressed secretome genes yielded two distinct clusters representing genes upregulated after only i) RTKI treatment, or ii) antibody treatment (Figure 2A). We compared ‘global’ and ‘secretome’ gene expression changes for each therapy and found that RTKIs share many gene changes compared to antibodies, which share few (Figures S2A and S2B). Pearson correlation for gene expression found positive correlations (i.e., having similar changes) between RTKIs, with the strongest between axitinib and crizotinib [Pearson Correlation Coefficient (PCC) = 0.93]. Similarly, antibodies were also positively correlated (to each other), with the strongest between PD-1 pathway inhibitors (PCC = 0.52). Interestingly, all RTKIs were negatively correlated (have opposite changes) with all antibodies, with the strongest between either axitinib or crizotinib and αVEGFR2 (PCC = -0.56) (Figure 2B). Secretome gene signatures were identified for each treatment separately (Table S2), with RTKIs yielding the strongest inter-group secretory genes upregulated (>70 comparted to <20 for antibodies) (Figure 2C). Next, we performed gene ontology analysis for biological processes and found that, similar to global gene changes (described in Figure S1B), RTKI-induced secretomes affect biological processes involved with migration, whereas antibody-induced secretomes primarily impact the immune system (Figure 2D). To further examine this separation between drug types, we identified common genes between antibodies and RTKIs (including VEGF inhibitors and αPD-L1) (Figure 2E). Venn diagrams show only 1 common gene between antibodies (CCL4), compared to 9 shared genes upregulated by αPD-L1 and αVEGFR2 antibodies (Figure 2E, left panel). Interestingly, VEGFR RTKIs shared no upregulated secretory genes with αVEGFR2 (Figure 2E, middle panel) or αPD-L1 (Figure 2E, right panel). Together these results indicate strong positive correlations (i.e., similar changes) for secretory profiles within one drug class (i.e., RTKIs or antibodies), but surprisingly suggest that, independent of drug target, drugs between different classes (RTKIs vs. antibodies) have mostly negative correlations (i.e., opposite changes).
Figure 2. Treatment-induced secretory gene expression changes are tumor-independent and differ between drug class and target.
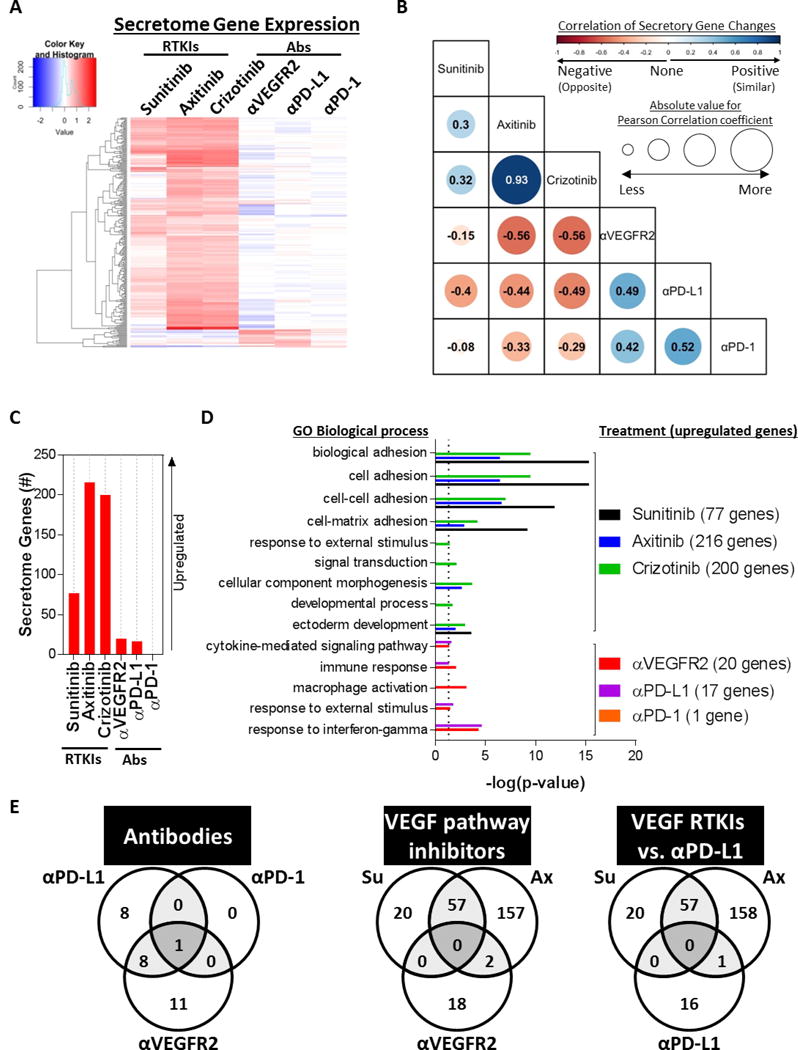
Secretome genes were identified by gene ontology analysis for cellular components (GO:0005576) (Balb/c mice; n=3). (A) Heat map of secretory genes expressed in animals treated with RTKIs or Abs after hierarchical clustering. (B) Table of Pearson correlation coefficients between drugs and secretory gene changes. Circle size corresponds to absolute Pearson correlation coefficient value, Similar changes (positive correlation) are blue while opposite changes (negative correlation) are red. (C) Bar graph of upregulated secretome genes in RTKI- and antibody-treated animals compared to their corresponding vehicle-treated controls. (D) Gene ontology analysis for biological processes for upregulated TIS genes. See Table S2 for a list of secretome genes after each treatment. (E) Venn diagrams of common upregulated secretome genes between Abs (left panel), VEGF pathway inhibitors (middle panel), and VEGF RTKIs and αPD-L1 (right panel). TIS, therapy-induced secretomes; RTKI, Receptor Tyrosine Kinase Inhibitor; Ab, Antibody; Su, Sunitinib; Ax, Axitinib; GO, Gene Ontology.
Host-mediated TIS are not always consistent between genes and circulating proteins
To test whether gene-based host TIS signatures correspond with circulating blood proteins, we examined mouse plasma after 7-days of treatment with multiple drugs. First, we examined several cytokines and growth factors previously shown (by us and others) to change after VEGF pathway inhibition(7,25). These included PlGF2, VEGF, sVEGFR2, HGF, OPN, SCF, TGFβ, SDF-1α, and G-CSF. Interestingly, we found that gene changes do not consistently correspond to plasma protein changes and vice versa (Figure 3A). For instance, αVEGFR2 treatment-induced significant upregulation of 7 of 9 plasma proteins but only 1 (OPN) was significantly upregulated at the gene level in the lungs of the same mice (Figure 3A-upper left). For RTKIs, plasma protein and gene TIS rarely coincided, suggesting that host TIS may be dominated by post-translational regulation of secretory products (Figure 3A). Next, we compared how multiple cancer therapies can affect 4 selected proteins related to angiogenesis (sVEGFR2, VEGF, and PlGF2) and immune-checkpoint control (sPD-L1). Therapies included 7 days of treatment with multiple VEGF pathway inhibitors (RTKIs, antibodies, or a VEGFR2 neutralizing adnectin), a PD-L1 antibody, or cytotoxic chemotherapy (CTX) given at maximum tolerated dose. Radiation (XRT) at 5Gy was also administered 24 hours prior to plasma analysis (see Table S3 for drug type and dosing). In total, we examined 15 cancer treatments and found significant changes induced by all therapies in at least one protein tested (Figures 3B and 3C – see S3A and S3B for full results). Interestingly, these systemic host TIS changes varied significantly between treatments, with inconsistent overlap in protein levels between treatment targets (such as between VEGF RTKIs and antibodies) and treatment types (i.e., chemotherapy and targeted therapy). Together, these findings show that protein changes are not consistently predicted by tissue gene expression levels. However, our results clearly indicate that systemic cancer treatments, both targeted and broadly cytotoxic, trigger diverse systemic secretory changes that are directly and indirectly related to the primary drug targets.
Figure 3. Host-mediated treatment-induced secretory gene changes are not always consistent with circulating protein changes.
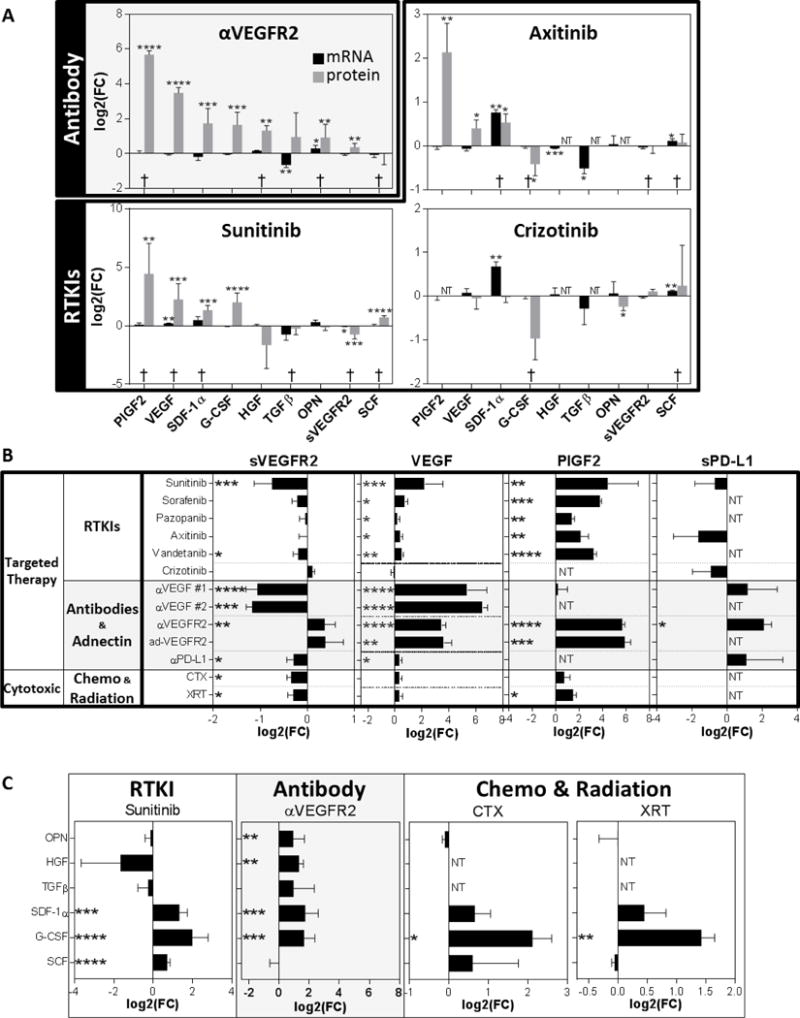
Protein analysis for circulating factors after 7-day treatment with different cancer therapies (RTKI, Ab, Chemotherapy, and Radiation). (A) Bar graph showing mRNA and protein levels changes after treatment with an Ab or RTKIs (Balb/c mice; n=3 for mRNA from microarray data; n=3-10 for protein data). (B) Circulating protein levels for sVEGFR2, VEGF, PlGF-2, and sPD-L1 after treatment with targeted therapies (RTKI, Ab, or Adnectin) or Cytotoxic therapies (Chemotherapy or Radiation) (Balb/c mice; n=3-10). (C) Sunitinib, αVEGFR2, CTX, and XRT effect on multiple plasma proteins. Protein levels of OPN, HGF, TGFβ, SDF1α, and SCF in plasma are shown (Balb/c mice; n= 3-10; 7-day treatments). See Table S3 for drug treatment and dosing schedule. Drugs used: αVEGF #1 (G6.31), αVEGF #2 (B20), αVEGFR2 (DC101), adVEGFR2 (CT322), αPD-L1 (10F.9G2). NT, not tested; Bar graphs showing log2-fold changes (FC) compared to vehicle treated animals; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 indicate significance; †: similar change for both mRNA and protein levels.
Drug dose and treatment duration impact host TIS
We next examined the impact of low/high doses over a 7-day period, and the effect of duration (i.e., single dose given over short/long periods) on host TIS in mouse plasma. First, we tested 3 plasma proteins (sVEGFR2, VEGF, and PlGF2) after 7 days of treatment with multiple VEGF RTKIs (sorafenib, pazopanib, axitinib, and vandetanib) at a range of doses both above and below the doses that have been previously shown to elicit anti-tumor effect in mice (Table S3). These studies aimed to expand upon findings previously reported by us involving sunitinib treatment in cancer-free mice (shown for comparison)(7). All host TIS proteins were elevated (or decreased in the case of sVEGFR2) with increasing doses (Figure 4A). Interestingly, sunitinib dose escalation did not yield corresponding changes in mRNA levels (using lung tissue), demonstrating again that mRNA expression can sometimes not be predictive of systemic protein changes after treatment (Figure S4). We next examined the effects of chronic drug treatment over a period of 40-60 days with plasma analyzed at multiple intervals. Interestingly, all RTKIs induced constant upregulation of plasma osteopontin (OPN), but for axitinib and crizotinib, these increases diminished over longer treatment periods, even leading to significant decreases in axitinib-treated mice (Figure 4B). Leptin was also found to be temporally upregulated, with treatment-induced increases observed in all RTKI treatment groups after 7 days, but then eventually decreasing over time. Together, these results indicate that dose impacts host TIS, and that physiological adaptations to treatment after prolonged periods may mute or negate host TIS changes in some instances. This suggests that compensatory responses to prolonged TKI inhibition may diminish overall host effects (Figure 4B).
Figure 4. Drug dose and treatment duration impact host TIS.
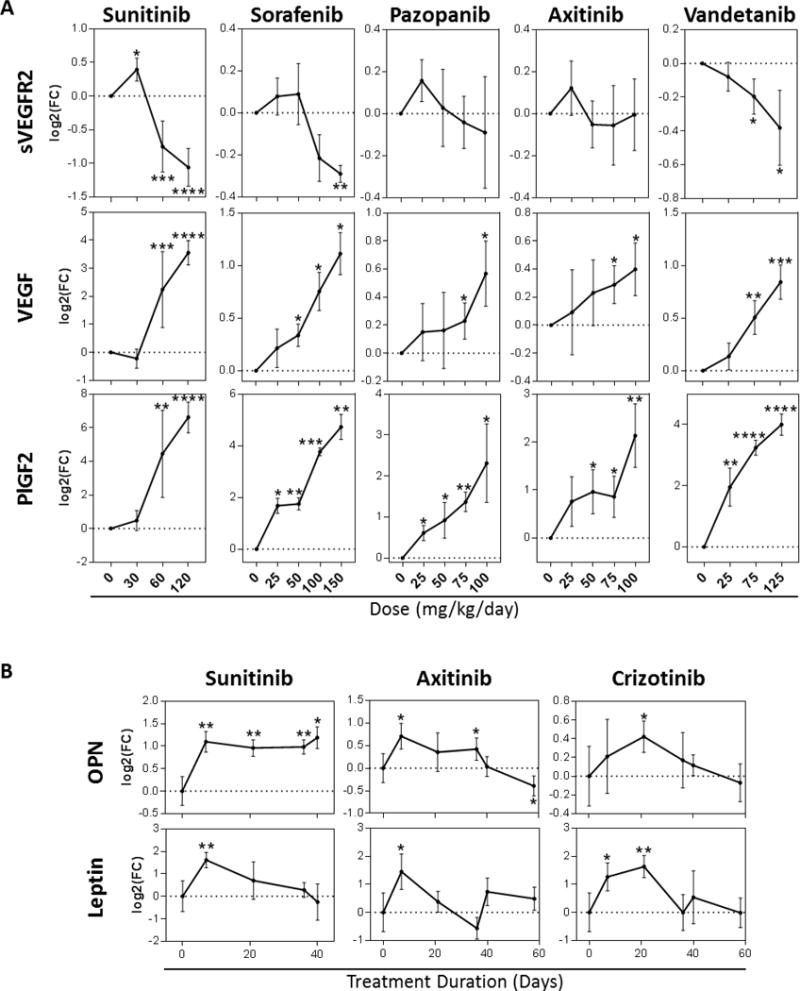
(A) Protein levels of circulating sVEGFR2, VEGF, and PlGF2 after treatment with varying doses of Sunitinib, Sorafenib, Pazopanib, Axitinib, and Vandetanib (Balb/c mice; n= 3-16; 7-day treatment). (B) Plasma protein levels of OPN, and Leptin after chronic treatment with Sunitinib (40 days), Axitinib, and Crizotinib (58 days) (SCID mice; n= 3-11). * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to corresponding vehicle treated animals or baseline (Day 0). Note: Sunitinib data shown in Fig 4A for VEGF and sVEGFR-2 is an expansion of results published in Ebos et al., PNAS, 2007 and is shown here for comparative purposes.
Tissue-based immune cell levels vary based on drug targets and class
Circulating and tissue-based secretory changes can impact immune cell population levels which can, in turn, directly modulate secretory protein levels locally and systemically(26,27). Using microarray data from mouse lung tissue treated with multiple targeted agents over a 7-day period (Fig 1A), we used CiberSort (21) with the mouse immune cell signature ImmuCC (20) to estimate the relative cell populations of 25 different immune cell types after treatment. Similar to secretory gene and protein TIS changes, we observed a broad array of immune cell population changes after treatment that differed between drug class and target (Figure 5). Interestingly, TKIs led to a broad decrease in CD8+ memory T and neutrophil cells, but trended toward an increase in naïve CD4+ T cells. Interestingly, all antibodies decreased Th2 cells, with significance reached following both PD-1 and PD-L1 antibody treatment. Together, these results suggest tumor-independent host effects can include multiple immune cell populations known to modulate, and be modulated by, circulating chemokines and growth factors induced by targeted therapy.
Figure 5. Tissue-based immune cell levels vary based on drug targets and class.
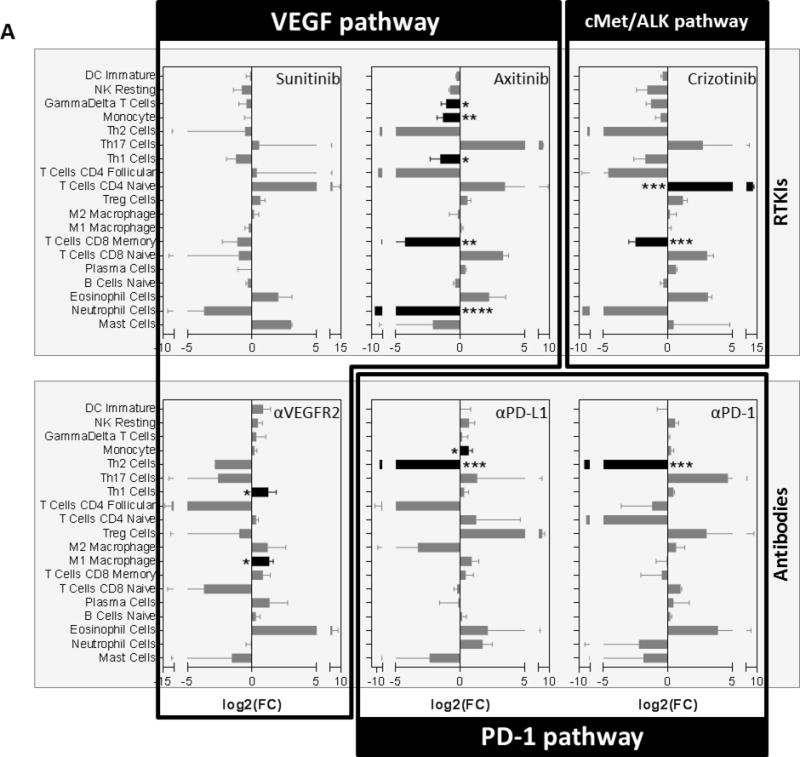
(A) Gene expression data derived from the lungs of tumor-free mice (as described in Fig. 1A) were evaluated for mouse immune cell gene signatures following treatment with RTKIs (Sunitinib; Axitinib; or Crizotinib) or Abs (αVEGFR2, αPD-L1, or αPD-1) (Balb/c mice; n=3). See methods for Cibersort deconvolution using ImmuCC mouse immune cell signature. Abs used: αVEGFR2 (DC101), αPD-L1 (10F.9G2), and αPD-1 (RMP1-14). RTKI; Receptor Tyrosine Kinase Inhibitor; Bar graphs showing log2 fold-changes (FC) compared to vehicle-treated animals; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 indicating significance are shown in black bars; Data shown as mean ± Standard Deviation.
Discussion
In this study, we investigated an often underappreciated side-effect of all cancer therapies that include the secretion of proteins independent of cancer type or stage. Fifteen different drugs were tested, including small molecule RTKIs (VEGFRs and cMET/ALK pathways); antibody and protein-based blockers (VEGF, VEGFR-2, PD-1, and PD-L1); and traditional cytotoxics (i.e., chemotherapy and radiation). We performed proteomic and transcriptomic analysis of tissues taken from cancer-free mice and identified consistent (yet diverse) secretory signatures that can be induced by treatment. Host TIS were highly drug-class specific and included varying degrees of overlap, despite divergent drug targets. Multi-targeted RTKIs produced a more robust host TIS than antibodies and, notably, tissue gene TIS changes were not consistently predictive of systemic (circulating) changes at the protein level. These studies are the first to compare the off-target collateral effects for TME inhibitors independent of cancer, and raise the potential for host TIS to be considered as ‘disease agnostic’ biomarkers in VEGF and PD-1 targeted drug development.
Does host TIS explain toxicity?
While this study is an important first step in identifying host TIS signatures, our results raise multiple questions. Perhaps foremost is whether these induced host-derived secretory changes contribute to treatment-related adverse events (TRAEs) seen in cancer patients(2). At least for antiangiogenic therapies, TRAEs are now well known. VEGF RTKI-treated patients experience decreased appetite, diarrhea, dysgeusia, fatigue, hypertension, and mucosal inflammation (among many others), yet these toxicities are not always cumulative(28). For example, in a retrospective analysis of 807 sunitinib-treated patients many TRAEs were found to peak after the first year but then steadily decrease(28). While we previously found that VEGF pathway inhibition (including RTKIs and antibodies) could elevate plasma levels of VEGF and plateau with repeated dosing(7), our current studies show that at least some of the host TIS changes could be temporal, and eventually decrease over time. Our current findings show that initial elevations of OPN and leptin decreased over extended periods (e.g., 40-60 days in mice), despite continued therapy. This diminishing host TIS effect may reflect a systemic tolerance or compensation of treatment effects, and may explain diminished severity of toxicity in some cancer patients over time. For immune-checkpoint blockade, the rapid clinical approval and expanded clinical testing of PD-1/PD-L1 antibodies in patients have led to multiple unexpected TRAEs that could be mediated by TIS. For example, cytokine release syndrome (CRS) has been noted following CAR-T and PD-1 therapy(29,30), and PD-L1 itself may be a direct regulator of pain signals controlled by neuronal tissues, which is linked to cytokine release(31). Related to this, recent studies involving PD-1 inhibitors nivolumab or pembrolizumab have examined several cytokines (including IL-8(32)) for changes after treatment as possible biomarkers of drug efficacy. Our results show that PD-L1 antibodies can induce multiple secretory changes - including CCL4, CCL5, CXCL9, and several others - but did not show a similarly robust host TIS in PD-1-treated mice. Interestingly, CCL4 was upregulated in all PD-1, PD-L1, and VEGFR-2 antibody-treated animals suggesting that host response may overlap in certain instances. CCL4 expression is a potent regulator of dendritic cell recruitment and T-cell activation, therefore future studies would be required to examine whether host CCL4 induction can affect tumor cell growth in vivo. Interestingly, our results also offer evidence that immune cell population levels are impacted by systemic treatments independent of tumor growth. Clinically, angiogenesis inhibitors have been shown to modulate immune cell populations in the blood (such as sunitinib downregulation of MDSCs and Treg cells(33,34)) and in the tumor (i.e., Treg cell recruitment in RCC after bevacizumab and sunitinib(35)). Similarly, immune-checkpoint blockade can potently modulate immune cell populations in cancer patients(36), and there is an established role for cytokine-mediated T cell differentiation which may impact both treatment response and disease progression(37). While tissue-based immune-cell gene signatures observed in our studies require broader confirmation in the circulation after treatment, as well in other tissues, it is nevertheless interesting to consider that chemokine-regulated immune cell populations may drive, or be directly affected by, the TIS observed in our studies. Taken together, our results show tumor-independent TIS may be pertinent to all patients treated with angiogenesis- and immunecheckpoint-targeted agents, and may help guide clinical biomarker development in determining what protein changes may occur independent of disease.
Can host TIS impact cancer response and progression?
One critical question raised by these studies is whether induced secretory changes may alter disease, potentially undercutting the net benefits of therapy or (conversely) even assisting overall anti-tumor effects. For the former, we and others have shown that a short-term 7-day treatment of VEGF RTKIs and other cytotoxics in mice prior to the intravenous implantation of tumor cells can act as a ‘primer’ of metastasis, allowing tumor growth to accelerate ((23,38) and summarized in (5)). Though these prior metastasis-promoting host effects were found to be dose-dependent and limited to VEGF RTKIs rather than antibodies, it is possible that similar off-target treatment effects of PD-1 pathway inhibition could also occur. In this regard, recent studies have explored treatment-induced disease, or ‘hyperprogression’ following PD-1 pathway inhibitors(39,40), but this topic remains to be more thoroughly investigated.
Is there a common mechanism controlling host TIS?
Another obvious question raised by our results is whether a single underlying mechanism may explain host TIS for different drug types. Cytotoxic chemotherapies and radiation are well known to induce DNA-damage secretory responses (DDSR), which include a diverse array of cytokines and growth factors(41). DDSRs share overlapping (yet confusingly distinct) mechanisms involving inflammation, stress, and senescence. Indeed, senescence-associated secretory phenotypes (SASPs) represent a broad list of secretory products that can be induced by several chemotherapies, and can paradoxically drive tumor progression in some instances (4,42–48). Related to this, recent work by Obenauf et al. showed that targeted inhibitors to BRAF, ALK or EGFR kinase can lead to TIS in tumor cells which, in turn, are capable of driving resistance and metastasis. In this case, and in other studies involving SASPs, a regulatory role for mTOR and NFκB signaling was found to be responsible for controlling secretory profiles. Future studies are required to determine whether the systemic host TIS after VEGF and PD-1 pathway inhibition may share similar treatment-induced mechanistic control.
Is there a host ‘therasome’ for every treatment?
An important consideration for the interpretation of our results involves the potential that experimental variables affect host TIS. Our studies utilized lung tissue and plasma from 8-10 week old female mice, including balb/c and SCID mice. While treatment dose and duration clearly impact TIS, the experimental variables of sex, age (i.e., age-related inflammation, or ‘inflammaging’), and mouse strain are likely to impact circulating blood protein levels(49). Even the time of day treatment is given, and the time of day sampling is performed, can influence blood-based protein levels. For example, circadian rhythms have been shown to lead to fluctuations in cytokine levels which, in turn, can influence the levels of cytokines after treatment (50). Importantly, it is also likely that the tissue-type tested will impact gene-based secretory analysis. While future studies are needed to examine the impact of these variables on host TIS, our limited studies do indicate that all cancer treatments are capable of inducing certain ‘off-target’ effects. For example, PlGF2 and SDF-1α levels were increased by most treatments in our study, despite not being direct targets of PD-1 or VEGF pathway activation. Also, sVEGFR2 was downregulated by all treatments (mainly by VEGF pathway inhibitors), whereas VEGF was upregulated by VEGF pathway inhibitors and PD-L1 antibody treatment, but not by cytotoxic agents. Together, these results show that treatment-induced secretory signatures lead to unexpected ‘collateral’ effects, perhaps owing to stress-related or compensatory reactions to therapy. However, these host TIS signatures vary considerably depending on the treatment given.
Can host TIS be impacted by combination therapy?
Finally, a question of immediate clinical relevance raised by our results is whether host TIS after monotherapy treatment is enhanced or attenuated when treatments are combined. For example, PD-1 pathway agents are now being tested in combination with a multitude of drugs, including VEGF pathway inhibitors. In some instances, evaluation of the efficacy of combining PD-1/PD-L1 treatments with some VEGF RTKIs (including sunitinib, pazopanib, and several others) has been limited by significant toxicities, while some combinations have not (i.e., pembrolizumab and axitinib resulted in >67% OOR in mRCC patients)(51). Interestingly, it may be antibody/antibody (i.e., bevacizumab with atezolizumab) combinations which could result in the most favorable toxicity profiles in patients, though the efficacy of these strategies is still being determined in the clinical testing(51). Since it is rare that healthy people are treated with cancer agents to test toxicities and secretory products (see example with sunitinib in healthy volunteers(52)), studies in mice to examine the impact of combination approaches on host TIS may be of clinical value. It is possible that TRAEs observed in patients may be reflected in host TIS responses in mice.
Taken together, our results identify several consequences of novel TME inhibitors on normal systems that occur independent of cancer, and may have potential for exploitation as surrogate biomarkers for drug dosing, toxicity, and/or to indicate drug resistance in patients.
Supplementary Material
Acknowledgments
We would like to thank A. Haninec for helpful comments during manuscript preparation. Funding: This work was supported by a Roswell Park Comprehensive Cancer Center (RPCCC) Support Grant from the National Cancer Institute (NCI) (P30CA016056). Additional support was received from the Roswell Park Alliance Foundation (RPAF) and the Department of Defense (DoD) through the Peer Reviewed Cancer Research Program (Award No. W81XWH-14-1-0210). Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the RPAF, RPCCC, or DoD. Materials used in this study were provided by Novartis, Lilly, Adnexus, Genentech, Pfizer, AstraZeneca, Bayer (to RSK or JMLE).
Abbreviations List
- RTKIs
receptor tyrosine kinase inhibitors
- TIS
therapy-induced secretomes
- VEGF
vascular endothelial growth factor
- PD-1
programmed cell death protein 1
- TME
tumor microenvironment
- PCC
pearson correlation coefficient
- PlGF2
placental growth factor 2
- sVEGFR2
soluble VEGF receptor 2
- HGF
hepatocyte growth factor
- OPN
osteopontin
- SCF
stem-cell factor
- TGFβ
transforming growth factor
- SDF-1α
stromal derived factor
- G-CSF
granulocyte-colony stimulating factor
- CTX
cyclophosphamide
- XRT
radiation therapy
Footnotes
Conflict of Interest Statement: No conflicts
References
- 1.Baldo BA, Pagani M. Adverse events to nontargeted and targeted chemotherapeutic agents: emphasis on hypersensitivity responses. Immunol Allergy Clin North Am. 2014;34(3):565–96, viii. doi: 10.1016/j.iac.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Shaked Y. Balancing efficacy of and host immune responses to cancer therapy: the yin and yang effects. Nat Rev Clin Onc. 2016;13(10):611–26. doi: 10.1038/nrclinonc.2016.57. [DOI] [PubMed] [Google Scholar]
- 3.Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15(16):5020–5. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520(7547):368–72. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebos JM. Prodding the Beast: Assessing the Impact of Treatment-Induced Metastasis. Cancer Res. 2015;75(17):3427–35. doi: 10.1158/0008-5472.CAN-15-0308. [DOI] [PubMed] [Google Scholar]
- 6.Daenen LG, Houthuijzen JM, Cirkel GA, Roodhart JM, Shaked Y, Voest EE. Treatment-induced host-mediated mechanisms reducing the efficacy of antitumor therapies. Oncogene. 2014;33(11):1341–7. doi: 10.1038/onc.2013.94. [DOI] [PubMed] [Google Scholar]
- 7.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA. 2007;104(43):17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerbel RS, Ebos JM. Peering into the aftermath: The inhospitable host? Nat Med. 2010;16(10):1084–5. doi: 10.1038/nm1010-1084. [DOI] [PubMed] [Google Scholar]
- 9.Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15(16):5020–5. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georgilis A, Gil J. Controlling secretion to limit chemoresistance. Genes Dev. 2016;30(16):1791–2. doi: 10.1101/gad.288571.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duda DG, Ancukiewicz M, Jain RK. Biomarkers of antiangiogenic therapy: how do we move from candidate biomarkers to valid biomarkers? J Clin Oncol. 2010;28(2):183–5. doi: 10.1200/JCO.2009.24.8021. [DOI] [PubMed] [Google Scholar]
- 12.Zurita AJ, Gagnon RC, Liu Y, Tran HT, Figlin RA, Hutson TE, et al. Integrating cytokines and angiogenic factors and tumour bulk with selected clinical criteria improves determination of prognosis in advanced renal cell carcinoma. Br J Cancer. 2017;117(4):478–84. doi: 10.1038/bjc.2017.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zurita AJ, Khajavi M, Wu HK, Tye L, Huang X, Kulke MH, et al. Circulating cytokines and monocyte subpopulations as biomarkers of outcome and biological activity in sunitinib-treated patients with advanced neuroendocrine tumours. Br J Cancer. 2015;112(7):1199–205. doi: 10.1038/bjc.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13(8):827–37. doi: 10.1016/S1470-2045(12)70241-3. [DOI] [PubMed] [Google Scholar]
- 15.Nikolinakos PG, Altorki N, Yankelevitz D, Tran HT, Yan S, Rajagopalan D, et al. Plasma cytokine and angiogenic factor profiling identifies markers associated with tumor shrinkage in early-stage non-small cell lung cancer patients treated with pazopanib. Cancer Res. 2010;70(6):2171–9. doi: 10.1158/0008-5472.CAN-09-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ott PA, Hodi FS, Buchbinder EI. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front Oncol. 2015;5:202. doi: 10.3389/fonc.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 18.Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. doi: 10.1093/nar/gku1179. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Huang A, Sun J, Jiang T, Qin FX, Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Sci Rep. 2017;7:40508. doi: 10.1038/srep40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–59. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebos JM, Mastri M, Lee CR, Tracz A, Hudson JM, Attwood K, et al. Neoadjuvant antiangiogenic therapy reveals contrasts in primary and metastatic tumor efficacy. EMBO Molecular Medicine. 2014 doi: 10.15252/emmm.201403989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bocci G, Man S, Green SK, Francia G, Ebos JM, du Manoir JM, et al. Increased plasma vascular endothelial growth factor (VEGF) as a surrogate marker for optimal therapeutic dosing of VEGF receptor-2 monoclonal antibodies. Cancer Res. 2004;64(18):6616–25. doi: 10.1158/0008-5472.CAN-04-0401. [DOI] [PubMed] [Google Scholar]
- 26.Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. 2013;2(8):e25961. doi: 10.4161/onci.25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. 2010;40(12):3317–20. doi: 10.1002/eji.201041170. [DOI] [PubMed] [Google Scholar]
- 28.Porta C, Gore ME, Rini BI, Escudier B, Hariharan S, Charles LP, et al. Long-term Safety of Sunitinib in Metastatic Renal Cell Carcinoma. Eur Urol. 2016;69(2):345–51. doi: 10.1016/j.eururo.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rotz SJ, Leino D, Szabo S, Mangino JL, Turpin BK, Pressey JG. Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatr Blood Cancer. 2017 doi: 10.1002/pbc.26642. [DOI] [PubMed] [Google Scholar]
- 30.Kroschinsky F, Stolzel F, von Bonin S, Beutel G, Kochanek M, Kiehl M, et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit Care. 2017;21(1):89. doi: 10.1186/s13054-017-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, Kim YH, Li H, Luo H, Liu DL, Zhang ZJ, et al. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci. 2017;20(7):917–26. doi: 10.1038/nn.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. 2017;28(8):1988–95. doi: 10.1093/annonc/mdx190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506–13. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Hoang A, Zhou L, Kalra S, Yetil A, Sun M, et al. Resistance to anti-angiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunology Research. 2015 doi: 10.1158/2326-6066.CIR-14-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. doi: 10.1038/ncomms12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol. 2013;3:63. doi: 10.3389/fonc.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharon E. Can an Immune Checkpoint Inhibitor (Sometimes) Make Things Worse? Clin Cancer Res. 2017;23(8):1879–81. doi: 10.1158/1078-0432.CCR-16-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res. 2017;23(8):1920–8. doi: 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Nelson PS. Molecular pathways: involving microenvironment damage responses in cancer therapy resistance. Clin Cancer Res. 2012;18(15):4019–25. doi: 10.1158/1078-0432.CCR-11-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bent EH, Gilbert LA, Hemann MT. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016;30(16):1811–21. doi: 10.1101/gad.284851.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 47.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 48.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 49.Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24(2):242–56. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Ebos J, Mastri M, Hudson JM, Lee CR, Tracz A, Attwood K, et al. Effect of the timing of sunitinib administration on the predictive value of biomarkers in renal cell cancer (mRCC) Journal of Clinical Oncology. 2015;33(15_suppl):11096. doi: 10.1200/jco.2015.33.15_suppl.11096. [DOI] [Google Scholar]
- 51.Kuusk T, Albiges L, Escudier B, Grivas N, Haanen J, Powles T, et al. Antiangiogenic therapy combined with immune checkpoint blockade in renal cancer. Angiogenesis. 2017;20(2):205–15. doi: 10.1007/s10456-017-9550-0. [DOI] [PubMed] [Google Scholar]
- 52.Lindauer A, Di Gion P, Kanefendt F, Tomalik-Scharte D, Kinzig M, Rodamer M, et al. Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin Pharmacol Ther. 2010;87(5):601–8. doi: 10.1038/clpt.2010.20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.