

Abstract
Tac (CD25) is expressed on multiple hematological malignancies and is a target for cancer therapies. LMB-2 is an extremely active anti-Tac recombinant immunotoxin composed of an Fv that binds to Tac and a 38kDa fragment of Pseudomonas exotoxin A (PE38). While LMB-2 has shown high cytotoxicity towards Tac-expressing cancer cells in clinical trials, its efficacy was hampered by the formation of anti-drug antibodies against the immunogenic bacterial toxin and by dose-limiting off-target toxicity. To reduce toxin immunogenicity and non-specific toxicity, we introduced six point mutations into domain III that were previously shown to reduce T cell immunogenicity and deleted domain II from the toxin, leaving only the 11aa furin cleavage site which is required for cytotoxic activity. While this strategy has been successfully implemented for mesothelin and CD22 targeting immunotoxins, we found that removal of domain II significantly lowered the cytotoxic activity of αTac immunotoxins. To restore cytotoxic activity in the absence of PE domain II, we implemented a combined rational design and screening approach to isolate highly active domain II deleted toxin variants. The domain II deleted variant with the highest activity contained an engineered disulfide-bridged furin cleavage site designed to mimic its native conformation within domain II. We found that this approach restored five-fold of the cytotoxic activity and dramatically improved the maximum tolerated dose. Both of these improvements led to significantly increased anti-tumor efficacy in vivo. We conclude that the next generation anti-Tac immunotoxin is an improved candidate for targeting Tac expressing malignancies.
Keywords: Tac, CD25, recombinant immunotoxin, Pseudomonas exotoxin A, PE domain II
Introduction
The alpha chain of the Il-2 receptor, also known as Tac or CD25, is expressed on activated T and B cells, on regulatory T cells and to a lesser degree on sub-populations of resting memory T cells (1–5). Tac is also expressed on multiple T and B cell malignancies and is therefore a target for cancer therapy (6). LMB-2 is an anti-Tac recombinant immunotoxin (RIT) containing a targeting scFv and a 38-kDa portion of Pseudomonas exotoxin A (PE38) that shows extremely high cytotoxic activity and has been evaluated in several clinical trials (6–9). One such clinical trial involved various hematological malignancies including adult T-cell leukemia, B-cell chronic lymphocytic leukemia, anaplastic large-cell lymphomas, B-cell non-Hodgkins lymphoma, Hodgkin’s disease and hairy cell leukemia (6). In this trial, a general 3% complete remission and a 20% partial remission rate was observed, with 37% of immunotoxin recipients generating anti-drug antibodies and with vascular leak syndrome (VLS) toxicity limiting the dosage.
Parental PE38 consists of PE domains II and III, with the cytotoxic catalytic activity residing within domain III. In order to partially address the problems of non-specific toxicity and immunogenicity of immunotoxins containing the PE38 fragment, we previously engineered a lysozyme resistant version of PE38, designated PE24 or LR (10–12). We showed that all of domain II can be replaced with a short furin cleavage site (FCS) (“RHRQPRGWEQL”), which is necessary for immunotoxin activation after internalization, and is natively found within domain II. These domain II truncated PE24s showed less proteolytic cleavage by lysosomal enzymes and contain less immunogenic sequences immunotoxins than the parental PE38 immunotoxins, and should theoretically be less immunogenic. In addition, PE24 immunotoxins exhibit less non-specific toxicity, allowing much higher doses of immunotoxin to be given (10–12). However, removal of domain II, which does not affect or enhance the cytotoxic activity of immunotoxins targeting the B cell marker CD22 or the mesothelioma marker mesothelin, causes a 30-fold reduction in the activity of αTac immunotoxins [see Results and (13)].
In order to further reduce the immunogenicity of the bacterial toxin part of the immunotoxin, we previously determined single amino acid point mutations within PE domains II and III that disrupt B (14–16) and T cell epitopes (17–20). While B cell epitope disrupting mutations are intended to avoid recognition by B cell receptors, the T cell epitope disrupting mutations are intended to reduce activation of T helper cells that are necessary for B cell activation. T cell epitopes within domain II and III were mapped by activating human PBMCs with a peptide array spanning domains II and III of PE38 and measuring T cell activation by Il-2 secretion. Once mapped, point mutations that disrupt each epitope without significantly reducing cytotoxic activity were determined. A combination of PE domain II truncation and T cell epitope disruption in PE domain III has previously been introduced into an anti-mesothelin immunotoxin and been found to reduce the activation of human PBMCs in vitro by 90% (19) and the formation of anti-drug antibodies (ADA) in mice by 99% (20).
Here we describe the rational design of a next generation version of LMB-2 that combines high cytotoxic activity with low immunogenicity and low general toxicity by introducing six T cell epitope disrupting point mutations into toxin domain III combined with a deletion of toxin domain II. We found that removal of domain II significantly lowered the activity of Tac mediated intoxication, but not transferrin mediated intoxication in the same cells. To restore Tac mediated cytotoxic activity, we screened a panel of rationally designed domain II truncation mutants. The most active domain II truncated mutant (αTac-M18-PE24) showed a 5-fold increase in cytotoxic activity, a 68-fold improvement in the maximum tolerated dose (MTD) and significantly increased anti-tumor efficacy in vivo. We conclude that the next generation anti-Tac immunotoxin is an improved candidate for targeting Tac expressing malignancies.
Materials and Methods
Vector construction
All protein-encoding sequences were constructed by ordering double stranded DNA fragments (gBlocks, Integrated DNA Technologies, Coralville, IA) codon optimized for E. coli and cloning them into the appropriate vector using Gibson Master Mix (New England Biolabs, Ipswich, MA). The ZZ fusion proteins were cloned into the pET22-NN-ZZ-PE38 (kindly provided by Itai Benhar, Tel Aviv University, Israel) which contains the pelB periplasmic leader peptide for periplasmic secretion followed by sequences encoding for two repeats of the Z protein and a multiple cloning site (21). The sequences encoding for refolded RITs were cloned into our standard protein production lab vector (22).
RIT production
All RITs were expressed and purified from E. coli inclusion bodies as previously described (23). Briefly, inclusion bodies from E. coli were washed and then solubilized using a guanidinium buffer (6M guanidine-HCl, 100mM Tris-HCl, 2 mM EDTA). The soluble denatured proteins were then diluted 1:100 into refolding buffer (100 mL Tris-HCl, 1 mM EDTA, 0.5M arginine, 0.5M NDSB-201, pH 10) and stirred for 32 hours at 4°C. The proteins solution was then dialyzed against 30 mM Tris-HCl, 0.1M urea for 40 hours. The refolded proteins were further purified using ion-exchange and size exclusion chromatography. Protein purity was assessed by SDS-PAGE on a 4-12% Bis-Tris gel (Novex, Life Technologies, Waltham, MA).
ZZ fusion protein production
The ZZ fusion proteins were induced by growing transformed E. coli BL21(DE3) in 50 ml of auto-inducing Magic Media (Thermo-Fisher Scientific, Waltham, MA) overnight at 37°C. After growth, the cells were spun for 10 minutes at 5,000 RPM and frozen over night. The pellets were then lysed by re-suspending the cells in BPER complete reagent (Thermo-Fisher Scientific) supplemented with 1 mM EDTA and gentle shaking for 30 minutes at room temperature. The lysate was then cleared by centrifuging for 20 minutes at 12,000 RPM at 4°C and the soluble fraction taken for further purification. Purification of ZZ fusion proteins was accomplished using IgG coated beads (GE Healthcare Life Sciences, Chicago, IL) according to manufacturer’s instructions. Finally, proteins were desalted using PD-10 columns (GE Healthcare Life Sciences) and concentrated using Ultracel-4 10-kD cutoff centricons (Millipore, Billerica, MA). Protein purity was assessed by SDS-PAGE on a 4-12% Bis-Tris gel (Novex, Life Technologies).
Cell growth inhibition assays
The cytotoxic activity of the RITs was evaluated in a cell growth inhibition assay using the WST-8 cell counting kit (Dojindo Molecular Technologies, Rockville, MD). Cells were incubated with varying concentrations of immunotoxin for 72 hours, after which the WST-8 reagent was added and the plates read at OD 450 nm. Readings were normalized to the PBS only positive control and the IC50 (concentration inhibiting growth by 50%) for each construct was calculated using a variable four-parameter slope, non-linear regression fit with Graphpad Prism vs. 6.01. One-hundred percent cell killing was achieved using 100 μg/ml of Cyclohexamide (Sigma, St. Louis, MO) as a control. Each assay contained four replicates of each concentration and the assays were repeated three to four times. Standard RITs were directly added to the cell culture. ZZ-PE fusion proteins were mixed at a 1:1 molar ratio with αTac human IgG1 (Dacluzimab, kindly provided by Thomas A. Waldmann, National Cancer Institute, Bethesda, MD) (24) for 30 minutes at room temperature and then added to the cell culture. HUT-102, the prototype HTLV-1-positive leukemic cell line, was also obtained from Thomas A. Waldmann. KARPAS-299 cells where obtained from Robert J. Kreitman (National Cancer Institute, Bethesda, MD). Identity of all cell lines was confirmed by short tandem repeat testing within the past 12 months and all cell lines were tested negative for mycoplasma. cells were seeded at a concentration of 1 million cells/T75 flask, split when confluent and used after 3-9 passages.
Non-specific toxicity
All animal experiments were performed in accordance with NIH guidelines and approved by the NCI Animal Care and Use Committee (protocol LMB-014). Non-specific toxicity was evaluated by single IV injections of indicated doses to Swiss mice. A dose was considered toxic if mice appeared morbid, loss >10% body weight or died. Morbid and dead mice were sent for necropsy or pathology analysis two days after treatment. Pathological examination included mice weight, hydration, muscularity and histopathological examination of the heart, lungs, spleen, kidneys, gastrointestinal and reproductive tract.
Mouse xenograft tumor model
Female SCID mice were injected subcutaneously in the flank with 4.0 × 106 KARPAS-299 cells. Tumor size was evaluated every other day using a caliper. When tumors reached a range of 62-152 mm4, mice were assembled in groups with similar average tumor sizes and treated with IV injections of LMB-2, αTac-dsFv-M18-PE24(T) or vehicle three times (on days 1, 3 and 6). Body weight and tumor size were evaluated in all mice until tumors reached 550 mm4. No animals were excluded from statistical analysis. Tumor growth delay was calculated by interpolation of the time point each tumor reached 200 mm3. Kaplan Meier survival curves were calculated based on the fact that mice were euthanized when tumors reached ≥550 mm3.
Graphs and statistics
Graphs were plotted and analyzed using Graphpad Prism vs. 6.0101 (GraphPad Software, Inc., La Jolla, CA). For differences in tumor growth a one-way ANOVA with Holm-Sidak multiple comparisons test was used, while a Mantel-Cox test was used for the Kaplan-Meier survival graph.
Results
Efficient intoxication through the Tac pathway requires PE domain II
LMB-2 is an αTac-scFv-PE38 immunotoxin. For this study, we used the same targeting αTac Fv in our immunotoxins, but in a dsFv format (αTac-dsFv-PE38) to improve the stability of the antibody fragment (Fig. 1). We compared the cytotoxic activity of LMB-2 and αTac-dsFv-PE38 by treating HUT-102 cells with the two RITs and found that the two had identical IC50s (Table 1), indicating that disulfide stabilization of the Fv does not affect overall activity.
Figure 1. Structural model of PE38 and illustrations of the different αTac domain II mutants.
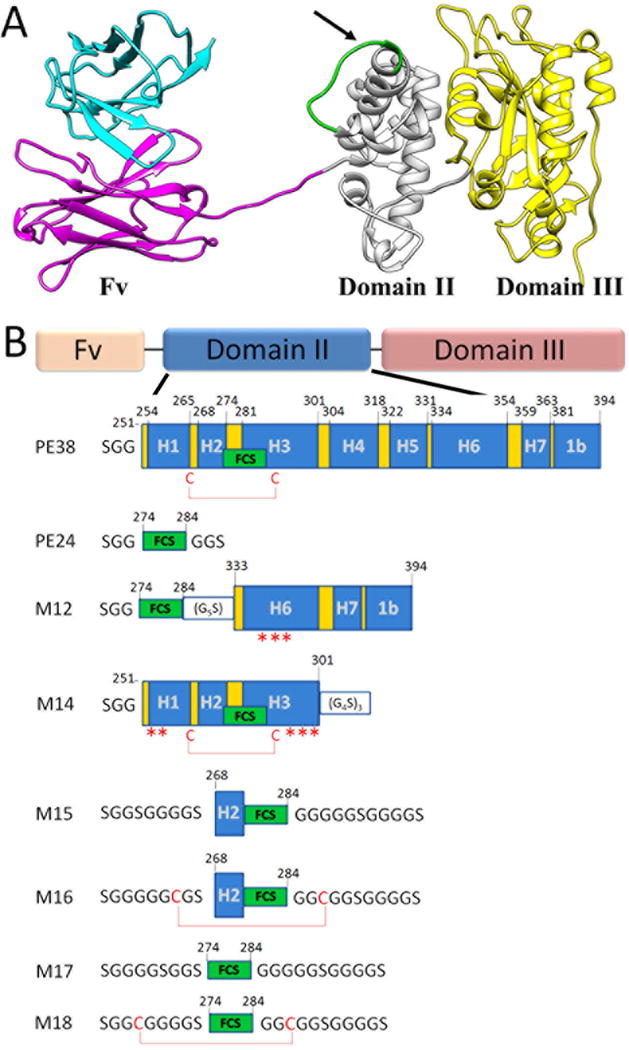
(A) αTac-dsFv-PE38 is composed of an αTac dsFv targeting moiety followed by the PE38 toxin comprising domains II and III. Note the FCS marked in green within PE domain II. The arrow signifies the cleavage site. (B) Illustrations of the rationally designed domain II truncation mutants. PE domain II is divided into seven helixes, designated H1-H7, with helixes H1, H3, H4, H5 and H6 bundled around a hydrophobic core. The FCS (residues 274-284, “RHRQPRGWEQL”) naturally resides in helixes H2 and H3 of domain II, and is constrained by a disulfide bond formed between Cys265 and Cys287. Mutants were designed based upon the crystal structure of the full PE38 (PDB ID: 1IKQ). Helixes are marked H1-H7, the FCS (“RHRQPRGWEQL”) is marked as a green box, disulfide bonds are shown as red lines, while asterisks denote solubilizing point mutations (hydrophobic to polar). The numbering system is based upon the mature PE38.
Table 1.
Cytotoxic activity of the domain II mutant immunotoxins
| Construct | IC50 (pM) | Fold reduction in activity (compared to PE38) |
|---|---|---|
| LMB-2 (αTac-scFv-PE38) | 0.1 | |
| αTac-dsFv-PE38 | 0.1 | |
| αTac-dsFv-PE24 | 3.5 | 34 |
| αTac-dsFv-M12-PE24 | 30.2 | 297 |
| αTac-dsFv-M14-PE24 | 5.4 | 53 |
| αTac-dsFv-M15-PE24 | 1.5 | 15 |
| αTac-dsFv-M16-PE24 | 4.2 | 42 |
| αTac-dsFv-M17-PE24 | 3.2 | 32 |
| αTac-dsFv-M18-PE24 | 0.7 | 7 |
Immunotoxins were tested for cytotoxicity in a cell growth inhibition assay on the HUT-102 strain. Averages of 3-6 assays are shown.
To reduce the non-specific toxicity and immunogenicity of domain II, we constructed αTac-dsFv-PE24 (Fig. 1B). The cytotoxic activity of αTac-dsFv-PE38 and αTac-dsFv-PE24 was compared by treating HUT-102 and KARPAS-299 cells with the two variants and evaluation of cell viability 72 hours later (Fig. 2A, 2C and Table 1). We found that on HUT-102 cells, the αTac-dsFv-PE24 immunotoxin was 34-fold less active than αTac-dsFv-PE38 (IC50s of 3.4 pM and 0.1 pM, respectively). This pattern was more profound on KARPAS-299 cells, where αTac-dsFv-PE24 was 45-fold less active than αTac-dsFv-PE38 (IC50s of 10.3 pM and 0.23 pM, respectively). This indicates that PE domain II is important either for killing through the Tac receptor or in general for killing Tac expressing cells.
Figure 2. The cytotoxic activity of αTac PE24 immunotoxins is significantly and specifically lower than αTac PE38 immunotoxins.
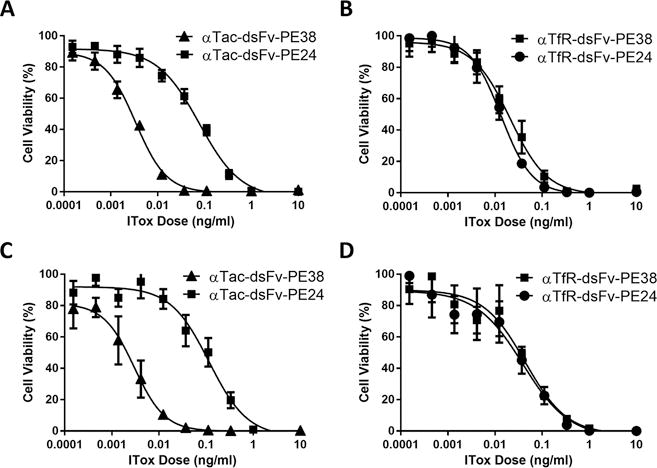
Immunotoxins targeting either Tac or the TfR containing either the PE38 or PE24 toxins were tested for cytotoxicity on the HUT-102 (A and B) or the KARPAS-299 (C and D) cell lines in a cell growth inhibition assay. (A) αTac-dsFv-PE38 and αTac-dsFv-PE24 were tested on the HUT-102 cell line. While αTac-dsFv-PE38 had an average IC50 of 0.1 pM, αTac-dsFv-PE24 exhibited an IC50 that was 34-fold lower (3.4 pM). (B) In contrast, targeting PE38 and PE24 immunotoxins against the TfR on the same HUT-102 cells yields a very similar IC50 for αTfR-dsFv-PE38 and αTfR-dsFv-PE24 (IC50s of 0.77 pM and 0.32 pM, respectively). This difference was also seen when αTac-dsFv-PE38, αTac-dsFv-PE24, αTfR-dsFv-PE38, and αTfR-dsFv-PE24 were tested on KARPAS-299 cells (C and D) (IC50s of 0.23 pM, 10.3 pM, 0.64 pM and 0.93 pM, respectively). Representative graphs are shown. Each assay contained four replicates for each data point and the assays were repeated three times.
To further study the role of domain II in Tac-mediated intoxication, we produced PE38 and PE24 immunotoxins that target the ubiquitous transferrin receptor (TfR). This allowed us to target Tac expressing cells through two different endogenous receptors. Comparison of the cytotoxic activity of αTfR-dsFv-PE38 and αTfR-dsFv-PE24 revealed similar cytotoxic activity of αTfR-dsFv-PE38 and αTfR-dsFv-PE24 (Fig. 2B and 2D) with IC50s of 0.77 pM and 0.32 pM, respectively in HUT-102 cells and 0.64 pM and 0.93 pM in KARPAS-299 cells, respectively. These results suggest that the large difference in activity between αTac PE38 and PE24 immunotoxins is specific to the Tac intoxication pathway and not a general property of Tac expressing cells.
Rational design and screening of PE domain II mutants
In order to isolate a PE domain II truncation mutant containing the minimal amount of domain II residues while retaining high cytotoxic activity, a panel of domain II truncation mutants were rationally designed based on the structure of the full PE38 (PDB ID: 1IKQ) (the most active mutants are described in Figure 1 while the full panel is described in Supplementary Figure S1). By expressing all of our constructs as ZZ-fusion proteins, we were able to produce them by secretion into the E. coli periplasm and bypass the laborious protein refolding process necessary to produce standard RITs (21, 25, 26). The production and screening of the ZZ-PE fusion proteins is detailed in Supplementary Figure S2. Briefly, a preliminary comparison of the cytotoxic activity of ZZ-PE38 and ZZ-PE24 mixed with an anti-Tac IgG mAb showed similar activities on HUT-102 cells, unlike the large difference previously seen between PE38 and PE24 standard RITs. We concluded that the ZZ fusion system does not recapitulate the internal cellular trafficking of RITs, and thus can only be used to discriminate between active/non-active mutants, which must then be produced as refolded immunotoxins and re-tested. Subsequently, the ZZ fusion domain II mutants were mixed with an anti-Tac mAb and screened for cytotoxic activity on HUT-102 cells. Of the active mutants found in this screen, we decided to focus on mutants M12, M14 and M15-M18 based upon on a combination of activity and a minimal amount of retained domain II residues (Fig. 1 and Supplementary Fig. S2). M12 was designed to retain helixes 6, 7 and a part of domain 1b. M14 retained helixes 1, 2 and 3. M15-M18 were designed with extended Gly-Ser linkers around the FCS. Of these, M16 and M18 also included a constraining disulfide bond around the FCS, intended to mimic the native constrained conformation found in domain II (naturally found as a constrained loop within helixes H2 and H3 of domain II).
Domain II mutant M18 containing immunotoxin shows improved cytotoxic activity
The selected six domain II mutants (M12, M14 and M15-M18) (Fig. 1) were produced as refolded RITs according to our standard lab protocol and tested for improved cytotoxic activity compared to αTac-dsFv-PE24 (Table 1). The most active of the domain II mutants, αTac-dsFv-M18-PE24 exhibited a 5-fold increase in cytotoxic activity compared to αTac-dsFv-PE24 (IC50 of 0.7 pM and 3.5 pM, respectively).
Disruption of T cell epitopes in PE domain III
We have previously published point mutations that disrupt T cell epitopes found within PE domain III (R427A, F443A, L477H, R494A, R505A and L552E) (20). These mutations were introduced into αTac-dsFv-M18-PE24, producing αTac-dsFv-M18-PE24(T). The low immunogenicity αTac-dsFv-M18-PE24(T) was tested for cytotoxic activity to ascertain the effect of the introduced de-immunizing mutations (Fig. 3). As shown in Figure 3, αTac-dsFv-M18-PE24(T) exhibited a 1.5-fold lower IC50 on HUT-102 cells (IC50 of 1.1 pM vs. 0.7 pM) and a 3-fold lower IC50 on KARPAS-299 cells IC50 of 5.25 pM vs. 16.1 pM) compared to the parental αTac-dsFv-M18-PE24.
Figure 3. Disruption of T cell epitopes in αTac-dsFv-M18-PE24.
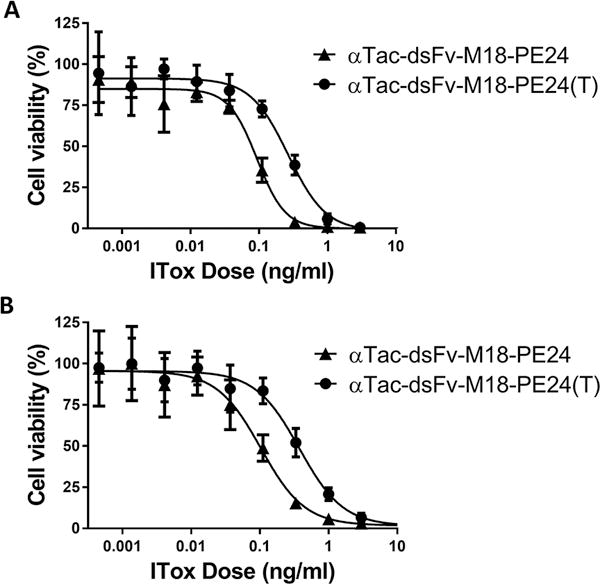
T cell epitope disrupting point mutations in PE domain III were introduced into αTac-dsFv-M18-PE24, creating αTac-dsFv-M18-PE24(T). αTac-dsFv-M18-PE24(T) was tested on (A) HUT-102 and (B) KARPAS-299 cells in a cell growth inhibition assay. On HUT-102 cells αTac-dsFv-M18-PE24(T) exhibited a normalized IC50 of 1.1 pM compared to an IC50 of 0.7 pM for the parental αTac-dsFv-M18-PE24, while on KARPAS-299 cells αTac-dsFv-M18-PE24 and αTac-dsFv-M18-PE24(T) exhibited an average IC50 of 5.25 pM and 16.1 pM, respectively. Representative graphs are shown. Assays were repeated 3-4 times.
αTac-dsFv-M18-PE24(T) exhibits low general toxicity
To evaluate the non-specific toxicity of LMB-2 (αTac-scFv-PE38) and αTac-dsFv-M18-PE24(T), Swiss mice were treated with single doses of LMB-2 or αTac-dsFv-M18-PE24(T) (Table 2). We found that LMB-2 was tolerated with no significant weight loss at a dose of 0.35 mg/kg (n=4), but that 0.4 mg/kg was toxic to 2/4 mice. We also evaluated the toxicity of multiple doses by giving three doses in tumor bearing SCID mice (see below). We found that 4/4 mice died after treatment with 0.3 mg/kg of LMB-2, while three doses of 0.25 mg/kg (n=5) were tolerated with only mild weight loss. It should be noted that the Swiss mice were generally about 20% larger than the SCID mice.
Table 2.
Maximum tolerated dose in mice
| Construct | Dose (mg/kg) |
Result | Weight loss (%) |
|---|---|---|---|
| αTac-dsFv-M18-PE24(T) | 18 | 4/4 healthy | 2 |
| 24 | 4/4 healthy | 1 | |
| 28 | 4/4 dead | – | |
|
| |||
| LMB-2 (αTac-scFv-PE38) | 0.3 | 4/4 healthy | 2 |
| 0.35 | 4/4 healthy | 3 | |
| 0.4 | 2/4 healthy | 2 | |
| 0.5 | 4/4 dead | 1 | |
Swiss mice (n=4) were given a single dose of immunotoxin and monitored for health to establish the maximum tolerated dose.
On the other hand, αTac-dsFv-M18-PE24(T) was tolerated even at a 68-fold higher dose of 24 mg/kg (n=4) (54-fold when corrected for molarity). We did not identify the MTD, however 4/4 mice treated at a dose of 28 mg/kg died 24 hours after injection, indicating that the MTD is between 24-28 mg/kg. Toxicity of multiple doses (three) in tumor bearing SCID mice indicated that the multiple dose MTD for αTac-dsFv-M18-PE24(T) was 10 mg/kg (40-fold higher than LMB-2, 32-fold when corrected for molarity).
Histopathological examination of morbid or dead mice recognized hepatotoxicity as the main cause of toxicity in 8/8 mice treated with a single dose at LD50 of either αTac-dsFv-M18-PE24(T) or αTac-dsFv-PE38. This toxicity has been previously reported in mice treated with LMB-2 (27). Interestingly, αTac-dsFv-M18-PE24(T) also induced nonspecific toxicity in the gastrointestinal track in 2/4 mice consisting of damage to the duodenum and jejunum.
αTac-dsFv-M18-PE24(T) exhibits improved anti-tumor activity
To evaluate the anti-tumor activity of αTac-dsFv-M18-PE24(T), we implanted KARPAS 299 cells into the flank of SCID mice (Fig. 4). Once each tumor reached 62-152 mm4, mice were arranged into groups with similar average tumor sizes and treated with IV injections of LMB-2 (αTac-scFv-PE38), αTac-dsFv-M18-PE24(T) or vehicle three times (on days 1, 3 and 6) (Fig. 4). We found that while the tumors treated with vehicle grew rapidly, reaching an average of 200 mm4 on day 3.1 (n=7), mice that were treated with three doses of 10 mg/kg of αTac-dsFv-M18-PE24(T) showed a significant delay in tumor growth compared with the vehicle group (p<0.0001 in a one-way ANOVA with Holm-Sidak’s multiple comparisons test) and with the LMB-2 group (p=0.0026), reaching an average of 200 mm4 on day 9.4 (Fig. 4A). Mice treated with three doses of 0.25 mg/kg of LMB-2 showed a milder delay in tumor growth reaching an average of 200 mm4 on day 5.5 (n=5) (p=0.04 compared to the vehicle group).
Figure 4. αTac-dsFv-M18-PE24(T) exhibits improved anti-tumor activity.
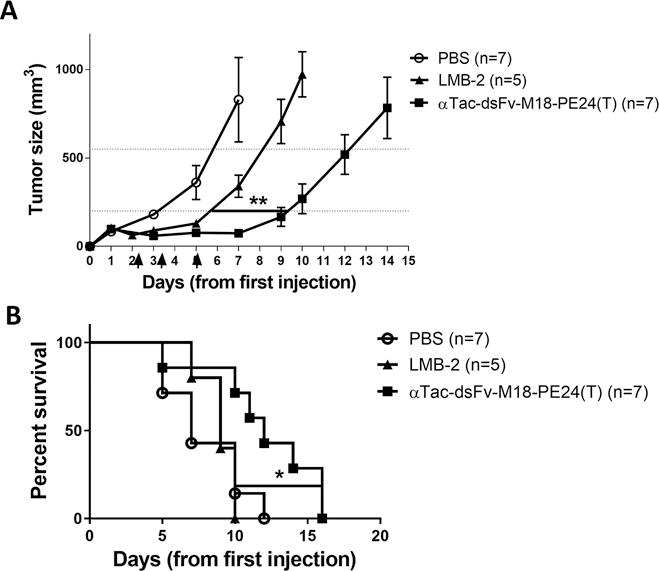
SCID mice were implanted with KARPAS-299 cells and then treated with three doses of vehicle (PBS), LMB-2 (αTac-scFv-PE38) or αTac-dsFv-M18-PE24(T). Treatment with αTac-dsFv-M18-PE24(T) causes a significant delay in tumor growth (p=0.0026, one-way ANOVA) and in survival (p=0.036, Mantel-Cox test) compared to treatment with LMB-2.
To model the effect of tumor growth delay on survival, we employed a Kaplan Meier survival analysis based on a euthanasia criterion when tumor size reached >550 mm4 (Fig. 4B). We found that mice treated with αTac-dsFv-M18-PE24(T) had a significantly longer median survival of 12 days compared to vehicle (7 days) and LMB-2 (9 days) (p=0.036 in Mantel-Cox test).
Discussion
Immunogenicity and dose-limiting toxicity severely hamper the efficacy of cancer targeting immunotoxins. PE based immunotoxins, being of bacterial origin, are extremely immunogenic, with ADA being produced in patients with an intact immune system after only one cycle of treatment (9). The production of ADA against immunogenic protein therapeutics can lead to a shorter serum half-life, loss of therapeutic efficacy and can cause serious adverse immunological reactions (28–31). The use of low immunogenicity immunotoxins should allow more treatment cycles to be given safely without the need for an additional immunosuppressive regimen, and increase the overall treatment efficacy. Similarly, addressing dose-limiting toxicity should also increase efficacy by allowing higher dosing. Here we have taken a combined approach to produce a low immunogenicity, low toxicity and yet highly active αTac immunotoxin. This was achieved by combining T cell disrupting mutations in toxin domain III, deletion of toxin domain II and engineering of a disulfide constrained FCS to replace domain II and restore cytotoxic activity.
Removal of PE domain II as a general strategy for reducing non-specific toxicity
Immunotoxins can cause either specific or non-specific general toxicities. Specific toxicities occur when the target receptor is expressed not only on the target cells but on additional tissues, usually at much lower levels. Specific toxicities targeting the liver, kidneys and the central nervous system have been described for individual immunotoxins (32–35). Non-specific toxicities, on the other hand, are associated with multiple immunotoxins against different targets. The most common non-specific toxicity for PE based immunotoxins in humans is VLS, caused by an unknown interaction between immunotoxins and endothelial cells that can frequently be dose limiting (35–37). Hepatic and renal non-specific toxicities have also been observed (35–37). Removal of PE domain II has been shown to reduce the non-specific toxicity of several immunotoxins, including those targeting CD22 on B cells (10), mesothelin on multiple solid tumors (12) and Tac on T and B cells (this study) in mice. This indicates that removal of PE domain II addresses some common non-specific toxicity and is a general strategy for lowering the toxicity of PE based immunotoxins. It remains to be seen whether this reduction in toxicity seen in mice will translate to humans, and if so, which human non-specific toxicity will be reduced.
The disulfide constrained M18 linker can increase PE24 immunotoxin serum half-life
Part of the reduction in general toxicity seen with dsFv/scFv-PE24 immunotoxins may be attributed to the lower half-life of PE24 based immunotoxins, as their smaller size (51-kD) allows them to be cleared by renal filtration. However, a Fab containing PE24 immunotoxin targeting CD22 (74-kD) that is too large for renal filtration also exhibited a much higher MTD in vivo than its dsFv-PE38 immunotoxin counterpart (63-kD) (22). This shows that the reduced half-life is not the main cause of the lower general toxicity and that increasing the serum half-life of PE24 immunotoxins is a viable strategy for increasing their efficacy.
We have previously shown that PE24 based anti-mesothelin immunotoxins containing the standard linear FCS between the targeting Fv and the toxin moiety are subjected to faster serum clearance than ones containing a disulfide constrained FCS (38). The disulfide constrained FCS sequence that showed a longer serum half-life was identical to the M18 mutant used in this study. It is therefore reasonable to expect that αTac-dsFv-M18-PE24(T) will also have a longer serum half-life than the parental αTac-dsFv-PE24 as a result of the addition of a protective disulfide bond around the FCS. Additional approaches to further extend immunotoxin serum half-life, such as PEGylation or using a larger Fab targeting moiety, may need to be incorporated into αTac-dsFv-M18-PE24(T) in the future.
Using αTac immunotoxins to target regulatory T cells
LMB-2 has also been tested for specific elimination of T regulatory cells (Treg) (9, 39). In melanoma patients, LMB-2 caused a 50% transient reduction in circulating and a 68% reduction in lesion associated Treg cells, but did not improve patient outcomes. As Tac (CD25) is expressed on activated B and T cells in addition to Treg cells (1–5), treatment with an αTac immunotoxin removes both the unwanted Treg cells and the crucial activated effector lymphocytes (9). Therefore, αTac immunotoxins are probably not suited for classic immunomodulation, but may be used to target Treg cells (along with bystander activated lymphocytes) prior to adoptive T cell transfer or administration of an immune checkpoint inhibitor. In such a scenario, several treatment cycles of an αTac immunotoxin will be given instead of lymphocyte depleting chemotherapy in order to deplete immunosuppressive Treg cells, after which the necessary effector T cells or the immune checkpoint inhibitor will be infused into the patient and be able to function better without Treg immune suppression. In all of these scenarios, the recipient will probably have a mostly intact immune system and will require multiple treatment cycles. Therefore, only a low immunogenicity immunotoxin could be repeatedly administered in order to effectively reduce Treg cells.
In summary, the described next generation immunotoxin αTac-dsFv-M18-PE24(T) combines high cytotoxic activity and low immunogenicity in vitro with low general toxicity and significantly higher anti-tumor efficacy in vivo making it an improved therapeutic candidate for treating Tac expressing malignancies.
Supplementary Material
Acknowledgments
Financial support: This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research
FUNDING: This research and all authors were supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research under Project ZO1 BC008753.
Abbreviations
- ADA
anti-drug antibodies
- FCS
furin cleavage site
- MTD
maximum tolerated dose
- PE38
Pseudmonas exotoxin A
- RIT
recombinant immunotoxin
- TfR
transferrin receptor
- VLS
vascular leak syndrome
Footnotes
Conflict of Interest: G.K., R.M. and I.P. are inventors on several patents on immunotoxins that have all been assigned to the NIH. All authors declare no conflict of interest.
References
- 1.Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–68. doi: 10.1146/annurev.iy.11.040193.001333. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 3.Herndler-Brandstetter D, Schwaiger S, Veel E, Fehrer C, Cioca DP, Almanzar G, et al. CD25-expressing CD8+ T cells are potent memory cells in old age. J Immunol. 2005;175:1566–74. doi: 10.4049/jimmunol.175.3.1566. [DOI] [PubMed] [Google Scholar]
- 4.Triplett TA, Curti BD, Bonafede PR, Miller WL, Walker EB, Weinberg AD. Defining a functionally distinct subset of human memory CD4+ T cells that are CD25POS and FOXP3NEG. Eur J Immunol. 2012;42:1893–905. doi: 10.1002/eji.201242444. [DOI] [PubMed] [Google Scholar]
- 5.Brisslert M, Bokarewa M, Larsson P, Wing K, Collins LV, Tarkowski A. Phenotypic and functional characterization of human CD25+ B cells. Immunology. 2006;117:548–57. doi: 10.1111/j.1365-2567.2006.02331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18:1622–36. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 7.Kreitman RJ, Wilson WH, Robbins D, Margulies I, Stetler-Stevenson M, Waldmann TA, et al. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood. 1999;94:3340–8. [PubMed] [Google Scholar]
- 8.Kreitman RJ, Stetler-Stevenson M, Jaffe ES, Conlon KC, Steinberg SM, Wilson W, et al. Complete remissions of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin Cancer Res. 2016;22:310–8. doi: 10.1158/1078-0432.CCR-15-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell DJ, Jr, Felipe-Silva A, Merino MJ, Ahmadzadeh M, Allen T, Levy C, et al. et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–28. doi: 10.4049/jimmunol.179.7.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113:3792–800. doi: 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen JK, Weldon JE, Xiang L, Beers R, Onda M, Pastan I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother. 2010;33:297–304. doi: 10.1097/CJI.0b013e3181cd1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weldon JE, Xiang L, Zhang J, Beers R, Walker DA, Onda M, et al. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol Cancer Ther. 2013;12:48–57. doi: 10.1158/1535-7163.MCT-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazor R, Kaplan G, Park D, Jang Y, Lee F, Kreitman R, et al. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cell Immunol. 2017;313:59–66. doi: 10.1016/j.cellimm.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci USA. 2008;105:11311–6. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci USA. 2011;108:5742–7. doi: 10.1073/pnas.1102746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. 2012;109:11782–7. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci USA. 2012;109:E3597–603. doi: 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USAmerica. 2014;111:8571–6. doi: 10.1073/pnas.1405153111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, et al. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther. 2015;14:2789–96. doi: 10.1158/1535-7163.MCT-15-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazor R, Crown D, Addissie S, Jang Y, Kaplan G, Pastan I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. 2017;14:432–42. doi: 10.1038/cmi.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazor Y, Barnea I, Keydar I, Benhar I. Antibody internalization studied using a novel IgG binding toxin fusion. J Immunol Methods. 2007;321:41–59. doi: 10.1016/j.jim.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Bera TK, Onda M, Kreitman RJ, Pastan I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk Res. 2014;38:1224–9. doi: 10.1016/j.leukres.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol. 2004;248:503–18. doi: 10.1385/1-59259-666-5:503. [DOI] [PubMed] [Google Scholar]
- 24.Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, et al. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci USA. 1989;86:10029–33. doi: 10.1073/pnas.86.24.10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, et al. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107–13. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 26.Mazor Y, Noy R, Wels WS, Benhar I. chFRP5-ZZ-PE38, a large IgG-toxin immunoconjugate outperforms the corresponding smaller FRP5(Fv)-ETA immunotoxin in eradicating ErbB2-expressing tumor xenografts. Cancer Lett. 2007;257:124–35. doi: 10.1016/j.canlet.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Onda M, Willingham M, Wang QC, Kreitman RJ, Tsutsumi Y, Nagata S, et al. Inhibition of TNF-alpha produced by Kupffer cells protects against the nonspecific liver toxicity of immunotoxin anti-Tac(Fv)-PE38, LMB-2. J Immunol. 2000;165:7150–6. doi: 10.4049/jimmunol.165.12.7150. [DOI] [PubMed] [Google Scholar]
- 28.Mazor R, Onda M, Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev. 2016;270:152–64. doi: 10.1111/imr.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan FR, Morton LD, Spindeldreher S, Kiessling A, Allenspach R, Hey A, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs. 2010;2:233–55. doi: 10.4161/mabs.2.3.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Yang C, Xia Y, Bertino A, Glaspy J, Roberts M, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98:3241–8. doi: 10.1182/blood.v98.12.3241. [DOI] [PubMed] [Google Scholar]
- 31.Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–75. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 32.Pai LH, Bookman MA, Ozols RF, Young RC, Smith JW, 2nd, Longo DL, et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J Clin Oncol. 1991;9:2095–103. doi: 10.1200/JCO.1991.9.12.2095. [DOI] [PubMed] [Google Scholar]
- 33.Pai-Scherf LH, Villa J, Pearson D, Watson T, Liu E, Willingham MC, et al. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin Cancer Res. 1999;5:2311–5. [PubMed] [Google Scholar]
- 34.Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–7. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 35.Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–65. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 36.Aruna G. Immunotoxins: a review of their use in cancer treatment. J Stem Cells Regen Med. 2006;1:31–6. doi: 10.46582/jsrm.0101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weidle UH, Tiefenthaler G, Schiller C, Weiss EH, Georges G, Brinkmann U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics. 2014;11:25–38. [PubMed] [Google Scholar]
- 38.Kaplan G, Lee F, Onda M, Kolyvas E, Bhardwaj G, Baker D, et al. Protection of the furin cleavage site in low-toxicity immunotoxins based on pseudomonas exotoxin A. Toxins. 2016;8:E217. doi: 10.3390/toxins8080217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attia P, Powell DJ, Jr, Maker AV, Kreitman RJ, Pastan I, Rosenberg SA. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J Immunother. 2006;29:208–14. doi: 10.1097/01.cji.0000187959.45803.0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.