

Abstract
Purpose
Folate receptor alpha (FR) is overexpressed in several cancers. Endogenous immunity to the FR has been demonstrated in patients and suggests the feasibility of targeting FR with vaccine or other immune therapies. CD4 helper T cells are central to the development of coordinated immunity and prior work shows their importance in protecting against relapse. Our previous identification of degenerate HLA-class II epitopes from human FR led to the development of a broad coverage epitope pool potentially useful in augmenting antigen-specific immune responses in most patients.
Experiment Design
We conducted a Phase I clinical trial testing safety and immunogenicity of this vaccine, enrolling patients with ovarian cancer or breast cancer who completed conventional treatment and who showed no evidence of disease. Patients were initially treated with low dose cyclophosphamide and then vaccinated 6 times, monthly. Immunity and safety were examined during the vaccine period and up to one year later.
Results
Vaccination was well tolerated in all patients. Vaccine elicited or augmented immunity in greater than 90% of patients examined. Unlike recall immunity to tetanus toxoid, FR T cell responses developed slowly over the course of vaccination with a median time to maximal immunity at 5 months. Despite slow development of immunity, responsiveness appeared to persist for at least 12 months.
Conclusions
The results demonstrate that it is safe to augment immunity to the FR tumor antigen and the developed vaccine is testable for therapeutic activity in the majority of patients whose tumors express FR, regardless of HLA genotype.
Keywords: folate receptor, cancer vaccine, ovarian cancer, breast cancer, antibodies, T cells
INTRODUCTION
Despite advances in surgery and adjuvant systemic therapy, distant relapse and subsequent mortality from breast (BC) and ovarian cancer (OC) remain considerable public healthcare problems, with an estimate of greater than 40,000 deaths from BC and 14,000 deaths from OC annually (1). Strategies for adjuvant therapy that complement current modalities are needed to decrease the recurrence rates for BC and OC. Two shared features of BC and OC that may be exploited in the adjuvant setting are 1) the majority of patients without overt distant metastatic disease at diagnosis achieve an initial complete remission from their cancers, and 2) women with BC and OC often generate endogenous immune responses to their tumors. Generation of anti-tumor immunity has been consistently associated with better outcomes (2–4), leading to the hypothesis that improving anti-tumor immune responses with strategies such as vaccination may improve disease-free and overall survival.
We have identified the folate receptor alpha (FRα) as a candidate therapeutic vaccine antigen for patients with BC and OC (5). FRα is normally expressed in areas of the body that need to retrieve folic acid prior to excretion (kidney proximal tubules) or to concentrate it in the cerebrospinal fluid (choroid plexus). Additionally, FRα is expressed in the lung, although its function there is unknown (6, 7). In contrast to this relatively restricted expression in healthy tissues, FRα is frequently highly overexpressed in several epithelial tumors, including BC and OC (8–12). We have previously demonstrated that many women with BC and OC have evidence of spontaneous T cell and antibody immune responses to FRα at levels higher than observed in women who had not experienced BC or OC (13). Importantly, spontaneous immunity to FRα was not associated with paraneoplastic syndrome or other symptoms of autoimmunity in our cases or controls (13).
Specifically, we have identified five putative HLA class II-binding peptides from the FRα sequence that elicited T cell immune responses from BC and OC patients (13). We estimated that > 88% of women will express HLA proteins that would recognize at least one of the peptides, such that the majority of unselected HLA diverse patients would have the potential to respond to vaccine (13). This is significantly higher than vaccines incorporating class I-binding peptides, which would be presented to CD8+ cells. The use of HLA class-II binding peptides specifically targets activation of CD4+ T helper cells, which are essential for coordinated memory immune responses to antigen. Thus, due to its degeneracy, this pool of could be effective at generating persistent and protective immunity in most ovarian or breast cancer patients, regardless of HLA genetic background.
Based on our prior demonstration of spontaneous anti-FRα immunity in a subset of BC and OC patients (13) we conducted a Phase I clinical trial study to assess the immunogenicity and safety profile of a multi-epitope FRα peptide vaccine comprised of FR30, FR56, FR76, FR113, and FR238 (Table 1) admixed with GM-CSF. This twenty-two patient trial enrolled women with ovarian, primary peritoneal, or breast cancer who had completed treatment (surgery, chemotherapy, radiation, or a combination of the above) at least 90 days previously and who showed no evidence of disease following their treatment. Perhaps the largest barrier to the generation of effective anti-tumor immunity is local and systemic tumor-induced immunosuppression, often mediated through T regulatory cells (Tregs) (14). Several strategies to inhibit immunosuppressive enzymes (IDO), surface markers (PD-L1, CTLA-4), and Tregs (Denileukin Diftitox, cyclophosphamide) (15–22) have or are currently undergoing testing to determine whether they improve responses to vaccines. For this trial, we selected cyclophosphamide (CTX) based on its long safety history, cost effectiveness, oral administration, and its status as an FDA-approved agent for OC and BC patients (23–27). The dose and schedule were selected based on a small previous study that showed decreased levels of circulating Tregs in 9 of 9 advanced cancer patients (28). Directly targeting immunosuppressive Tregs, plus careful timing of vaccination for when disease burden is lowest, provide an opportunity to generate meaningful immune responses with the goal of preventing or delaying disease recurrence. Thus, the primary objectives were to determine the safety of the combination therapy protocol (CTX followed by vaccine) and the ability of this regimen to generate anti-FRα immune responses.
Table 1.
Vaccine FRα peptides
| Sequence | Position | Designation | Length (aa) |
|---|---|---|---|
|
| |||
| RTELLNVCMNAKHHKEK | 30-46 | FR30 | 17 |
| QCRPWRKNACCSTNT | 56-70 | FR56 | 15 |
| KDVSYLYRFNWNHCGEMA | 76-93 | FR76 | 18 |
| LGPWIQQVDQSWRKERV | 113-129 | FR113 | 17 |
| PWAAWPFLLSLALMLLWL | 238-255 | FR238 | 18 |
MATERIALS AND METHODS
Eligibility and enrollment
This single arm Phase I study enrolled women 18 years of age or older with histologically confirmed stage II-III breast cancer or stage II-IV ovarian (including primary peritoneal and fallopian tube) cancer who had completed systematic therapy (except for hormonal treatment or bisphosphonates) at least 90 days prior to registration and were without evidence of disease. Patients could enroll after initial diagnosis or after a complete response to therapy for recurrent disease. Additional eligibility criteria included Eastern Cooperative Oncology Group (ECOG) performance status of 0-1, and adequate hematologic, renal and hepatic function. Exclusion criteria included active infection, immunocompromising conditions, autoimmunity, systemic steroid use within 30 days of registration, concurrent thyroid replacement therapy, NYHA class III/IV congestive heart failure, myocardial infarction or stoke within 6 months of registration, another invasive malignancy within 5 years of enrollment, pregnancy, or breast feeding. A decision was made not to require FRα expression in the primary tumor because of the (1) this was a phase I safety trial, (2) lack of a validated antibody IHC test for expression and, (3) lack of data that FRα would or would not be expressed on recurrent tumors, regardless of primary tumor expression. With respect to the latter, we have previously shown that it is possible for recurrent ovarian lesions to express FRα even though the primary lesion may not have shown positivity (29). All patients were at Mayo Clinic in Rochester MN. Protocol chairs obtained study approval from the institutional review board and filed assurances with the Department of Health and Human Services. The study was conducted in accordance with the U.S. Common Rule. Written, informed consent was required for enrollment. The study was registered at Clinicaltrials.gov as NCT01606241.
Preparation of the vaccine
Peptides were produced by Polypeptide Systems (San Diego, CA). After reconstituting, the five peptides were pooled into single vials containing 550 μg of each peptide (representing a 10% overfill to allow complete retrieval of the appropriate dose), lyophilized, and stored at −80°C until use. On the day of vaccination, vials were brought to room temperature over 2 hours. 1.1 ml of 1% DMSO in sterile water was added and the vial swirled until the peptides solubilized. GM-CSF was added and mixed gently. Three syringes were prepared with ~ 0.4 ml of vaccine.
For stability testing of the vaccine, reconstituted peptides were separated chromatographically using an Agilent Zorbax 300SB C18 RP column with a water:acetonitrile:formic acid gradient to assess stability quarterly during the first year after mixing, then every six months to the present time, with no significant degradation observed during the vaccination period. Concentrations (mg/ml) at manufacture and 2 years following reconstitution were 0.58 and 0.74 for FR30, 0.6 and 0.63 for FR56, 0.68 and 0.61 for FR76, 0.75 and 0.73 for FR112, 0.76 and 0.78 for FR238.
Vaccinations
Prior to registration, patients underwent baseline screening evaluation. Within 14 days of study registration, prior to each treatment cycle, 4 weeks after completion of treatment, and 3, 6 and 12 months thereafter until disease recurrence or a maximum of 18 months post-registration, patients underwent a complete physical exam, assessment of performance status, blood chemistries and metabolic panel, urinalysis for proteinuria, TSH, ANA, and RF testing, toxicity assessments (CTCAE v. 4.0), and research blood draws for immune monitoring. Disease status was radiographically evaluated at registration and as per standard of care by the treating physician.
Patients received a standard tetanus booster (DT, Sanofi, Bridgewater, NJ) just prior to FRα vaccination unless they indicated that they had received a booster within one year of registration. This was done as a positive control to verify that the patients demonstrated a measurable immune response and prior human observations that concurrent administration of different vaccines did not appear to influence immunogenicity (30). All patients received one cycle of priming with 50 mg of cyclophosphamide administered orally twice daily on days 1-7 and 15-21 of a 28-day cycle (cycle 1). Patients were vaccinated intradermally at 3 sites in forearm (on a side where no axillary lymph node dissection [ALND] was performed) or the outer thigh (if bilateral ALNDs were performed) with a mixture of 500 μg each of FR30, FR56, FR76, FR113, and FR238 admixed with 125 μg of GM-CSF (Sargramostim, trade name Leukine from Genzyme) on day 1 of a 28 day cycle for a maximum of 6 vaccination cycles (cycles 2-7).
Treatment was discontinued if a patient developed grade ≥3 toxicity or grade ≥2 neurologic problems during cyclophosphamide that did not resolve within 3 weeks. Vaccinations were discontinued if the patient developed a grade ≥2 allergic reaction, autoimmune reaction, neurologic difficulties, any grade 3+ adverse events; antinuclear antibody value >3, or urine protein ≥2.0 gm/24 hours for more than 4 weeks.
Immunohistochemistry
Expression of FRα was ascertained as previously described (5). Briefly, tissue sections from the primary tumors of patients on the study were incubated with the monoclonal antibody mab343 (kindly provided by Dr. Phillip Low, Purdue University) or an isotype-matched control antibody after antigen retrieval. The mMACH3 system (Biocare Medical, Concord, California) with horseradish peroxidase detection was used. Tissue samples were scored by a pathologist specializing in breast and ovarian cancers (DV). Samples were considered positive for FRα expression if at least 10% of the epithelial cells expressed FRα. Expression of FRα was not used an eligibility criteria. Staining was scored as negative (0), weak (1+), moderate (2+) and strong (3+) membrane staining. The percent of cells within each section stained at each intensity was recorded to calculate an H-score for each sample. The H-score is a weighted score that captures both the proportion of positive staining and its intensity, and thus more representative of the staining of the entire tumor section. The H-score for staining each sample was defined as: H-score = 3 _ (% at 3 +) + 2 _ (% at 2 +) + 1 _ (% at 1 +) + 0 _ (% at 0). H-score values can range from zero (no membrane staining) to a maximum of 300 (100% membrane staining at 3+).
PBMC preparation, and cryopreservation
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation, as previously described (31). PBMCs were cryopreserved in liquid nitrogen at 10 million PBMCs/ml in RPMI containing 10% (vol/vol) DMSO, 110 mg/ml human serum albumin, and 2.7 mg/ml HEPEs. For serum preparation, whole blood was allowed to clot and then the serum was immediately frozen at −80°C. Peripheral blood samples were also obtained from 12 healthy volunteer women for comparison of Tregs levels.
Flow cytometry
Treg frequencies were assessed using flow cytometry. PBMCs obtained at baseline (i.e. before CTX treatment) and at the end of cycle 1 were surface-labeled with fluorochrome-conjugated antibodies against CD4, CD25, and CD127 (32–34). Cells were then permeabilized and labeled with anti-FoxP3. Samples were analyzed by flow cytometry (BD LSRII), and data were analyzed using FlowJo software (v.10.0.8). Within the lymphocyte gate, CD4+25+ cells were assessed for CD127 and FoxP3, and Tregs (defined as CD4+25+127loFoxP3+) were calculated as a percentage of total cells in the lymphocyte gate.
IFN-γ Enzyme-linked immunosorbent spot (ELISpot) assay
A 2-day, direct ex vivo, IFN-γ enzyme-linked immunosorbent spot (ELIspot) assay was used to determine frequencies of peptide-specific T lymphocytes, a modification of that which we have described previously (35). Initially a fetal bovine serum (FBS)-based assay was used for assessing immunity in the first 15 patients but then we switched to a human serum (HS)-based assay because concerns that low signal-to-noise ratio (SNR) may interfere with reliable detection of antigen-specific immune responses. The SNR was 0.92 ± 0.22 for the FBS-based assay and 1.74 ± 0.19 for the HS-based assay (p=0.043). On day 1, 2.5 ×105 PBMCs/well were plated in triplicate into 96-well round bottom plates in 200 μl of RPMI-1640 containing L-glutamine, penicillin, streptomycin, and 10% AB (of FBS) serum (T cell medium) along with cyclin D1 (control) peptide (MELLLVNKLKWNLAA)(35), FR30, FR56, FR76, FR113, FR238, FRα protein (Sino Biological Inc., Beijing, China), tetanus toxoid (TT) (List Biological Laboratories, Campbell, CA), or Phytohemagglutinin (PHA) (Sigma-Aldrich, St. Louis, MO). Peptides were plated at 10 μg/ml. PHA and tetanus were used at 1 μg/ml and FRα protein at 100 ng/ml. The cells were incubated at 37°C at 5% CO2. On the same day as plating the cells, nitrocellulose-backed (NC-plates) plates were coated with 10 μg/ml anti–IFN-γ antibody (MabTech USA, Mariemont, OH) in PBS at 50 μl/well for 24 hours. After 24 hours, the cells were then transferred to the washed and blocked NC plates and further incubated at 37°C for a further 24 hours followed by washing three times using PBS containing 0.05% Tween-20. The plate was then incubated for 2.5 hours at room temperature in 50 μl/well PBS containing 5 μg/ml biotinylated anti–IFN-γ Ab, washed three times with PBS, and further incubated with 100 μl/well streptavidin-alkaline phosphatase at a dilution of 1:1,000 in PBS for 2 hours at RT. After washing three times in PBS, the plate was incubated with 100 μl/well AP-colorimetric substrate for 20–30 minutes, rinsed with cool tap water, and allowed to dry completely. After drying overnight, the plates were read on an AID ELIspot reader (San Diego, CA), which provides quantitative spot information based on the number of stimulated cells that secrete IFN-γ. Antigen-specific T cell frequencies were defined as the average number of spots elicited by a given antigen minus the average number of spots with no added antigen.
Enzyme-linked immunosorbent assay (ELISA)
Antibody (IgG) responses were assessed using a typical ELISA. Briefly, 96-well microplates (Fisher Scientific) were coated with 20 ng/ml of FR30, FR56, FR76, FR113, FR238, cyclin D1 peptides, or with 100 ng/ml TT or FRα protein. Human IgG was used as a protein standard that was added at a concentration range of 250 to 1.95 μg/ml. After blocking the plates with 1% BSA, patient serum samples were added at a 1:100 dilution incubated for 2 hours at room temperature. Anti-human IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA) and ELISA substrate (BD Bioscience) were then added in standard fashion, and absorbance was read at 450 nm on a plate reader.
Statistical considerations
The study was designed to assess the immunogenicity and safety profile of a multi-epitope FRα peptide vaccine comprised of FR30, FR56, FR76, FR113, and FR238 admixed with GM-CSF. The sample size was powered for the immune monitoring endpoint. Assuming that half of the participants would develop immune responses, 22 patients were required to achieve a lower 95% confidence interval of 30% true responses, which was deemed the lowest proportion of participant responses that would support continued development of the vaccine. The primary clinical endpoint was the percentage of patients who developed a severe toxicity (defined as a Grade 3-5 AE by NCI-CTC version 4.0 criteria that was possibly, probably or definitely related to treatment). The sample size of 22 patients resulted in a maximum width of the 90% binomial confidence interval for the percentage of patients who develop severe toxicity of ± 17.5%.
Mean levels of antigen-specific T cells and antibodies were compared using the Paired Student’s t test (Microsoft Excel 2010; Microsoft Inc., Bellevue, Washington, USA). A patient was considered to have responded to vaccination if they had developed a 3-fold or greater increase in FRα-specific T cells or antibodies at any point during the vaccine or observation periods. For comparing the relative immunogenicity of the peptides, if patients had undetectable T cell immunity, a positive response was defined as was ≥ 50 antigen-specific T cells/million PBMC or at any point during the trial period. None of the patients had undetectable levels of antibodies prior to vaccination. Mean levels of Tregs in patients at baseline were compared with Tregs in normal healthy women using an unpaired Student’s T test. Correlations between the baseline levels and the magnitude of change of T cell frequency were examined using linear regression analysis (Prism, v.6, GraphPad Software, Inc.).
Rigid protocol criteria for moving the vaccine forward into future clinical testing (e.g. phase II or phase I combinations) was established. For this, mean values derived from triplicates with coefficients of variations of greater than 20% were excluded from further analysis. Post-immunization values were thereafter considered elevated relative to pre-immunization values if they were at least double and with non-overlapping standard deviations. If pre-immunization values were zero or below after subtracting the background, the post-immunization mean values must have been above zero with a standard deviation that was non-overlapping with the pre-immunization values. If at least 30% of patients developed an immune response (T cell or antibody) following vaccination, as measured in the vaccine period, then the vaccine was considered a candidate for future investigation.
RESULTS
Patient characteristics
Twenty-two female patients were treated in this phase I trial at Mayo Clinic in Rochester, MN. Table 2 summarizes the patient characteristics including the disease type, grade, histology. Fourteen patients had been previously diagnosed with ovarian cancer and eight with breast cancer. Four of the ovarian cancer patients had recurrent disease at the time of study enrollment. Patients were enrolled regardless of FRα (Materials and Methods). All 8 breast cancer patients were diagnosed with ER+PR+HER2/neu− cancers consistent with the high frequency of this phenotype in the patient population diagnosed with breast cancer.
Table 2.
Patient and tumor characteristics
| Breast cancer
cohort N=8 |
Ovarian cancer
cohort N=14 |
||
|---|---|---|---|
|
| |||
| median age (range) | 48 (39-61) | median age (range) | 57 (35-68) |
|
| |||
| T stage | Site | 13 (92.9%) | |
| T1 | 3 (37.5%) | ovary | 1 (7.1%) |
| T2 | 4 (50.0%) | fallopian tubes | |
| T4 | 1 (12.5%) | ||
|
| |||
| N stage | FIGO stag | 2 (14.3%) | |
| N1 | 5 (62.5%) | II | 12 (85.7%) |
| N2 | 2 (25.0%) | III | |
| N3 | 1 (12.5%) | ||
|
| |||
| Histology | Histology | ||
| Invasive ductal carcinoma | 5 (62.5%) | serous | 10 (71.4%) |
| Invasive lobular carcinoma | 1 (12.5%) | endometroid | 1 (7.1%) |
| mix of IDC and ILC | 2 (25.0%) | clear cell | 1 (7.1%) |
| mixed | 2 (14.3%) | ||
|
| |||
| Prior systemic therapy | Prior systemic therapy | ||
| Neo-adjuvant chemotherapy | 4 (50.0%) | Neo-adjuvant carboplatin/paclitaxel | 3 (21.4%) |
| Adjuvant chemotherapy | 4 (50.0%) | Adjuvant carboplatin based regimen | 9 (64.3%) |
| Adjuvant endocrine therapy | 8 (100.0%) | Adjuvant IP cisplatin/paclitaxel -> carboplatin/paclitaxel | 3 (21.4%) |
| Adjuvant IP cisplatin/paclitaxel -> BEV | 1 (7.1%) | ||
|
| |||
| Grade 1 fatigue | 3 (37.5%) | Grade 1 fatigue | 6 (42.9%) |
|
| |||
| Race | Race | ||
| White | 7 (88%) | White | 13 (93%) |
| Not reported | 1 (13%) | Not reported | 0 |
| Unknown | 0 | Unknown | 1 (7%) |
|
| |||
| FR H score | FR H score | ||
| 0 | 8 (100%) | 0 | 0 |
| 1-100 | 0 | 1-100 | 7 (50.0%) |
| 101-200 | 0 | 101-200 | 3 (21.4%) |
| 201-300 | 0 | 201-300 | 2 (14.3%) |
| no tumor available | 0 | no tumor available | 2 (14.3%) |
Vaccination against the FRα is safe
Twenty-two patients were evaluable for toxicity assessments. Overall, vaccination was well tolerated and was not associated with significant grade ≥ 3 toxicity (Table 3). One patient developed a treatment-related grade 3 injection site reaction. Grade 2 reactions observed during the active vaccination phase included lymphopenia (3 patients), fatigue (3 patients), neutropenia (2 patients), leukopenia (2 patients) and injection site reactions (2 patients). Two patients developed grade 3-4 infections during priming (CTX) phase that were not treatment-related. Tests for autoimmunity (rheumatoid factor, anti-nuclear antibodies, and thyroid stimulating hormone) did not show elevation for any participants throughout the vaccination schedule (data not shown because all were negative and below limits of detection of an abnormal response).
Table 3.
Adverse events at least possibly related to study treatment
| Grade | 2 | 3 | 4 |
|---|---|---|---|
| During Priming with Cyclophosphamide (N=21) | |||
| Decreased lymphocyte counts | 3 | 0 | 0 |
| Decreased white blood cell counts | 3 | 0 | 0 |
| Decreased neutrophil counts | 1 | 0 | 0 |
| Fatigue | 2 | 0 | 0 |
| Nausea | 1 | 0 | 0 |
| Sinus Pain | 1 | 0 | 0 |
| Sepsis | 0 | 0 | 1 |
| During Vaccination and Observation period (N=21) | |||
| Eye disorders | 1 | 0 | 0 |
| Arthralgia | 1 | 0 | 0 |
| Fatigue | 1 | 0 | 0 |
| Myalgia | 1 | 0 | 0 |
| Injection site reaction | 2 | 1 | 0 |
| Neutrophil count decreased | 2 | 0 | 0 |
| Dyspnea | 1 | 0 | 0 |
| White blood cell decreased | 1 | 0 | 0 |
| Lymphocyte count decreased | 3 | 0 | 0 |
| General disorder, admin site conditions | 1 | 0 | 0 |
Circulating Tregs are not decreased following CTX treatment
Frequencies of Tregs from pre-treatment and post-CTX blood samples were compared in 17 evaluable patients (Figs. 1A–B). Prior to priming with CTX, the mean frequency of Tregs was 0.6 ± 0.1% (± s.e.m.) of the total lymphocyte population. After CTX, the mean frequency was 0.7 ± 0.1%, which was not significantly different than mean pre-treatment levels (p=0.16). In contrast to prior studies, in this patient population with no evidence of disease, Tregs were not significantly elevated (127 ± 13% (± s.e.m.) of normal, p=0.2) relative to levels seen in 12 normal healthy women.
Fig. 1. Vaccination generates T cell but not antibody immunity to FRα following immunization.
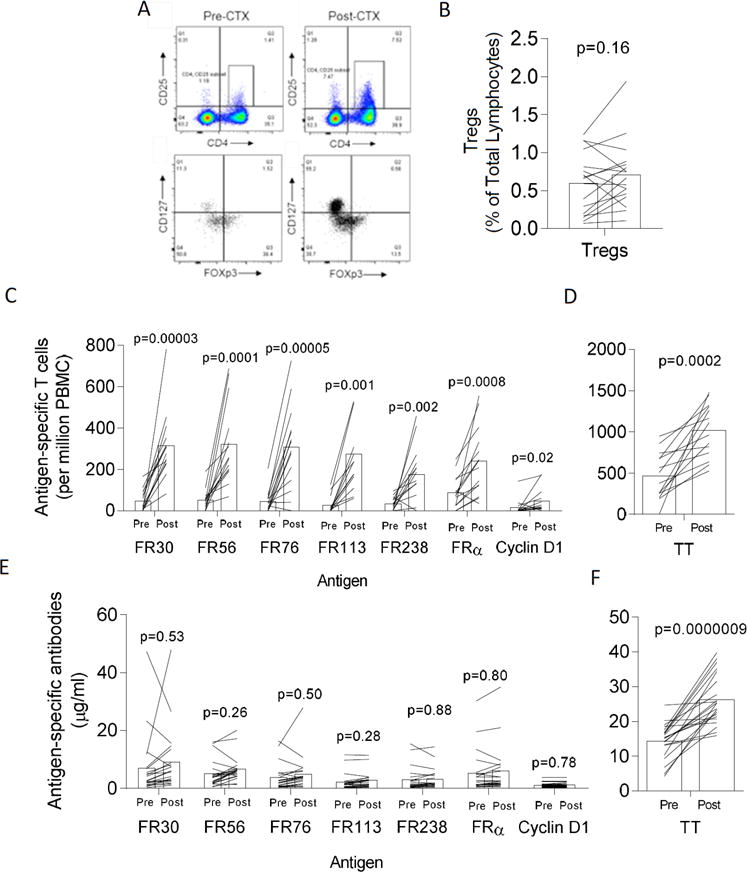
Panel A shows a representative set of flow cytometry dot plots demonstrating detection of Tregs using CD4, CD25, FoxP3, and CD127. Panel B: Bars show the mean levels of Tregs in 17 evaluable patients measured on time points 0 and 1 month before and after treatment with CTX just prior to vaccination. Each line traces the pre- and post-CTX Treg levels for a single unique patient. Panel C shows the mean (n=14 patients) pre-immunization (Pre) and highest post-vaccination (Post) frequency of antigen-specific T cells frequencies (per million PBMC plated) that recognize vaccine antigens, FR30, FR56, FR76, FR113, and FR238. Also shown are frequencies to control cyclin D1 and the FRα protein. Shown are the high post-vaccination values for the vaccine period which includes measurements up to 30 days following final injection. Panel D shows the mean pre-immunization (Pre) and highest post-vaccination frequency of tetanus toxoid-specific T cells for same patients in Panel C. Panel E shows the mean pre-immunization (Pre) and highest post-vaccination (Post) frequency of antigen-specific antibodies (μg/ml) that recognize vaccine antigens, FR30, FR56, FR76, FR113, FR238, control cyclin D1 and the FRα protein. Panel F shows the mean pre-immunization (Pre) and highest post-vaccination (Post) frequency of tetanus toxoid-specific antibodies (μg/ml) for same patients in Panel C. p values shown were calculated using the paired student’s T test. For panels C-F, each line traces the pre- and post-antigen-specific T cell levels for a single unique patient measured during the vaccine period.
Vaccination generates T cell but not antibody immunity to FRα following immunization
The frequency of IFN-γ-producing T cells specific for FRα peptides, FRα protein, TT, and control peptide were compared between pre-treatment samples and multiple post-treatment samples, during vaccine treatment (up to 30 days following the last vaccination). PBMCs from the vaccine period were available from 14 of 21 patients. In Fig. 1C, the pre-immunization and highest post-immunization (as observed during the vaccination period only) T cell frequencies, are plotted as previously described (36, 37), excluding values from the observation period at 3, 6, and 12 months following the final vaccination. The median number of post-vaccination (not including observation) period samples was 5.5 (range 1-6). The pre-immunization IFN-γ T cell frequency to FR30 was 47 ± 14 (± s.e.m., n=14) T cells/million PBMCs which increased to 315 ± 14 to T cells/million PBMCs (p=0.00003). To FR56, T cells increased from 52 ± 13 to 322 ± 52 (p=0.0001), to FR76 from 43 ± 14 to 307 ± 55 (p=0.00005), to FR113 from 27 ± 11 to 274 ± 68 (p=0.001), to FR238 from 33 ± 8 to 176 ± 37 (p=0.002). Analysis of data from the FBS-based assay yielded similar information (Supplementary Fig. S1).
There was a minor, albeit statistically significant, increase in the reactivity to the pan-DR binding cyclin D1 peptide from 15 ± 10 to 45 ± 15 (p=0.02). Although preexistent immunity appeared to be high for FRα, which has many potential epitopes, the mean frequency increased significantly from 88 ± 21 to 239 ± 44 T cells (p=0.0008), which indicates that the vaccine is generating T cells that are recognizing naturally processed antigens. Consistent with tetanus boosters in several of the patients prior to FRα vaccination, the mean TT T cell frequency increased from 541 ± 67 to 1007 ± 68 TT-specific T cells per million PBMC (p=0.00005) (Fig. 1D).
In contrast to increased T cell immunity, increased mean antibody responses to the peptides encompassing the vaccine were not observed following immunization (Fig. 1E). Serum for antibody assessments were available from 21 patients. The median number of post-vaccination (not including observation) period serum samples was 6.0 (range 3-6) and the maximum was 6. The pre-immunization mean antibody concentration to FR30 was 7.0 ± 2.3 μg/ml (± s.e.m., n=21) which increased to 9.1 ± 2.4 to μg/ml (p=0.53). To FR56, mean antibodies increased from 5.1 ± 1 to 6.7 ± 1.0 (p=0.26), to FR76 from 3.8 ± 1.0 to 4.9 ± 1.3 (p=0.5), to FR113 from 2.3 ± 0.6 to 2.8 ± 68 (p=0.28), and to FR238 from 3.1 ± 1.6 to 5.9 ± 1.8 (p=0.88). These slight non-significant increases did not lead to an overall increase in antibodies which could bind the whole FRα protein (5.3 ± 1.6 to 5.9 ± 1.8 ng/ml, p=0.8). Despite the lack of immune responses to the FRα, the mean TT serum antibody concentration increased from 14.4 ± 1.1 to 26.2 ± 1.6 (p=0.0000009), also consistent with tetanus boosters in several of the patients prior to FRα vaccination series (Fig. 1F).
T cell immunity slowly increases over the vaccine course
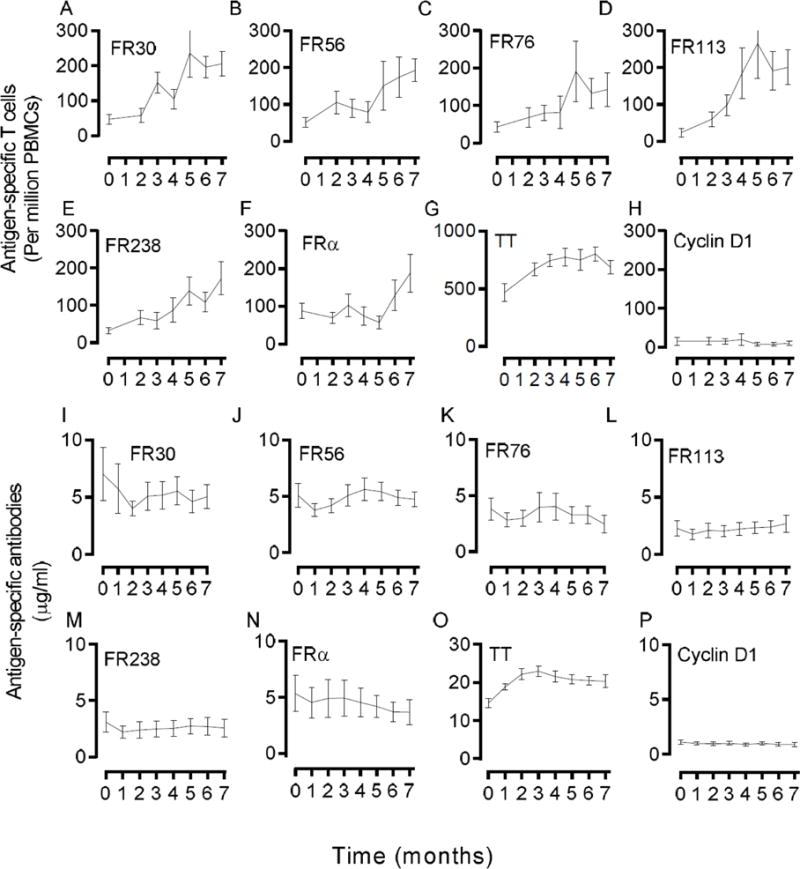
The T cell data was also examined at each individual time point over the course of vaccination, enabling an analysis of the duration to maximal immunity over the vaccine period (Figs. 2A–H). For this analysis, there were samples from 14, 12, 9, 11, 10, 11, and 10 patients for time points 0, 2, 3, 4, 5, 6, and 7, respectively. Time to maximal immunity was estimated to be 5 months for FR30, 7 months for FR56, 5 months for FR76, 5 months for FR113, and 7 months for FR238 with a median time to maximal immunity at 5 months. Excluding a patient who received only four of the six vaccines did not change the curves or conclusions (data not shown). In contrast, the levels of FRα-specific antibodies tended to remain even over the vaccine period with a slight downward trend observed at month 1 at the time of first vaccine (Figs. 2I–P). TT-specific antibodies increased to a maximum at the time of the second FRα immunization, a rapid response consistent with immunologic memory.
Fig. 2. T cell immunity increases slowly over the vaccine course.
Shown in Panels A-H are times courses of antigen-specific T cell frequencies (T cells per million PBMC, mean ± s.e.m.) for FR30, FR56, FR76, FR113, FR238, FRα protein, TT, and control cyclin D1 peptide, respectively, in 14 evaluable patients. Shown in Panels I-H are times courses of antigen-specific antibody frequencies (μg/ml, mean ± s.e.m.) for 21 evaluable patients. For all panels, time 0 months represents the baseline levels prior to CTX treatment. T cell frequencies were not measured at time point 1 month.
T cell immunity to FRα is durable
One of the key hallmarks of an efficacious vaccine is the ability to generate durable immunity to prevent disease. In order to determine whether immunologic memory was achieved, we collected and analyzed blood specimens at 10, 13 and 18 months following start of vaccination (observation period). Similar to that depicted in Fig. 1, we determined the frequency of IFNγ-producing T cells specific for FRα peptides, FRα protein, TT, and control peptide which were compared between pre-treatment samples and multiple post-treatment observation period samples. Assays were done separately from the vaccine period. PBMCs from the observation period were available from 16 of 21 patients, 11 of which overlapped with samples from the vaccine period.
In Fig. 3A, the pre-immunization and highest observation period (i.e. not including vaccination period samples) T cell frequencies are plotted. The median number of observation period samples was 2 (range 2-3) and the maximum was 3. The pre-immunization IFN-γ T cell frequency to FR30 was 10 ± 5 (± s.e.m., n=16) T cells/million PBMCs compared with the highest observation frequency of 169 ± 32 to T cells/million PBMCs (p=0.0002). To FR56, T cells increased from 16 ± 6 to 191 ± 40 (p=0.0004), to FR76 from 16 ± 6 to 178 ± 38 (p=0.0004), to FR113 from 5 ± 2 to 197 ± 52 (p=0.001), to FR238 from 20 ± 8 to 160 ± 32 (p=0.0002). There was a minor but insignificant increase in the reactivity to the pan-DR binding cyclin D1 peptide from 8 ± 5 to 21 ± 5 (p=0.09). The mean frequency of FRα-specific T cells increased 58 ± 14 to 222 ± 51 T cells (p=0.0008), which indicates that the vaccine is generating persistent T cells that recognize naturally processed antigens. The mean TT T cell frequency increased from 535 ± 62 to 1034 ± 71 TT-specific T cells per million PBMC (p=0.00005) (Fig. 3B).
Fig. 3. T cell immunity to the FRα is durable after immunization.
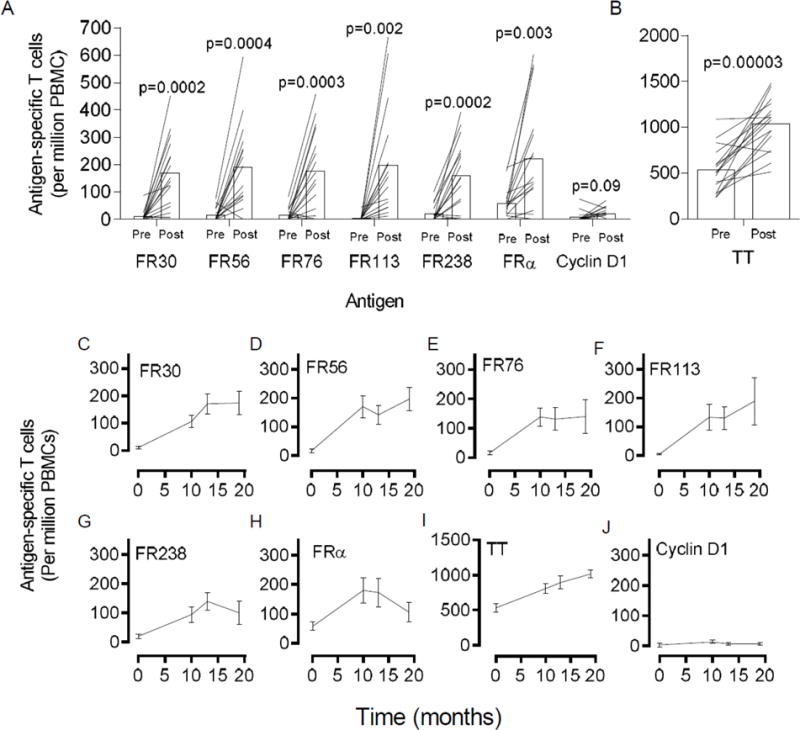
Panel A shows the mean (n=16 patients) pre-immunization (Pre) and highest observation (Post) frequency of antigen-specific T cells frequencies (per million PBMC plated) that recognize vaccine antigens, FR30, FR56, FR76, FR113, and FR238. Also shown are T cell frequencies to control cyclin D1 and the FRα protein. Shown are the high post-vaccination values for the observation period which includes measurements at months 10, 13, and 18. Panel B shows the mean pre-immunization (Pre) and highest post-vaccination frequency of tetanus toxoid-specific T cells for same patients in Panel A. Panels C-J are times courses of antigen-specific T cell frequencies (T cells per million PBMC, mean ± s.e.m.) for FR30, FR56, FR76, FR113, FR238, FRα protein, TT, and control cyclin D1 peptide, respectively, in 16 evaluable patients. p values shown were calculated using the paired student’s T test. For panels A-B, each line traces the pre- and highest observation period (Post) antigen-specific T cell levels for a single unique patient measured during the vaccine period.
The T cell data was also examined at each individual time point over the course of observation, enabling an analysis of the duration of immunity over the observation period (Figs. 3C–J). For this analysis, there were samples from 16 patients at each of the time point. As shown FRα-peptide-specific generally appeared stably high during the observation period demonstrating immunologic memory.
All of the peptides in the vaccine appear to be immunogenic in most patients
To determine the relative immunogenicity of the vaccine component we analyzed the percentage of patients that responded (either in the vaccine or observation period, n=19 patients) to each of the individual peptides based on having a 3 fold increased T cell frequency over baseline. In case of zero baselines, a minimum frequency of 50 antigen-specific T cells was the cutoff for response. T cell immunity to all of the peptides was frequently observed with 79% to FR30, 74% to each of FR56, FR76, and FR113, and 58% to FR238 (Fig. 4A). Antibody response rates were minimal at 14% for FR30, 14% for FR56, 5% for each of FR76 and FR113, and 10% for FR238 (Fig. 4B). All patients in this analysis responded to vaccine with a T cell response with the majority (17/19) of patients responding to multiple epitopes (Fig. 4C). The median number of epitopes that patients demonstrated T cell immunity was 3. Six of 19 patients responded with antibody responses for a median of 0 epitopes to which there was antibody responses. Based on the more rigid protocol criteria for advancing the vaccine (Materials and Methods) 19/21 (~91%) patients demonstrated an antigen-specific response (either antibody or T cell response) in the vaccine period.
Fig. 4. All of the vaccine epitopes are immunogenic in most patients.
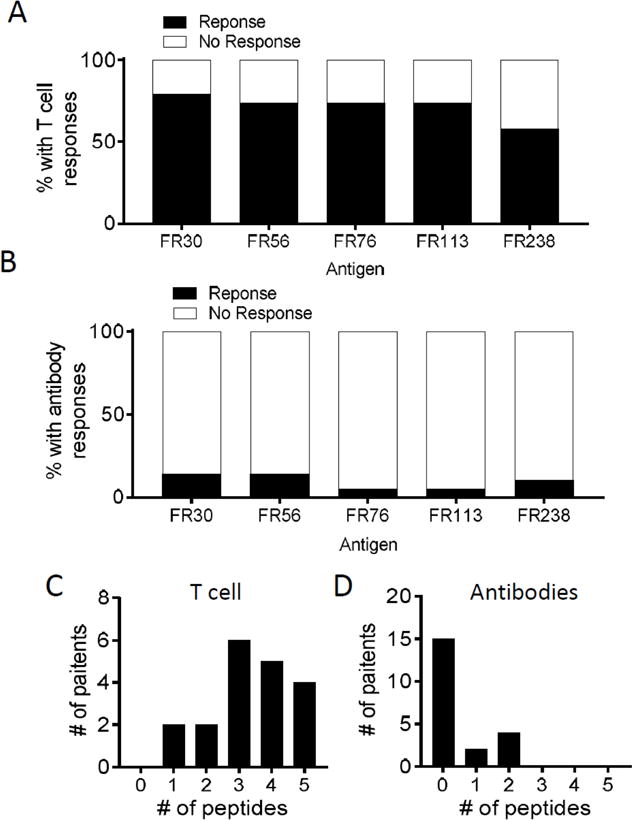
Panels A–B shows the % of patients that responded to vaccine epitopes with either T cell responses (Panel A) or antibody responses (Panel B). Panels C-D shows the distribution of T cell and antibody responses to the individual epitopes.
Tumor expression of FRα
FRα protein expression was observed on pre-enrollment archived tumor specimens from 12 of 12 assessable OC patients and 0 of 8 BC patients (Table 2). Two ovarian cancer tissue samples did not contain evaluable tumor. Finally, the T cell response to vaccine peptides did not appear to correlate with FRα positivity or negativity in the primary tumor (FR30, p=0.36; FR56, p=0.31; FR76, p=0.73; FR113, p=0.69; FR238, =0.82).
Clinical outcomes
Although the trial was not designed to evaluate clinical outcomes, all patients remained alive at last follow-up, at least two years following initiation of immunizations. OC patients in first remission demonstrated a median of relapse-free survival of 528 days, whereas median survival was not reached for the OC patients in second remission and the BC patients.
DISCUSSION
There were two main goals of this phase I clinical trial using a multi-epitope FRα peptide vaccine. The first was to assess the safety, since this approach was the first-in-human trial of the FRα vaccine and second was to assess the ability of this vaccine to generate T cell and antibody immunity against FRα With respect to the first goal, the vaccine immunizations was found to be uniformly safe overall and associated only with mild to moderate toxicities, despite the fact that some normal healthy tissues, notably the kidney and the choroid plexus, express FRα (10, 38). One potential reason for the reasonable safety profile is that the vaccine targets CD4 T cells whose activity is regulated by HLA class II which is typically only expressed on hematopoietic cells. Since normal tissues apparently shed very little FRα relative to cancer tissues, it is likely that there is very little presentation of FRα in normal healthy tissue for the vaccine-generated T cell to respond to (39). The trial enrolled adults who had previously been diagnosed with either breast or ovarian cancer and who had completed curative intent treatment (surgery, chemotherapy). Participants were allowed to continue concurrent hormonal therapy, if indicated. This might explain the musculoskeletal-related side effect profile (myalgias, arthralgias) seen in some of the breast cancer patients, since all breast cancer patients enrolled in the study were noted to be hormone positive (ER/PR positive, HER2/neu negative)(40). This biased enrollment was secondary to competing studies at our institution for patients with hormone negative and HER2/neu positive breast cancers, respectively.
Contradictory to what was expected and what was seen in other studies using metronomic CTX (28), Tregs did not decrease consistently with exposure to CTX. In some patients Tregs actually increased for unclear reasons despite a cumulative CTX dose of 1400 mg. Comparisons with previously published values are complicated by the fact that Tregs have been quantitated and reported in literature in numerous ways. The method reported here (CD127lo/FoxP3+ cells within the CD4+/CD25+ gate) is more rigorous than studies using fewer markers (41). Assessment was done one week after the last CTX dose; other studies have reported on continuing CTX treatment until the administration of vaccine. It is likely that the variation in Tregs levels is quite high and that the relatively minor increases or decreases observed reflect normal variation. Additionally, many studies in the literature assess CTX effects on Tregs in patients with advanced metastatic disease, whereas this clinical trial enrolled patients who did not have any evidence of disease at the time of treatment with CTX. Levels and function of peripheral Tregs may differ with disease stage, noting our finding that the presently described patient population did not have elevated Tregs relative to control.
ELIspots were utilized to estimate peptide antigen-specific T cell responses to the 5 FRα peptides. Most patients responded to at least 3 out of the 5 peptides resulting in the generation of immunity able to recognize naturally processed antigen. However, despite having established a cutoff, it is notable that it was an arbitrary cutoff because at present there is no way to know what levels of antigen-specific T cells are required for disease protection. The high frequency of responses observed is consistent with the degeneracy of the epitopes with respect to predicted HLA-DR binding (13). Antigen-specific T cell responses during the observation period were relatively similar to those observed during active priming with the FRα vaccine. Despite the high prevalence of immunity to the FRα, it is important to note that the assay used was optimized to detect immunity but was not validated to the extent to provide an assured accurate assessment of the frequency of T cells, including rigorous control of all of the constituent assay components. Future implementation of the vaccine in clinical trials, thus, needs to be accompanied by validation programs to better depict the longitudinal increase in antigen-specific immune effectors. (42, 43). Additional analyses could also include cell separations to specifically determine if CD4 T cells or the primary responding population or whether CD8 T cells are also activated as a result of exposure to class I binding peptides embedded within the longer vaccine peptides as we have previously suggested in earlier studies (13).
Assessment of antibody responses revealed that some, albeit a small number, of patients demonstrated a vaccine-induced antibody immune response to one or more of the peptides. However, there were no responses to intact FRα protein, which is the only response that would be expected to have clinical relevance. It is not currently known whether directing both cellular and humoral immune responses against FRα are necessary for efficacy. In some situations (e.g. HER2/neu positive breast cancer), antibody responses appear to be helpful (44). However, anti-FRα based antibody treatment strategies did not show great responses in clinical trials (45).
Given the enrollment of several patient populations and a small total number of subjects, this trial cannot assess the clinical efficacy of the vaccine, nor was it designed for that purpose. While it is noteworthy that the OC patients that were in first remission demonstrated a median progression-free survival of 528 days following the initiation of the immunization and that all patients are still alive, a carefully controlled clinical trial in which patients are enrolled within a defined period following conventional treatment should enable a better understanding of the role of vaccine in delaying or preventing disease recurrence. Alternatively, there are other possible uses of vaccines in the course of a patient’s treatment. One potential example is combination with checkpoint blockade, a setting in which we and others have demonstrated synergistic activity in murine models with respect to tumor regression and lasting protection from disease relapse (46, 47).
Identified over three decades ago, GM-CSF has been widely as a vaccine adjuvant with notable immune promoting properties including increasing cytotoxic T cell activity, upregulation of co-stimulatory molecules (e.g. CD80 and CD86), and increasing the migratory and antigen-presentation properties of antigen-presenting cells (reviewed in (48, 49)). In our study we chose to use GM-CSF as the vaccine adjuvant primarily due to (1) our past successful experience with the cytokine in human trials, (2) its outstanding safety profile, and (3) its association with clinically effective peptide vaccination strategies (50–55). Thorough examination of GM-CSF as a vaccine adjuvant has demonstrated that its use is not straightforward and under certain conditions, such as concurrent delivery with montanide, the cytokine may actually be immune suppressive and detrimental to the generation of immunity (56). Other adjuvants have recently emerged as alternatives to GM-CSF and other commonly used adjuvants (e.g. montanide), including poly-ICLC, STING agonists, CpG ODN, and even checkpoint modulators such as anti-PD-1 (46, 57–59). Future clinical testing should reveal whether these newer vaccine adjuvants demonstrate superior activity relative to GM-CSF.
Tumor expression of FRα was not an eligibility requirement for this trial. This decision was made based on the high proportion of patients with serous ovarian cancer whose tumors express FRα, the observation that breast cancers noted to be initially negative for FRα expression may develop FRα expression at the time of recurrence. In the current cohort, 12 out of 12 (100%) evaluable ovarian cancer patients and 0 out of 8 breast cancer patients were noted to have FRα expression in their tumor samples. These results are comparable to previous studies of the expression of FRα in these tumors (5). FRα expression in hormone positive breast cancer is estimated to be around 20%, with higher reported rates in hormone negative (triple negative) breast cancers (60). Whether tumor expression of FRα should be an inclusion criterion in future studies is debatable, as there is rationale to vaccinating against antigens that are not expressed by the tumor at disease onset but are expressed at the time of recurrence.
In summary, this first-in-human phase I study targeting FRα with a multi-peptide vaccine after priming with metronomic CTX showed minimal untoward and no severe unanticipated toxicities. Projected clinical efficacy of vaccine-induced anti-tumor immunity cannot easily be derived from the data but supports continued development of the vaccine perhaps as a means to prevent recurrence or to be used in combination with immune checkpoint blockade (46).
Supplementary Material
TRANSLATIONAL RELEVANCE.
FRα, in its native form, has recently been identified as a potential tumor antigen due to its aberrant expression in a variety of cancers such as ovarian, lung and breast cancer. Patients with these diseases often demonstrate pre-existing immunity to the protein indicating it is a natural target of the immune response. This pre-existing immune response and its previously defined role in tumor aggressiveness suggest that it may be useful in vaccine preparations designed to augment pre-existing immunity to levels that maybe therapeutic. In this first report, we demonstrate that immune responses to the FRα can safely generated in patients previously diagnosed with cancer. Our findings illustrate the potential for the safe use of vaccines targeting the FRα to prevent disease recurrence. Additional uses to consider would be incorporating newer adjuvant and use of the vaccine in combination with new immunotherapeutic strategies such as immune checkpoint blockade.
Acknowledgments
This work was supported by grants from the US National Institutes of Health (P50-CA136393-Mayo Clinic SPORE in Ovarian Cancer to Dr. Scott Kaufmann and P30-CA015083-Mayo Comprehensive Cancer Center Grant to Dr. Robert Diasio), the Fred C. and Katherine B. Andersen Foundation to (K.L. Knutson), the Mayo Clinic Discovery Translation Research Fund (K.L. Knutson), Department of Defense Breast Cancer Research Program grants W81XWH-15-1-0292 (K.L. Knutson) and the Minnesota Ovarian Cancer Alliance (K.L. Knutson). The authors would like to acknowledge the contributions (conception, design and acquisition of data) of Dr. Lynn Hartmann, previously of Mayo Clinic in Rochester MN. The authors would also like to gratefully acknowledge the statistical assistance provided by Dr. Vera Suman, Dr. Amylou Dueck and Travis Dockter of the Mayo Clinic Comprehensive Cancer Center. This study has previous been published in Abstract for at 2015 Annual Meeting of the American Society for Clinical Oncology. The citation is: Kasi PM, Kalli K, Block MS, Hobday TJ, Dockter TJ, Suman VJ, Erskine CL, Visscher DW, Wilson G, Shreeder B, Knutson KL. A phase I trial of the safety and immunogenicity of a multi-epitope folate receptor alpha peptide vaccine used in combination with cyclophosphamide in subjects previously treated for breast or ovarian cancer.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
Drs. Kalli and Knutson are joint inventors on a patent, filed by the Mayo Clinic, entitled Immunity to Folate Receptors (US8486412), which is currently licensed to TapImmune, Inc. of Jacksonville, FL. Dr. Glynn Wilson is an employee of TapImmune, Inc. of Jacksonville. Dr. Block receives research financial support from TapImmune, Inc. for a related clinical trial.
AUTHOR’S CONTRIBUTIONS
Conception and design: KL Knutson, KL Kalli, MS Block and DJ Puglisi-Knutson
Development of methodology: KL Knutson, DJ Puglisi-Knutson, B Shreeder, KL Kalli, MS Block, D Padley, AB Dietz, and MP Gustafson
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): KL Knutson, B Shreeder, KL Kalli, MS Block, D Padley, DW Visscher, TJ Hobday, TK Mangskau
Analysis and Interpretation of data: KL Knutson, KL Kalli, MS Block, CL Erskine
Writing, review, and/or revision of the manuscript: KL Knutson, KL Kalli, MS Block, PM Kasi, G Wilson
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32:2959–66. doi: 10.1200/JCO.2013.55.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 5.Kalli KR, Oberg AL, Keeney GL, Christianson TJ, Low PS, Knutson KL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108:619–26. doi: 10.1016/j.ygyno.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev. 2004;56:1067–84. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski VR, Jr, et al. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992;52:3396–401. [PubMed] [Google Scholar]
- 8.Toffoli G, Cernigoi C, Russo A, Gallo A, Bagnoli M, Boiocchi M. Overexpression of folate binding protein in ovarian cancers. Int J Cancer. 1997;74:193–8. doi: 10.1002/(sici)1097-0215(19970422)74:2<193::aid-ijc10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 9.O’Shannessy DJ, Somers EB, Smale R, Fu YS. Expression of folate receptor-alpha (FRA) in gynecologic malignancies and its relationship to the tumor type. Int J Gynecol Pathol. 2013;32:258–68. doi: 10.1097/PGP.0b013e3182774562. [DOI] [PubMed] [Google Scholar]
- 10.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338:284–93. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 11.Hartmann LC, Keeney GL, Lingle WL, Christianson TJ, Varghese B, Hillman D, et al. Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer. 2007;121:938–42. doi: 10.1002/ijc.22811. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Wang J, Tacha DE, Li P, Bremer RE, Chen H, et al. Folate receptor alpha associated with triple-negative breast cancer and poor prognosis. Arch Pathol Lab Med. 2014;138:890–5. doi: 10.5858/arpa.2013-0309-OA. [DOI] [PubMed] [Google Scholar]
- 13.Knutson KL, Krco CJ, Erskine CL, Goodman K, Kelemen LE, Wettstein PJ, et al. T-cell immunity to the folate receptor alpha is prevalent in women with breast or ovarian cancer. J Clin Oncol. 2006;24:4254–61. doi: 10.1200/JCO.2006.05.9311. [DOI] [PubMed] [Google Scholar]
- 14.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 15.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Lyerly HK, et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood. 2008;112:610–8. doi: 10.1182/blood-2008-01-135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:9331–6. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swanson MS, Sinha UK. Rationale for combined blockade of PD-1 and CTLA-4 in advanced head and neck squamous cell cancer-review of current data. Oral Oncol. 2015;51:12–5. doi: 10.1016/j.oraloncology.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A Systematic Review of Immunotherapy in Urologic Cancer: Evolving Roles for Targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. 2015;68:267–79. doi: 10.1016/j.eururo.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 21.Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One. 2013;8:e61895. doi: 10.1371/journal.pone.0061895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94:41–53. doi: 10.1189/jlb.1212631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bass KK, Mastrangelo MJ. Immunopotentiation with low-dose cyclophosphamide in the active specific immunotherapy of cancer. Cancer Immunol Immunother. 1998;47:1–12. doi: 10.1007/s002620050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G, Gupta R, Petrik S, Laiko M, Leatherman JM, Asquith JM, et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res. 2014;2:949–61. doi: 10.1158/2326-6066.CIR-14-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009;27:5911–8. doi: 10.1200/JCO.2009.23.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–83. doi: 10.1007/s00281-011-0245-0. [DOI] [PubMed] [Google Scholar]
- 27.Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother. 2012;61:353–62. doi: 10.1007/s00262-011-1106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–8. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalli KR, Oberg AL, Keeney GL, Christianson TJ, Low PS, Knutson KL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108:619–26. doi: 10.1016/j.ygyno.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bock HL, Kruppenbacher JP, Bienzle U, De Clercq NA, Hofmann F, Clemens RL. Does the concurrent administration of an inactivated hepatitis A vaccine influence the immune response to other travelers vaccines? J Travel Med. 2000;7:74–8. doi: 10.2310/7060.2000.00025. [DOI] [PubMed] [Google Scholar]
- 31.Disis ML, Grabstein KH, Sleath PR, Cheever MA. Generation of immunity to the HER-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clin Cancer Res. 1999;5:1289–97. [PubMed] [Google Scholar]
- 32.Hartigan-O’Connor DJ, Poon C, Sinclair E, McCune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods. 2007;319:41–52. doi: 10.1016/j.jim.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Beyer M, Classen S, Endl E, Kochanek M, Weihrauch MR, Debey-Pascher S, et al. Comparative approach to define increased regulatory T cells in different cancer subtypes by combined assessment of CD127 and FOXP3. Clin Dev Immunol. 2011;2011:734036. doi: 10.1155/2011/734036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dasgupta A, Mahapatra M, Saxena R. Flow cytometric immunophenotyping of regulatory T cells in chronic lymphocytic leukemia: comparative assessment of various markers and use of novel antibody panel with CD127 as alternative to transcription factor FoxP3. Leuk Lymphoma. 2013;54:778–89. doi: 10.3109/10428194.2012.730614. [DOI] [PubMed] [Google Scholar]
- 35.Karyampudi L, Krco CJ, Kalli KR, Erskine CL, Hartmann LC, Goodman K, et al. Identification of a broad coverage HLA-DR degenerate epitope pool derived from carcinoembryonic antigen. Cancer Immunol Immunother. 2010;59:161–71. doi: 10.1007/s00262-009-0738-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Disis ML, Wallace DR, Gooley TA, Dang Y, Slota M, Lu H, et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J Clin Oncol. 2009;27:4685–92. doi: 10.1200/JCO.2008.20.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peoples GE, Holmes JP, Hueman MT, Mittendorf EA, Amin A, Khoo S, et al. Combined clinical trial results of a HER2/neu (E75) vaccine for the prevention of recurrence in high-risk breast cancer patients: U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin Cancer Res. 2008;14:797–803. doi: 10.1158/1078-0432.CCR-07-1448. [DOI] [PubMed] [Google Scholar]
- 38.Wollack JB, Makori B, Ahlawat S, Koneru R, Picinich SC, Smith A, et al. Characterization of folate uptake by choroid plexus epithelial cells in a rat primary culture model. J Neurochem. 2008;104:1494–503. doi: 10.1111/j.1471-4159.2007.05095.x. [DOI] [PubMed] [Google Scholar]
- 39.Basal E, Eghbali-Fatourechi GZ, Kalli KR, Hartmann LC, Goodman KM, Goode EL, et al. Functional folate receptor alpha is elevated in the blood of ovarian cancer patients. PLoS One. 2009;4:e6292. doi: 10.1371/journal.pone.0006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez EA. Safety profiles of tamoxifen and the aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Ann Oncol. 2007;18(Suppl 8):viii26–35. doi: 10.1093/annonc/mdm263. [DOI] [PubMed] [Google Scholar]
- 41.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Hakansson L, Janetzki S, et al. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clin Cancer Res. 2011;17:3064–76. doi: 10.1158/1078-0432.CCR-10-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knutson KL, dela Rosa C, Disis ML. Laboratory analysis of T-cell immunity. Front Biosci. 2006;11:1932–44. doi: 10.2741/1936. [DOI] [PubMed] [Google Scholar]
- 44.Knutson KL, Clynes R, Shreeder B, Yeramian P, Kemp KP, Ballman K, et al. Improved Survival of HER2+ Breast Cancer Patients Treated with Trastuzumab and Chemotherapy Is Associated with Host Antibody Immunity against the HER2 Intracellular Domain. Cancer Res. 2016;76:3702–10. doi: 10.1158/0008-5472.CAN-15-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vergote I, Armstrong D, Scambia G, Teneriello M, Sehouli J, Schweizer C, et al. A Randomized, Double-Blind, Placebo-Controlled, Phase III Study to Assess Efficacy and Safety of Weekly Farletuzumab in Combination With Carboplatin and Taxane in Patients With Ovarian Cancer in First Platinum-Sensitive Relapse. J Clin Oncol. 2016;34:2271–8. doi: 10.1200/JCO.2015.63.2596. [DOI] [PubMed] [Google Scholar]
- 46.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, et al. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 2014;74:2974–85. doi: 10.1158/0008-5472.CAN-13-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74:4042–52. doi: 10.1158/0008-5472.CAN-13-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clive KS, Tyler JA, Clifton GT, Holmes JP, Mittendorf EA, Ponniah S, et al. Use of GM-CSF as an adjuvant with cancer vaccines: beneficial or detrimental? Expert Rev Vaccines. 2010;9:519–25. doi: 10.1586/erv.10.40. [DOI] [PubMed] [Google Scholar]
- 49.Zhao W, Zhao G, Wang B. Revisiting GM-CSF as an adjuvant for therapeutic vaccines. Cell Mol Immunol. 2018;15:187–9. doi: 10.1038/cmi.2017.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, et al. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 2002;20:2624–32. doi: 10.1200/JCO.2002.06.171. [DOI] [PubMed] [Google Scholar]
- 51.Disis ML, Schiffman K, Guthrie K, Salazar LG, Knutson KL, Goodell V, et al. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein--based vaccine. J Clin Oncol. 2004;22:1916–25. doi: 10.1200/JCO.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 52.Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Invest. 2001;107:477–84. doi: 10.1172/JCI11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–61. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 54.Slingluff CL, Jr, Lee S, Zhao F, Chianese-Bullock KA, Olson WC, Butterfield LH, et al. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602) Clin Cancer Res. 2013;19:4228–38. doi: 10.1158/1078-0432.CCR-13-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mittendorf EA, Clifton GT, Holmes JP, Clive KS, Patil R, Benavides LC, et al. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients: from US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer. 2012;118:2594–602. doi: 10.1002/cncr.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slingluff CL, Jr, Petroni GR, Olson WC, Smolkin ME, Ross MI, Haas NB, et al. Effect of granulocyte/macrophage colony-stimulating factor on circulating CD8+ and CD4+ T-cell responses to a multipeptide melanoma vaccine: outcome of a multicenter randomized trial. Clin Cancer Res. 2009;15:7036–44. doi: 10.1158/1078-0432.CCR-09-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res. 2014;20:1355–65. doi: 10.1158/1078-0432.CCR-13-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khong H, Overwijk WW. Adjuvants for peptide-based cancer vaccines. J Immunother Cancer. 2016;4:56. doi: 10.1186/s40425-016-0160-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Necela BM, Crozier JA, Andorfer CA, Lewis-Tuffin L, Kachergus JM, Geiger XJ, et al. Folate receptor-alpha (FOLR1) expression and function in triple negative tumors. PLoS One. 2015;10:e0122209. doi: 10.1371/journal.pone.0122209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.