

Abstract
Among the metal-dependent histone deacetylases, the class IIb isozyme HDAC6 is remarkable due to its role in the regulation of microtubule dynamics in the cytosol. Selective inhibition of HDAC6 results in microtubule hyperacetylation, leading to cell cycle arrest and apoptosis, which is a validated strategy for cancer chemotherapy and the treatment of other disorders. HDAC6 inhibitors generally consist of a Zn2+-binding group such as a hydroxamate, a linker, and a capping group; the capping group is a critical determinant of isozyme selectivity. Surprisingly, however, even “capless” inhibitors exhibit appreciable HDAC6 selectivity. To probe the chemical basis for this selectivity, we now report high-resolution crystal structures of HDAC6 complexed with capless cycloalkylhydroxamate inhibitors 1–4. Each inhibitor hydroxamate group coordinates to the catalytic Zn2+ ion with canonical bidentate geometry. Additionally, the olefin moieties of compounds 2 and 4 bind in an aromatic crevice between the side chains of F583 and F643. Reasoning that similar binding could be achieved in the representative class I isozyme HDAC8, we employed isothermal titration calorimetry to study the thermodynamics of inhibitor binding. These measurements indicate that the entropy of inhibitor binding is generally positive for binding to HDAC6 and negative for binding to HDAC8, resulting in up to 313-fold selectivity for binding to HDAC6 relative to HDAC8. Thus, favorable binding entropy contributes to HDAC6 selectivity. Notably, cyclohexenylhydroxamate 2 represents a promising lead for derivatization with capping groups that may further enhance its impressive 313-fold thermodynamic selectivity for HDAC6 inhibition.
Graphical Abstract
Introduction
Since the discovery of histone acetylation more than 50 years ago,1 the reversible acetylation of lysine has emerged as a post-translational modification equally important to the reversible phosphorylation of serine, threonine, and tyrosine for the regulation of protein function.2 Thousands of acetylation sites have been identified in proteins from all domains of life, in which acetylation regulates diverse processes and pathways such as the cell cycle, central carbon metabolism, and cellular signaling networks.3–5 Lysine acetylation is catalyzed by a histone acetyltransferase (HAT) using acetyl-CoA as a co-substrate, and acetyllysine deacetylation is catalyzed by a histone deacetylase (HDAC) to yield products lysine and acetate.6–9 Members of the HDAC family are also referred to more generally as lysine (K) deacetylases or KDACs, since their substrates also include non-histone proteins. Aberrant HDAC activity is associated with numerous diseases, and various HDAC isozymes serve as validated targets for drug design.10–14
Phylogenetic analysis15 suggests the classification of metal-dependent HDAC isozymes as follows: the class I HDACs 1, 2, 3, and 8; the class IIa HDACs 4, 5, 7, and 9; the class IIb HDACs 6 and 10 (although the latter is a polyamine deacetylase16); and the sole class IV isozyme, HDAC11, which is a fatty acid deacylase.17 The class I, II, and IV HDACs are metal-dependent enzymes (Zn2+ or Fe2+)18 that adopt the arginase-deacetylase fold and likely share the same catalytic mechanism for amide hydrolysis.9,19–21 Class III HDACs, better known as sirtuins, exhibit a different fold and utilize a different, NAD+-dependent mechanism in comparison with metal-dependent HDACs.22,23
HDAC6 is unique among the metal-dependent HDACs in that it contains a serine/glutamate-rich tetradecapeptide directing localization in the cytosol,24 where HDAC6 acts primarily upon cytoskeletal protein substrates tubulin and tau,25,26 as well as proteins involved in the aggresome-autophagy pathway.27,28 HDAC6 contains two catalytic domains, CD1 and CD2, the crystal structures of which have recently been solved.29,30 While both domains are required for optimal biological activity,31–34 the high-activity CD2 domain is primarily responsible for lysine deacetylase activity in vitro and in vivo.33–35 Numerous diseases are associated with upregulated HDAC6 activity, including various cancers, Alzheimer’s disease, and immune disorders.36–40 Current drug discovery efforts focus mainly on blocking catalytic activity in the HDAC6 CD2 domain; moreover, there is particular interest in the development of inhibitors selective for HDAC6 over other HDAC isozymes to minimize off-target side effects.
Typical HDAC inhibitors consist of a zinc-binding group such as a hydroxamate moiety, a capping group capable of interacting with residues in the outer active site cleft, and a linker group connecting the two (Figure 1). Appreciable selectivity for HDAC6 is observed for inhibitors containing aromatic linker groups and bulky capping groups such as Tubastatin A,41 Nexturastat,42 N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB),43 and N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB).44 Crystal structures of HDAC6 complexed with some of these inhibitors have recently been reported,29,30,45 revealing that these sterically bulky inhibitors exploit an unusual monodentate hydroxamate-Zn2+ coordination mode that is nearly isoenergetic with the more commonly observed bidentate hydroxamate-Zn2+ coordination mode (Figure 1).29,45 Monodentate hydroxamate-Zn2+ coordination could conceivably result from steric constrictions in the HDAC6 active site that prevent phenylhydroxamates with bulky capping groups from making a closer interaction with the catalytic Zn2+ ion. However, since even simple “capless” inhibitors (Scheme 1) retain nanomolar potency and 10-fold or greater selectivity for HDAC6 relative to class I HDACs based on IC50 assays (Scheme 1),46 the bulky capping group is not the sole determinant of selectivity. What, then, determines the isozyme selectivity of these low-molecular weight, capless inhibitors?
Figure 1.
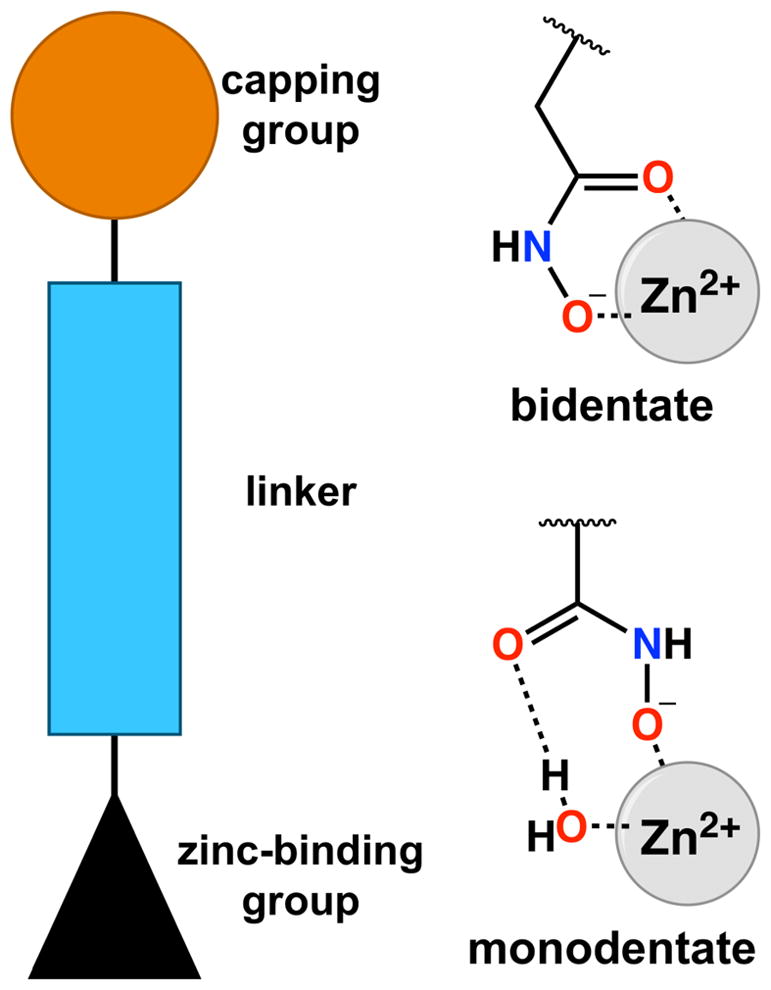
Generalized description of an HDAC inhibitor. A hydroxamate moiety is often utilized as a zinc-binding group and can coordinate to the catalytic Zn2+ ion with bidentate or monodentate geometry. Based on the 70:30 mixture of bidentate:monodentate coordination modes observed for trichostatin A, the monodentate coordination mode is only 0.5 kcal/mol less stable than the bidentate coordination mode.44 Parenthetically, an alternative monodentate hydroxamate-Zn2+ coordination mode is possible through the ionized NH group, as observed for hydroxamate binding to human carbonic anhydrase II;65 however, this binding mode has not been observed for hydroxamate binding to HDACs.
Scheme 1.
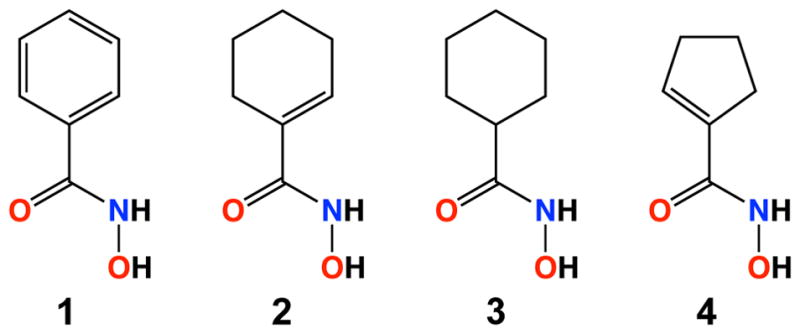
As the first step in answering this question, we now report the X-ray crystal structures of the high-activity CD2 domain from Danio rerio HDAC6 (henceforth simply “HDAC6”) complexed with the capless inhibitors phenylhydroxamate (1), cyclohexenylhydroxamate (2), cyclohexylhydroxamate (3), and cyclopentenylhydroxamate (4) (Scheme 1). These compounds have been previously profiled as HDAC6 inhibitors with sub-micromolar potencies and selectivities of up to 36-fold based on IC50 measurements made by Wagner and colleagues.46 Additionally, we report thermodynamic measurements of enzyme-inhibitor complexation using isothermal titration calorimetry (ITC). These studies reveal that favorable entropy of binding contributes to the selectivity of inhibitor binding to the class IIb enzyme HDAC6 over the class I enzyme HDAC8.
Materials and Methods
Reagents
In general, chemicals used in buffers and crystallization conditions were purchased from Fisher, Millipore Sigma, or Hampton Research and used without further purification. Compounds 1–4 were synthesized as described.46
Protein Preparation
Catalytic domain 2 from Danio rerio HDAC6 (herein designated simply “HDAC6”) was recombinantly expressed using His6-MBP-TEV-HDAC-pET28a(+) vectors and purified as previously described with minor modifications.45 Briefly, HDAC6 was expressed using E. coli BL21(DE3) (Stratagene) grown in 2× YT medium under the selection of 50 mg/mL kanamycin. Expression was induced with 250 μM isopropyl β-L-1-thiogalactopyranoside (IPTG; Gold Biotechnology) along with the addition of 500 μM ZnCl2 at 18°C. Cells were collected by centrifugation and stored at −80 °C prior to purification.
Pellets were thawed and resuspended in purification buffer [50 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (HEPES; pH 7.5), 300 mM KCl, 10% glycerol (v/v), 1 mM tris(2-carboxyethyl)phosphine (TCEP)] and lysed by sonication. Lysate was clarified by centrifugation at 38,000g for 1 h at 4 °C. The supernatant was applied to amylose resin (New England BioLabs). His6-MBP-TEV-HDAC6 fusion protein was either eluted using 10 mM maltose or digested on-column with 2 mg/mL recombinant His-TEV protease. Eluted fusion protein was digested using recombinant His-TEV protease overnight at 4 °C while dialyzing in purification buffer. The digest was applied to an equilibrated Ni-NTA resin column (Qiagen) to remove His-MBP and His-TEV, which were subsequently eluted using a 0–500 mM imidazole gradient in purification buffer. The HDAC6-containing fractions were concentrated to <10 mL over a 10,000 molecular weight cut-off filter unit (Millipore) and applied to a HiLoad Superdex 200pg column in size exclusion buffer [50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP]. Fractions containing pure HDAC6 were identified using SDS-PAGE, pooled, and concentrated to 14–20 mg/mL. Protein was flash cooled in liquid nitrogen and stored at −80 °C prior to usage.
HDAC8 was expressed and purified as previously described,47 with minor modifications. Briefly, a 500 mL culture [Lysogeny Broth (LB) at 100 μg/mL ampicillin] was grown overnight at 37 °C with shaking. Aliquots of this culture (30 mL) were used to inoculate 12 × 1 L of M9 minimal medium supplemented with 100 μg/mL ampicillin. Cells were grown until the OD600 ≈ 1.0, at which point the temperature was reduced to 18 °C. After cooling for 30 min, protein expression was induced with 100 μM ZnCl2 and 100 μM IPTG. Protein was expressed overnight and pellets were harvested via centrifugation and stored at −80 °C until they were purified. The purification was carried out as previously described using a Co2+-TALON column followed by size exclusion chromatography.47 All protein was concentrated to 10–20 mg/mL, flash-cooled in liquid nitrogen, and stored at −80 °C until use.
Crystallization
All HDAC6-inhibitor complexes were crystallized in sitting drops by the vapor diffusion method at 4 °C.
For cocrystallization of the HDAC6–1 and HDAC6–2 complexes, a 350 nL drop of protein solution [5 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, 5 mM 1 or 2, and 5% dimethyl sulfoxide (DMSO) (v/v)] was added to 350 nL of precipitant solution [200 mM ammonium chloride and 20% PEG 3,350] and equilibrated against 80 μL of precipitant solution. Rhomboid plate crystals appeared within 2 days.
For cocrystallization of the HDAC6–3 complex, a 2 μL drop of protein solution [2.5 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, 5 mM 3, and 5% dimethyl sulfoxide (DMSO) (v/v)] was added to a 2 μL drop of precipitant solution [40 mM sodium citrate/60 mM Bis-Tris propane (pH 6.4) and 25% PEG 3,350] and was streak-seeded with a seed stock of crushed HDAC6–3 crystals previously generated under the same conditions but with 5 mg/mL enzyme in the protein solution. This was equilibrated against 80 μL of precipitant solution. Rhomboid plate crystals appeared within 2 days.
For cocrystallization of the HDAC6–4 complex, a 350 nL drop of protein solution [5 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, 5 mM 4, and 5% dimethyl sulfoxide (DMSO) (v/v)] was added to 350 nL of precipitant solution [200 mM ammonium tartrate dibasic and 20% PEG 3,350] and equilibrated against 80 μL of precipitant solution. Rhomboid plate crystals appeared within 2 days.
All crystals were soaked in a cryoprotectant solution containing mother liquor supplemented with 20% ethylene glycol prior to flash-cooling in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected from crystals on beamline 9-2 at the Stanford Synchrotron Radiation Laboratory (SSRL), Stanford University for the HDAC6 complexes with inhibitors 1, 2, and 3. Diffraction data for the HDAC6–4 complex were collected on beamline 17-ID-2 (FMX) at the National Synchrotron Light Source II (NSLS2), Brookhaven National Lab. Data were indexed and integrated using iMosflm48 and scaled using Aimless in the CCP4 program suite.49 Data collection statistics are recorded in Table 1.
Table 1.
Data collection and refinement statistics for HDAC6-inhibitor complexes
| Inhibitor | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Unit Cell | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| a, b, c (Å) | 74.8, 91.9, 96.4 | 74.7, 91.8, 96.5 | 74.7, 91.8, 96.5 | 74.8, 92.0, 96.6 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Data Collection | ||||
| Wavelength (Å) | 0.97946 | 0.97946 | 0.97946 | 0.97933 |
| Resolution (Å) | 48.2 – 1.62 | 59.08 – 1.24 | 96.60 – 2.03 | 96.64 – 1.70 |
| Total/unique no. of reflections | 528011 / 84908 | 1156727 / 188513 | 271026 / 43532 | 507763 / 74006 |
| Rmergea,b | 0.189 (0.816) | 0.101 (0.593) | 0.228 (0.538) | 0.224 (1.558) |
| Rp.i.m.a,c | 0.082 (0.346) | 0.044 (0.257) | 0.099 (0.229) | 0.91 (0.670) |
| CC1/2a,d | 0.969 (0.682) | 0.997 (0.871) | 0.988 (0.850) | 0.991 (0.605) |
| I/σ(I)a | 6.7 (2.2) | 10.0 (3.0) | 6.3 (3.6) | 8.6 (3.4) |
| Redundancya | 6.2 | 6.1 (6.2) | 6.2 (6.4) | 6.9 (6.3) |
| Completeness (%)a | 99.5 (99.7) | 99.7 (99.9) | 99.8 (100) | 100 (100) |
| Refinement | ||||
| No. of reflections used in refinement/test set | 84783 (8378) | 188358 (18683) | 43445 (4284) | 73824 (7278) |
| Rworka,e | 0.170 (0.262) | 0.127 (0.177) | 0.176 (0.221) | 0.169 (0.220) |
| Rfreea,e | 0.219 (0.338) | 0.153 (0.219) | 0.223 (0.266) | 0.194 (0.256) |
| No. of nonhydrogen atoms: | ||||
| protein | 5721 | 5838 | 5563 | 5689 |
| ligand | 74 | 125 | 26 | 60 |
| solvent | 764 | 935 | 648 | 654 |
|
|
||||
| Average B-factors (Å2) | ||||
|
|
||||
| protein | 11 | 10 | 10 | 10 |
| ligand | 17 | 20 | 7 | 19 |
| solvent | 23 | 24 | 17 | 23 |
|
|
||||
| Root-mean-square deviation from ideal geometry | ||||
|
|
||||
| bonds (Å) | 0.006 | 0.008 | 0.003 | 0.003 |
| angles (°) | 0.8 | 1.0 | 0.6 | 0.7 |
| Ramachandran plot (%)f | ||||
| favored | 97.75 | 97.61 | 97.60 | 97.61 |
| allowed | 2.25 | 2.39 | 2.40 | 2.39 |
|
| ||||
| PDB accession code | 6CSR | 6CSP | 6CSQ | 6CSS |
Values in parentheses refer to the data from the highest resolution shell.
Rmerge = ΣhklΣi|Ii,hkl − 〈I〉 hkl|/ΣhklΣi Ii,hkl, where 〈I〉hkl is the average intensity calculated for reflection hkl from replicate measurements.
Rp.i.m.= (Σhkl(1/(N-1))1/2 Σi|Ii,hkl − 〈I〉hkl|)/ΣhklΣi Ii,hkl, where 〈I〉hkl is the average intensity calculated for reflection hkl from replicate measurements and N is the number of reflections
Pearson correlation coefficient between random half-datasets.
Rwork = Σ||Fo| − |Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated using the same expression for reflections contained in the test set held aside during refinement.
Calculated with PROCHECK.
Each crystal structure was solved by molecular replacement using the atomic coordinates of unliganded HDAC6 (PDB 5EEM)29 as a search model for rotation and translation function calculations using the program Phaser.50 Atomic models were constructed and modified using the graphics program Coot51 and crystallographic structure refinement was performed using Phenix.52 Inhibitor molecules were added when clearly resolved electron density was observed for each. Occasionally, maps displayed spurious electron density peaks that could not be satisfactorily modeled by ordered solvent, ligand, or cryoprotectant, in which case these peaks were left uninterpreted. The quality of each model was assessed using MolProbity53 and PROCHECK.54 Final refinement statistics are recorded in Table 1.
Isothermal Titration Calorimetry
Thermograms were measured for inhibitor binding to HDAC6 and HDAC8 using a MicroCal iTC 200 isothermal titration calorimeter (GE Healthcare). For each compound, 300 μM inhibitor was titrated against 30 μM enzyme in size exclusion buffer with 0.3% DMSO for compounds 1–4. Thirty-eight 1-μL injections were made over 80 min. Integration, curve fitting, and figure generation were performed using Origin (OriginLab, Northampton, MA).
Results and Discussion
X-ray crystallography
Crystal structures were solved and refined with Rwork and Rfree values ranging 0.127–0.176 and 0.153–0.223, respectively (Table 1). For each HDAC6–inhibitor complex, there are no major conformational changes relative to unliganded HDAC6 (PDB 5EEM; root-mean-square deviation (rmsd) = 0.16–0.17 Å for 290–305 Cα atoms).
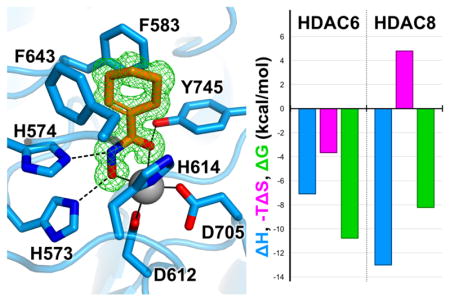
In the crystal structure of the HDAC6–phenylhydroxamate 1 complex determined at 1.62 Å resolution, the inhibitor hydroxamate group adopts canonical bidentate Zn2+ coordination, forming a 5-membered ring chelate complex with Zn2+---O distances of 2.0 Å and 2.4 Å for the N–O− and C=O groups, respectively (Figure 2a). This coordination geometry was first observed in the binding of hydroxamate inhibitors to the Zn2+ hydrolase thermolysin.55 In the HDAC6 active site, the hydroxamate oxyanion, NH group, and C=O group also form hydrogen bonds with H573, H574, and Y745, respectively (intermolecular distances are recorded in Table 2). The phenyl ring of the inhibitor is firmly nestled in an aromatic crevice formed by the side chains of F563 and F643; the phenyl ring of the inhibitor is offset so that the partial positive charges of its ring hydrogen atoms interact with the partial negative charges of the ring π electrons of F583 and F643. The dihedral angle between the inhibitor phenyl ring and the hydroxamate moiety is twisted 34° away from planarity.
Figure 2.
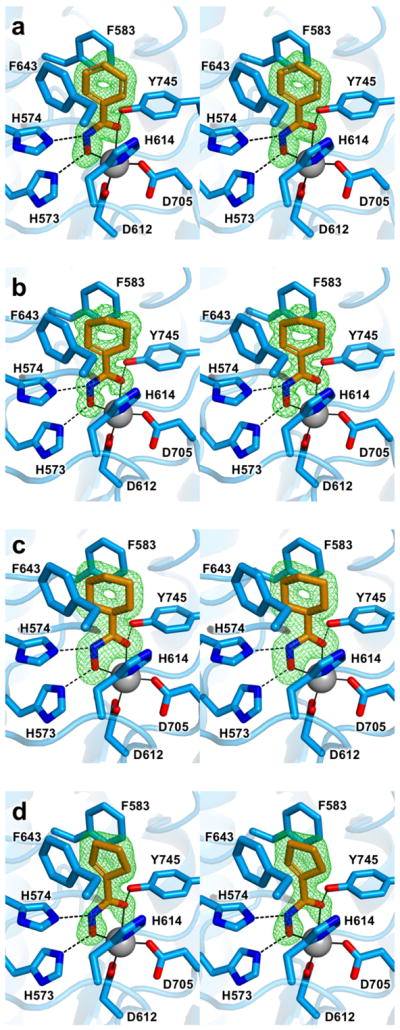
Stereoviews of Polder omit maps (green) contoured at 4.0σ for inhibitors 1 (a), 2 (b) 3 (c), and 4 (d) bound to HDAC6. Atoms are color-coded as follows: C = orange (inhibitor) or light blue (HDAC6), N = blue, O = red, Zn2+ = gray sphere. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
Table 2.
Interatomic separations for selected HDAC6–inhibitor interactions (Å)
| Inhibitor | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| C=O---Zn2+ | 2.4 | 2.2 | 2.2 | 2.4 |
| N–O−---Zn2+ | 2.0 | 2.0 | 2.1 | 2.0 |
| N---N(H574) | 2.7 | 2.6 | 2.8 | 2.6 |
| O−---N(H573) | 2.8 | 2.7 | 2.6 | 2.6 |
| C=O---O(Y745) | 2.4 | 2.6 | 2.5 | 2.4 |
The crystal structure of the HDAC6–cyclohexenylhydroxamate 2 complex determined at 1.24 Å resolution similarly reveals bidentate hydroxamate-Zn2+ coordination, with Zn2+---O distances of 2.0 Å and 2.2 Å for the ionized hydroxyl and carbonyl groups, respectively (Figure 2b). Hydrogen bond interactions with the Zn2+-bound hydroxamate are similar to those observed for phenylhydroxamate 1 (Table 2). The cyclohexenyl ring of the inhibitor is bound such that the olefin moiety is firmly nestled in the F583–F643 aromatic crevice. The dihedral angle between the olefin moiety and the hydroxamate is twisted 18° away from planarity.
The crystal structure of the HDAC6–cyclohexylhydroxamate 3 complex determined at 2.03 Å resolution reveals nearly perfect bidentate hydroxamate-Zn2+ coordination, with Zn2+---O distances of 2.1 Å and 2.2 Å for the ionized hydroxyl and carbonyl groups, respectively (Figure 2c). Hydrogen bond interactions with the Zn2+-bound hydroxamate are similar to those observed for phenylhydroxamate (Table 2). The cyclohexyl group adopts a chair conformation and packs between the side chains of F583 and F643. The dihedral angle between the adjacent C-C bond in the cyclohexyl ring and the hydroxamate is twisted 8° away from planarity.
Finally, the crystal structure of the HDAC6–cyclopentenylhydroxamate 4 complex determined at 1.70 Å resolution reveals canonical bidentate hydroxamate-Zn2+ coordination geometry with Zn2+---O distances of 2.0 Å and 2.4 Å for the ionized hydroxyl and carbonyl groups, respectively (Figure 2d). Hydrogen bond interactions with the Zn2+-bound hydroxamate are similar to those observed for phenylhydroxamate (Table 2). The cyclopentenyl ring of the inhibitor is bound such that the olefin moiety is firmly nestled in the F583–F643 aromatic crevice. The cyclopentenyl-hydroxamate dihedral angle is twisted 26° away from planarity.
Three interesting structural features emerge from analysis of the binding modes of capless inhibitors 1–4. First, each inhibitor binds with its hydroxamate group coordinated to Zn2+ with canonical bidentate geometry. Notably, many HDAC6-selective inhibitors such as HPB (Figure 3) contain a core phenylhydroxamate moiety as represented by inhibitor 1. Depending on the capping group attached to the phenylhydroxamate, bidentate or monodentate hydroxamate coordination to the catalytic Zn2+ ion will result, with the monodentate coordination mode being only 0.5 kcal/mol less stable than the bidentate coordination mode (Figure 1).45 For example, HPB44 exhibits monodentate Zn2+ coordination;45 least-squares superposition of the crystal structures of the HDAC6–HPB (PDB ID: 5WGK) and HDAC6–1 complexes (rmsd = 0.12 over 283 Cα atoms) reveals slight differences in the orientation of the aromatic ring (Figure 3). Specifically, the aromatic ring of the capless phenylhydroxamate 1 is tilted by approximately 10°, which allows a closer approach to the catalytic Zn2+ ion, which in turn enables bidentate hydroxamate-Zn2+ coordination.
Figure 3.

Chemical structure of HPB and stereoview of the superposition of the experimentally determined structure of the HDAC6–HPB complex (PDB 5WGK; blue) with a model of HPB in the same conformation with its phenylhydroxamate moiety superimposed with that observed in the HDAC6–1 complex. Phenylhydroxamate 1 is shown in yellow while the modeled HPB cap is shown in orange.
Superposition of the HPB phenylhydroxamate moiety with compound 1 demonstrates that no particular interaction of the capping group appears to govern the hydroxamate-Zn2+ binding mode. If HPB were tilted so as to superimpose its phenylhydroxamate moiety with that of capless phenylhydroxamate 1 in the HDAC6–1 complex (Figure 3), the bulky capping group would not clash with any active site residues. Thus, it is not clear how the geometry of hydroxamate-Zn2+ coordination might be influenced by interactions of the capping group of HPB.
Second, while compounds 1, 2, and 4 contain C=C bonds potentially capable of conjugation with the π system of the hydroxamate moiety, their respective C=C–C=O dihedral angles are distorted 36°, 11°, and 46° from planarity. These values lie within the broad distribution observed for similar compounds retrieved from the Cambridge Structural Database (Figure S1), so there does not appear to be a strong energetic driving force to maintain planarity of the extended π system.
Finally, the binding orientations of cyclohexenylhydroxamate 2 and cyclopentenylhydroxamate 4 unambiguously place the C=C bond in the F583–F643 aromatic crevice, as also observed for the aromatic ring of phenylhydroxamate 1. Therefore, this aromatic crevice preferentially accommodates a planar olefin moiety as contained in a 6- or 5-membered ring, including a 6-membered aromatic ring. The chair-conformation 6-membered ring of cyclohexylhydroxamate 3 is not as readily accommodated based on previously measured46 IC50 values (Figure 4). Of the linker groups represented in the current study, it appears that the cyclohexenyl group of inhibitor 2 would be most ideal for further derivatization to enhance HDAC6 selectivity.
Figure 4.

IC50 values compared with Kd values derived from ITC measurements for “capless” HDAC6-selective inhibitors 1–4 and “capped” inhibitors SAHA, Ricolinostat, and HPB. IC50 values are abstracted from ref. 46 (1–4), ref. 64 (SAHA), ref. 62 (Ricolinostat), and ref. 44 (HPB). Kd data derive from isothermal titration calorimetry.
Isothermal titration calorimetry (ITC)
To gain additional insight on the molecular basis of affinity and selectivity for inhibitor binding to HDAC6, we studied the thermodynamics of enzyme-inhibitor association using ITC. We also studied inhibitor binding to the representative class I isozyme HDAC8 using ITC to evaluate the extent of HDAC6 selectivity against class I HDACs. Titrations of inhibitors 1–4 against HDAC6 and HDAC8 reveal that the ITC-derived dissociation constants (Kd) generally correlate (within a factor of 4) with previously measured46 IC50 values for each enzyme-inhibitor complex (Figure 4, Figure S2).
The selectivity of the best capless inhibitors for HDAC6 compared with HDAC8 appears to be rooted in a substantial and favorable entropic gain for HDAC6 association. Specifically, the –TΔS term is negative and favorable for the binding of inhibitors 1–4 to HDAC6, whereas –TΔS is positive and unfavorable for the binding of these inhibitors to HDAC8 with the exception of compound 3 (Figure 5, Figure S2). In other words, the binding of cycloalkenyl hydroxamates 1, 2, and 4 to HDAC6 is accompanied by a substantial entropy gain, whereas the binding of these cycloalkenyl hydroxamates to HDAC8 is accompanied by an entropic loss. The outlier is the cycloalkane hydroxamate 3, which exhibits a modest and favorable entropy of binding to HDAC8; however, this compound also exhibits the weakest affinities for HDAC8 and HDAC6 and the poorest selectivity for HDAC6 (Figure 4). Thus, planar cycloalkenyl hydroxamates yield optimal affinities and HDAC6/HDAC8 selectivities of 16-fold up to 313-fold, and binding entropy appears to drive selectivity.
Figure 5.

Thermodynamic values derived from ITC measurements for the binding of inhibitors 1–4, SAHA, Ricolinostat (R’stat), and HPB to HDAC6 CD2 (left) and HDAC8 (right).
Presuming identical conformations for the binding of compounds 1–4 in the active sites of other HDAC isozymes, we speculate that differences in the entropy of inhibitor binding to HDAC6 and HDAC8 could be rooted in differences in conformational entropy and/or desolvation entropy involving the F583–F643 aromatic crevice of HDAC6, which is conserved in HDAC8 as F152 and F208. Crystal structures of the class I isozymes HDAC1, HDAC2, and HDAC3,56–58 and the class IIa isozymes HDAC4 and HDAC7,59,60 reveal that this aromatic crevice is similarly conserved. However, structural comparisons of all isozymes reveal that relative to HDAC6, the aromatic crevice is approximately 1 Å wider in class I and class IIa HDACs. Moreover, there is evidence of conformational flexibility for one wall of this crevice: upon the binding of a bulky phenylhydroxamate inhibitor to HDAC8, the side chain of F152 rotates to adopt a conformation similar to that of the corresponding phenylalanine residue, F679, in the aromatic crevice of HDAC7.60,61 It is conceivable that inhibitor binding rigidifies the conformation of the aromatic crevice in HDAC8, thereby accounting for a conformational entropy loss. This possibility may also extend to other class I and class IIa HDACs. If the narrower aromatic crevice of HDAC6 is more rigid, then inhibitor binding would not incur a conformational entropic cost and the favorable entropy gain from active site desolvation could dominate the binding thermodynamics.
To assess the influence of a capping group on the thermodynamics of HDAC6/HDAC8 selectivity, we additionally studied the binding of the phenylhydroxamate derivative HPB,44 the aliphatic hydroxamate inhibitor Ricolinostat,62 and the aliphatic hydroxamate inhibitor suberoylanilide hydroxamic acid (SAHA).63 The ITC-derived inhibitor dissociation constants (Kd) generally correlated with IC50 values for HDAC8 (within a factor of 4), but those for HDAC6 do not (7–32-fold variations are observed). Regardless, each of these inhibitors exhibits selectivity for binding to HDAC6 compared with HDAC8 (Figure 4),44,62,64 and selectivity is characterized by a favorable entropic gain upon binding to HDAC6 and an unfavorable entropic loss upon binding to HDAC8 (Figure 5, Figure S3). The results for HPB binding indicate that the HDAC6 selectivity inherent in the binding of capless cycloalkenyl hydroxamate inhibitors is generally maintained, but no inhibitor studied here surpasses the 313-fold thermodynamic selectivity based on Kd values measured for cyclohexenylhydroxamate 2 (Figure 4). Even so, it is interesting that favorable binding entropy to HDAC6 is not exclusive to inhibitors bearing aromatic or cycloalkenyl linker groups, but also includes inhibitors bearing aliphatic linker groups such as Ricolinostat and SAHA.
Conclusions
The binding thermodynamics of compounds 1–4 to HDAC6 versus HDAC8 reflect that entropy is a key contributor to HDAC6-inhibitor binding selectivity; moreover, compounds bearing a single double bond in the ring adjacent to the hydroxamate moiety are more selective for HDAC6 than their aromatic or saturated capless counterparts. In particular, cycloalkenyl hydroxamate 2 exhibits 313-fold selective tighter binding to HDAC6 compared with HDAC8 (Figure 4).
The olefin moiety of 2 is preferentially accommodated in the F583–F643 aromatic crevice of HDAC6. It is not clear, however, whether the entropic favorability contributing to HDAC6 selectivity is associated with binding in this crevice, since these aromatic residues are also conserved in HDAC8 as well as other class I HDAC isozymes. However, the active site of HDAC8 is slightly larger than that of HDAC6, with an 8 Å separation between F152 and F208 in the aromatic crevice, compared with the 7 Å separation between F583 and F643 in the aromatic crevice of HDAC6. Additionally, F152 in HDAC8 exhibits conformational flexibility in complex with a bulky phenylhydroxamate inhibitor, which expands the F152–F208 separation to 8.5 Å.61 Possibly, the conformational flexibility of the aromatic crevice in HDAC8 contributes to the generally unfavorable entropy of inhibitor binding evident in Figure 5. In HDAC6, there is no evidence for conformational flexibility in the aromatic crevice, so the favorable entropy of inhibitor binding may be linked solely to desolvation.
Notably, the F583–F643 aromatic crevice of HDAC6 preferentially accommodates planar olefins, and this crevice does not accommodate the chair-conformation cyclohexyl hydroxamate 3 as readily. This inhibitor exhibits the lowest inhibitory potency, affinity, and HDAC6 selectivity among the compounds studied (Figure 4). Thus, hydroxamate inhibitor designs using core cyclohexenyl hydroxamate 2 would represent an ideal starting point for the design of HDAC6 inhibitors with high affinity and selectivity.
Supplementary Material
Acknowledgments
Funding
We thank the National Institutes of Health for grant GM49758 in support of this research.
We thank the NIH for grant GM49758 (D.W.C.) in support of this research, and we thank the NIH for Chemistry-Biology Interface training grant T32 GM071339 (University of Pennsylvania and the Wistar Institute) in support of N.J.P. This research utilized the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393). This research also utilized the Frontier Microfocusing Macromolecular Crystallography Beamline (FMX) of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.
Footnotes
Notes
N.J.P. and D.W.C. declare no competing financial interests. F.F.W. is a consultant and scientific advisory board member for Eikonizo Therapeutics, Inc.
Accession Codes
The atomic coordinates and crystallographic structure factors of HDAC6 complexes with inhibitors 1, 2, 3, and 4 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 6CSR, 6CSP, 6CSQ, and 6CSS, respectively.
The Supporting Information is available free of charge on the ACS Publications website. Histogram of phenylhydroxamic acid dihedral angles; isothermal titration calorimetry thermograms for the binding of inhibitors 1–4, SAHA, Ricolinostat, and HPB to HDAC6 and HDAC8.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signaling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 6.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. J Mol Biol. 2001;311:433–444. doi: 10.1006/jmbi.2001.4859. [DOI] [PubMed] [Google Scholar]
- 7.McCullough CE, Marmorstein R. Molecular basis for histone acetyltransferase regulation by binding partners, associated domains, and autoacetylation. ACS Chem Biol. 2016;11:632–642. doi: 10.1021/acschembio.5b00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Disc. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 11.Penney J, Tasi LH. Histone deacetylases in memory and cognition. Sci Signal. 2014;7:re12. doi: 10.1126/scisignal.aaa0069. [DOI] [PubMed] [Google Scholar]
- 12.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 13.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Disc. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 14.Ganai SA, Ramadoss M, Mahadevan V. Histone deacetylase (HDAC) inhibitors – emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14:55–71. doi: 10.2174/1570159X13666151021111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implication of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Hai Y, Shinsky SA, Porter NJ, Christianson DW. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat Commun. 2017;8:15368. doi: 10.1038/ncomms15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kutil Z, Novakova Z, Meleshin M, Mikesova J, Schutkowski M, Barinka C. Histone deacetylase 11 is a fatty-acid deacylase. ACS Chem Biol. 2018;13:685–693. doi: 10.1021/acschembio.7b00942. [DOI] [PubMed] [Google Scholar]
- 18.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 19.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 20.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structure of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 21.Ash DE, Cox JD, Christianson DW. Arginase: a binuclear manganese metalloenzyme. Metal Ions in Biological Systems. 2000;37:407–428. [PubMed] [Google Scholar]
- 22.Yuan H, Marmorstein R. Structural basis for sirtuin activity and inhibition. J Biol Chem. 2012;287:42428–42435. doi: 10.1074/jbc.R112.372300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bheda P, Jing H, Wolberger C, Lin H. The substrate specificity of sirtuins. Annu Rev Biochem. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. [DOI] [PubMed] [Google Scholar]
- 24.Bertos NR, Gilquin B, Chan GKT, Yen TJ, Khochbin S, Yang XJ. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem. 2004;279:48426–48254. doi: 10.1074/jbc.M408583200. [DOI] [PubMed] [Google Scholar]
- 25.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 26.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VMY. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2010;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26:5648–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- 28.Yan J. Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014;33:567–580. doi: 10.1089/dna.2013.2300. [DOI] [PubMed] [Google Scholar]
- 29.Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol. 2016;12:748–754. doi: 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- 31.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;86:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem. 1999;274:2440–2445. doi: 10.1074/jbc.274.4.2440. [DOI] [PubMed] [Google Scholar]
- 33.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Gilquin B, Khochbin S, Matthias P. Two catalytic domains are required for protein deacetylation. J Biol Chem. 2006;281:2401–2404. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- 35.Zou H, Wu Y, Navre M, Sang BC. Characterization of the two catalytic domains in histone deacetylase 6. Biochem Biophys Res Commun. 2006;341:45–50. doi: 10.1016/j.bbrc.2005.12.144. [DOI] [PubMed] [Google Scholar]
- 36.Bazzaro M, Lin Z, Santillan A, Lee MK, Wang MC, Chan KC, Bristow R, Mazitschek R, Bradner J, Roden RB. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin Cancer Res. 2008;14:7340–7347. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanno K, Kanno S, Nitta H, Uesugi N, Sugai T, Masuda T, Wakabayashi G, Maesawa C. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion of hepatocellular carcinoma cells. Oncol Rep. 2012;28:867–873. doi: 10.3892/or.2012.1898. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Sheng S, Qin C. The role of HDAC6 in Alzheimer’s disease. J Alzheimers Dis. 2013;33:283–295. doi: 10.3233/JAD-2012-120727. [DOI] [PubMed] [Google Scholar]
- 39.de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, Bradner JE, Mazitschek R, Kozikowski AP, Matthias P, Hancock WM. Histone Deacetylase 6 and Heat Shock Protein 90 Control the Functions of Foxp3+ T-Regulatory Cells. Mol Cell Biol. 2011;31:2066–2078. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zali Y, Kossenkev AV, Vara-Ailor A, Rauscher FJ, III, Zou W, Speicher DW, Huntsman DG, Conejo-Garcia JR, Cho KR, Christianson DW, Zhang R. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962–973. doi: 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butler KV, Kalin J, Brochler C, Vistoli G, Langley B, Kozikowski AP. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem. 2012;55:9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JH, Mahendran A, Yao Y, Ngo L, Venta-Perez G, Choy ML, Kim N, Ham WS, Breslow R, Marks PA. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci USA. 2013;110:15704–15709. doi: 10.1073/pnas.1313893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Yao Y, Mahendran A, Ngo L, Venta-Perez G, Choy ML, Breslow R, Marks PA. Creation of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci USA. 2015;112:12005–12010. doi: 10.1073/pnas.1515882112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porter NJ, Mahendran A, Breslow R, Christianson DW. Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc Natl Acad Sci USA. 2017;114:13459–13464. doi: 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner FF, Olson DE, Gale JP, Kaya T, Weïwer M, Aidoud N, Thomas M, Davoine EL, Lemercier BC, Zhang YL, Holson EB. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J Med Chem. 2013;56:1772–1776. doi: 10.1021/jm301355j. [DOI] [PubMed] [Google Scholar]
- 47.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMosflm: a new graphical interface for diffraction-image processing with Mosflm. Acta Cryst. 2011;D67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Cryst. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwillinger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB, Arendal WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 55.Holmes MA, Matthew BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 56.Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BV, Schwabe JW. Insights in the activation mechanism of class I HDAC complexes by inositol tetraphosphates. Nat Commun. 2016;7:11262. doi: 10.1038/ncomms11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, Gangloff AR. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 58.Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of Hdac3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkuhler C, Gallinari P, Carfi A. Structural and functional analysis of the human Hdac4 catalytic domain reveals a regulatory zinc-binding domain. J Biol Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schuetz A, Min J, Allia-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabackman AA, Frankson R, Marsan ES, Perry K, Cole KE. Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J Struct Biol. 2016;195:373–378. doi: 10.1016/j.jsb.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang YL, Hennig KM, Gale J, Zhao W, Reis S, Barker DD, Berry-Scott E, Kim SW, Clore EL, Hooker JM, Holson EB, Haggarty SJ, Petryshen TL. A Selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related sets. Plos One. 2013;8:e71323. doi: 10.1371/journal.pone.0071323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scolnick LR, Clements AM, Liao J, Crenshaw L, Hellberg M, May J, Dean TR, Christianson DW. Novel binding mode of hydroxamate inhibitors to human carbonic anhydrase II. J Am Chem Soc. 1997;119:850–851. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.