

Abstract
The microbiome of sea urchins plays a role in maintaining digestive health and innate immunity. Here, we investigated the effects of long-term (90 day) exposure to elevated seawater temperatures on the microbiome of the common, subtropical sea urchin Lytechinus variegatus. The community composition and diversity of microbes varied according to the type of sample collected from the sea urchin (seawater, feed, intestines, coelomic fluid, digested pellet and faeces), with the lowest microbial diversity (predominately the order Campylobacterales) located in the intestinal tissue. Sea urchins exposed to near-future seawater temperatures maintained the community structure and diversity of microbes associated with their tissues. However, marginal, non-significant shifts in microbial community structure with elevated temperature resulted in significant changes in predicted metagenomic functions such as membrane transport and amino acid and carbohydrate metabolism. The predicted changes in key metabolic categories suggest that near-future climate-induced increases in seawater temperature could shift microbial community function and impact sea urchin digestive and immune physiology.
Keywords: Lytechinus variegatus, climate change, PICRUSt, microbial ecology, 16S rRNA, next-generation sequencing
1. Introduction
Ocean temperatures are warming due to anthropogenic-induced climate change. Since the 1970s, the global average seawater temperature has increased 0.11°C per decade, and it is predicted that the average surface seawater temperature will increase an additional 0.6°C (Representative Concentration Pathway [RCP] 2.6) to 2.0°C (RCP 8.5) by 2100 [1]. Although many eurythermal marine organisms are capable of withstanding fluctuating temperature on a daily or seasonal cycle, climate-induced elevated seawater temperatures are certain to challenge the thermal tolerance limits of many marine invertebrates, especially those living near the upper threshold of their thermal limit [2].
The common sea urchin Lytechinus variegatus (Toxopneustidae) is one such ectothermic organism that in certain geographical regions is already living near the high end of its thermal tolerance. In shallow bays of the northern Gulf of Mexico this sea urchin occurs in high densities. For example, mean densities of individuals living in association with the seagrass Thalassia testudinum can be as high as 13.4 individuals m−2 and periodically reach up to 35 individuals m−2 [3]. These sea urchins are generalist omnivores and common components of their diet include seagrasses, macroalgae, decaying seagrass blades, and seagrass epibionts [4]. As such, the grazing activity of L. variegatus directly affects the density of the seagrass beds, and individuals may consume up to 100% of the above-ground annual biomass of T. testudinum [5].
Seawater temperatures in these shallow, semi-enclosed bays currently range annually between 16°C and 30°C, with brief, extreme weather events pushing temperatures as high as 34°C [3,6]. Most models predict that mean seawater temperature in the Gulf of Mexico will increase at least 2°C by the end of the century [7], which will expose L. variegatus to higher seawater temperatures for more extended periods of time. A recent study indicated that L. variegatus held in the laboratory at 32°C for 14 days exhibited a 50% mortality rate [8], evidence that the species suffers stress at even slightly elevated chronic temperatures. Furthermore, in the northern Gulf of Mexico, geography prevents this species from gaining a thermal refuge by migrating northward and shallow depths limit vertical migration. Accordingly, it is important to evaluate whether near-future seawater temperatures may induce sub-lethal impacts on this ecologically important species.
Feed ingested by L. variegatus is formed into a pellet inside the buccal cavity and enclosed in a mucus membrane. Once the digested pellet is formed, it proceeds through the pharynx and oesophagus and then moves to the stomach and lower intestinal tract where digestion and nutrient uptake occurs. The bacterial load of the contents of the gut of the temperate sea urchin Strongylocentrotus droebachiensis has been estimated at up to 6 × 109 cells ml−1 [9]. In L. variegatus, the intestines of sea urchins collected from the field [10] and held in the laboratory while being fed a formulated feed [11] had low microbial diversity, dominated by the order Campylobacterales. Digestive enzymes produced by intestinal bacteria may expand upon the native digestive capabilities of the intestines [12]. For example, bacteria isolated from the intestines of L. variegatus and the sea urchin Arbacia punctulata were implicated in the digestion of a variety of algal polysaccharides [13] and bacteria isolated from the intestines of the sea urchin Asterechinus elegans may facilitate the digestion of wood fragments [14]. Intestinal [9] and subcuticular [15,16] bacteria also provide essential amino acids and nitrogen by nitrogen fixation and uptake from the surrounding environment, respectively.
The role that the sea urchin microbiome plays in immunity has received less attention [17,18]. In other invertebrate taxa, such as insects, the microbiome plays a role in protecting against pathogenic bacteria, while at the same time hosting commensal bacteria [19]. Reduced microbial diversity and/or changes in microbial diversity may reduce this ‘protective’ function and leave organisms more vulnerable to infection [20]. Echinoderms possess a relatively sophisticated innate immune system which mediates a generalized non-specific response to marine pathogens [21]. Much of the immune activity is centralized in the coelomic fluid surrounding the internal organs of the sea urchin. The coelomic fluid and coelomocytes (cells within the coelomic fluid) perform a variety of functions including oxygen transport, antibacterial activity, clotting, chemotaxis and phagocytosis [21]. In coelomic fluid from the sea star Asterias amurensis, a Helicobacter-related taxon was the most abundant bacterial taxon [22].
Establishing the predicted functionality of the sea urchin microbiome in the immune system is important not only because little is known about this relationship, but because interactions between the host and its microbiome may change under ocean warming and acidification conditions. To date, most studies on the microbiome of marine invertebrates under near-future conditions have focused on reef-building taxa. In the tropical hard corals (Porites spp., Siderastrea spp. and Agaricia spp.), elevated seawater temperature alters the microbial community structure from a microbiome dominated by a photoautotrophic symbiont species (Synechococcus spp.) to a bacterial taxon commonly associated with stress and disease in corals [23]. The purpose of the present study was to characterize the microbial community of sea urchins held in the laboratory and fed a plant-based diet and to determine if chronic exposure to predicted near-future elevated seawater temperature has an impact on microbial composition and predicted metagenomic functionality in microbes associated with the common nearshore sea urchin L. variegatus.
2. Methods
(a). Collection and maintenance
Lytechinus variegatus (n = 12; 42.7 ± 2.9 g wet weight; 34.6 ± 0.7 mm diameter; 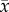 ± s.e.) were collected by hand from seagrass beds at Eagle Harbour (29°45′ N, 85°24′ W; 24.8°C, pH 8.12, salinity of 31.2) during May 2015. Sea urchins were transported to The University of Alabama at Birmingham and placed into twelve 19-litre aquaria containing artificial seawater (Instant Ocean® prepared with reverse-osmosis filtered water, 24°C, pH 8.10, salinity of 31.0). Each aquarium was aerated continuously via an airstone plumbed to a CORALIFE® Super LuftSL-65 high pressure aquarium air pump and equipped with its own recirculating aquarium filter pump (Aqueon Power Filter 10) and heater (EHEIM Jager Aquarium Thermostat Heater 50 W). Sea urchins were held for two weeks prior to the experiment at ambient temperature (24°C) and a constant photoperiod of 12 L : 12D to further pre-expose them to laboratory conditions and reduce any stress induced by collection and transport.
± s.e.) were collected by hand from seagrass beds at Eagle Harbour (29°45′ N, 85°24′ W; 24.8°C, pH 8.12, salinity of 31.2) during May 2015. Sea urchins were transported to The University of Alabama at Birmingham and placed into twelve 19-litre aquaria containing artificial seawater (Instant Ocean® prepared with reverse-osmosis filtered water, 24°C, pH 8.10, salinity of 31.0). Each aquarium was aerated continuously via an airstone plumbed to a CORALIFE® Super LuftSL-65 high pressure aquarium air pump and equipped with its own recirculating aquarium filter pump (Aqueon Power Filter 10) and heater (EHEIM Jager Aquarium Thermostat Heater 50 W). Sea urchins were held for two weeks prior to the experiment at ambient temperature (24°C) and a constant photoperiod of 12 L : 12D to further pre-expose them to laboratory conditions and reduce any stress induced by collection and transport.
At the beginning of the experiment the aquaria were randomly divided into two temperature treatments (n = 6 sea urchins per treatment). The seawater temperature in the two treatments was gradually increased every week over a month to either 26°C (ambient in situ early summer temperature in the northern Gulf of Mexico) or 30°C (predicted near-future temperature). While the gradual temperature increase was occurring (and for the duration of the experiment), sea urchins were fed an ad libitum diet of plant-based feed every other day. The feed was prepared from fresh blades of the seagrass T. testudinum (with epibionts) that were collected from Eagle Harbour at the same time L. variegatus were collected. The blades of T. testudinum were freeze-dried, ground with a mortar and pestle into a fine powder, embedded in agar, and cut into blocks measuring approximately 1.5 cm3 to serve as a standardized feed (10% dried, powdered plant material in 3% agar) [4]. Before each feeding, uneaten feed and faeces were removed from each tank using a siphon and a 30% water exchange was performed to maintain consistent salinity (30.75 ± 0.21; 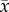 ± s.e.), dissolved oxygen (91.56 ± 0.32%), and pH (8.21 ± 0.01), and low levels of ammonia (≤0.25 ppm) and nitrate (≤5 ppm). Temperature (26.01 ± 0.04° and 29.85 ± 0.05°C) was monitored daily throughout the experiment using a YSI™ ProPlus meter.
± s.e.), dissolved oxygen (91.56 ± 0.32%), and pH (8.21 ± 0.01), and low levels of ammonia (≤0.25 ppm) and nitrate (≤5 ppm). Temperature (26.01 ± 0.04° and 29.85 ± 0.05°C) was monitored daily throughout the experiment using a YSI™ ProPlus meter.
After 90 days at the two experimental temperatures (26° and 30°C), the six sea urchins held at each temperature were sacrificed and samples for microbiome analysis were collected. Two ml aliquots of coelomic fluid were collected from each individual by syringe via puncture of the peristomial membrane. The Aristotle's lantern was excised followed by the intestines (excluding the pharynx). The latter was rinsed with sterile seawater and any feed pellets were removed from the interior of the tract. Samples of the surrounding seawater from each aquarium, as well as the faeces of each sea urchin were also collected. All samples were immediately frozen at −80°C and stored until DNA extraction.
(b). DNA isolation and sequence analysis
Metacommunity DNA was extracted using a Faecal DNA isolation kit from Zymo Research (catalogue #D6010). PCR employed the use of unique barcoded primers modified from the standard protocol of the Earth Microbiome project (www.earthmicrobiome.org) [24,25] to amplify the V4 region of the 16S rRNA gene (forward primer [515F] V4: 5′-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA-3′ and reverse primer modified from 806R to incorporate a uniquely barcoded 5′ region and adaptor sequence V4: 5′CAAGAGAAGACGGCATACGAGATNNNNNNAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT-3′ [Eurofins Genomics, Inc., Huntsville, AL, USA]) [10,11,26]. PCR products were obtained using New England Biolabs LongAmp Taq PCR kits (Cat# E5200S), electrophoresed on an agarose gel, and visualized by UV illumination [26]. The PCR product was excised from the gel using a sterile scalpel and purified using the Qiagen QIAquick Gel Extraction Kit (Cat# 2870) [26]. Although six sea urchins were sacrificed in each temperature treatment, not all samples were of sufficient mass to perform metacommunity DNA extraction and/or yield useable PCR products. Accordingly, actual sample numbers for each sample type for each treatment are given in the table heading of table 1.
Table 1.
Shannon's diversity indices ( ± s.e.) of samples from the sea urchin Lytechinus variegatus exposed to 26° and 30°C for 90 days. Samples taken were seawater (n = 4,4), feed (freeze-dried Thalassia testudinum; n = 3), intestines (n = 4,5), coelomic fluid (n = 5,5), digested pellet (sampled from inside the intestines; n = 3,3) and faeces (n = 6,5). The asterisk indicates feed samples were taken at the beginning of the experiment and the same feed source was used for both temperature treatments. p-values indicate post hoc analyses between temperature treatments.
± s.e.) of samples from the sea urchin Lytechinus variegatus exposed to 26° and 30°C for 90 days. Samples taken were seawater (n = 4,4), feed (freeze-dried Thalassia testudinum; n = 3), intestines (n = 4,5), coelomic fluid (n = 5,5), digested pellet (sampled from inside the intestines; n = 3,3) and faeces (n = 6,5). The asterisk indicates feed samples were taken at the beginning of the experiment and the same feed source was used for both temperature treatments. p-values indicate post hoc analyses between temperature treatments.
| Shannon's diversity index |
|||
|---|---|---|---|
| 26°C | 30°C | Sidak's p-value | |
| seawater | 6.63 ± 0.32 | 6.07 ± 0.32 | 0.986 |
| feed* | 5.28 ± 1.30 | ||
| intestines | 0.25 ± 0.05 | 0.18 ± 0.04 | >0.999 |
| coelomic fluid | 5.08 ± 1.19 | 5.62 ± 1.02 | 0.981 |
| digested pellet | 3.53 ± 1.02 | 4.07 ± 0.46 | 0.994 |
| faeces | 4.81 ± 0.80 | 5.77 ± 0.40 | 0.784 |
The NextGen Illumina MiSeq platform was used to sequence the PCR product and produced sequences of 251 bases. The rarefaction curves did reach an asymptote, demonstrating that sampling was sufficient to represent the total diversity of the microbial community (electronic supplementary material, figure S1). The QWRAP (QIIME wrapper) package was used to analyse the de-multiplexed dataset [26]. The most updated protocol is available at https://github.com/QWRAP/QWRAP and the dependencies of this package and scripts are described in detail by Kumar et al. [26]. In brief, quality analysis of the raw data was performed by trimming sequences to 250 bases and retaining only sequences with a QScore >20 over at least 80% of the bases. Chimera filtering was performed using the program ‘USEARCH’, sequences were assigned to operational taxonomic units (OTUs) at a similarity threshold of 97% using ‘UCLUST’, and taxonomic assignments were made (threshold 0.8) using the Greengenes v. 13.8 database [26]. The OTU table was rarefied to a minimum sampling depth of 31 036 sequences per sample and rare OTUs were filtered out at an abundance level less than 0.0005%. The alpha and beta diversity of all rarefied, filtered OTUs were calculated using the Shannon's diversity index and Bray–Curtis cluster analysis, respectively. All rarefied, filtered, and normalized OTUs at the most resolved taxonomic level were used to compare microbial community structure.
(c). Predicted microbial functioning
The bioinformatics software Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt v. 1.0.0) [27] was used to predict metagenome functions from the V4 region of the 16S rRNA genes. All rarefied, filtered OTUs were close-referenced picked against the Greengenes v. 13.5 database at 97% similarity, normalized by the 16S copy number predictions (as suggested by PICRUSt) [27], and assigned to the Kyoto Encyclopaedia of Genes and Genomes Orthology [28] at both level two (general predicted functional pathways) and three (more specific predicted functional pathways) categories. The weighted nearest sequenced taxon index (NSTI) was determined for each sample type to evaluate the prediction accuracy of PICRUSt [27].
(d). Statistical analysis
Microbial alpha diversity was calculated using the Shannon's diversity index and compared between the two temperature treatments (26° and 30°C) within sample type (seawater, intestines, coelomic fluid, digested pellet and faeces) using a two-way Analysis of Variance (ANOVA) and Sidak's post hoc test. Bray–Curtis similarity values were used to calculate beta diversity and generate a non-metric multidimensional scaling (nMDS) plot using the ggplot2 package in R software [29]. The Adonis function and similarity percentage (SIMPER) analysis [29] were used to compare the relative abundance of OTUs and predicted metagenomics functions at levels two and three between samples collected at 26° and 30°C. A significant p-value of less than 0.05 was used for all analyses.
3. Results
(a). Microbial diversity and community structure
Sequencing yielded 6 418 546 raw sequences assigned to 1073 OTUs after being rarefied and filtered. These OTUs are summarized in electronic supplementary material, table S1. Our results demonstrate that the microbial alpha diversity of samples collected from sea urchins held at 26° or 30°C for 90 days differed among the type of sample collected (seawater, sea urchin feed [freeze-dried T. testudinum], intestines, coelomic fluid, digested pellet and faeces). Seawater possessed the highest levels of microbial diversity, and the intestines possessed the lowest diversity levels (table 1). However, temperature did not have an overall significant effect on the microbial diversity of any sample type (two-way ANOVA; sample: F4,33 = 23.09, p < 0.0001; temperature: F1,33 = 0.412, p = 0.525; interaction: F4,33 = 0.403, p = 0.805; table 1) or significant pairwise comparisons (table 1).
Non-metric multidimensional scaling with Bray–Curtis-based cluster analysis revealed distinct microbiota composition clustering according to type of sample (Permutational Multivariate Analysis of Variance using Bray–Curtis distances, p = 0.001) rather than temperature treatment (p = 0.056; figure 1). The noticeable differences in microbial composition between the different sample types (seawater, feed, intestines, coelomic fluid, digested pellet and faeces) can be observed by comparing the 10 most abundant OTUs at the most resolved taxonomic level (figure 2). In seawater, Polaribacter sp. was the most abundant OTU at 26°C, representing 8.1% of the microbiota (figure 2). In the feed, family Exiguobacteraceae was the most abundant OTU representing 15.3% of the microbiota. The intestines and coelomic fluid shared the most abundant OTU, with the order Campylobacterales comprising 97.5% of the intestinal microbiota and 24.1% of the coelomic fluid microbiota at 26°C. In the digested pellet, the family Marinilabiaceae was the most abundant OTU, representing 46.5% of the microbiota at 26°C, whereas in the faeces, the most abundant OTU was order Campylobacterales at 26°C (14.3%).
Figure 1.

Non-metric multidimensional scaling (nMDS) ordination plot with Bray–Curtis-based cluster analysis of microbiota revealed distinct microbiota compositions from each sample type. Samples were collected from the sea urchin Lytechinus variegatus exposed to 26° and 30°C for 90 days. The 2D stress level was 0.135 and the ellipses confidence limit was 0.95.
Figure 2.

Relative abundance of the 10 most abundant microbial taxa in samples collected from the sea urchin Lytechinus variegatus exposed to 26° and 30°C for 90 days. Samples taken were: (a) seawater, (b) feed (freeze-dried Thalassia testudinum), (c) intestines, (d) coelomic fluid, (e) digested pellet (sampled from inside the intestines), and (f) faeces. Taxa are shown at the most resolved taxonomic level.
Elevated seawater temperatures were not associated with statistically significant changes in the composition of the microbial taxa. This was consistent across all sample types (Adonis; seawater [F1,6 = 1.262, R2 = 0.174, p = 0.250], intestines [F1,7 = 1.301, R2 = 0.157, p = 0.135], coelomic fluid [F1,8 = 0.611, R2 = 0.071, p = 0.912], digested pellet [F1,4 = 2.020, R2 = 0.336, p = 0.200] and faeces [F1,9 = 1.027, R2 = 0.102, p = 0.431]). Nonetheless, modest shifts in microbial community structure were observed between 26° and 30°C (figure 2). For example, in seawater, as temperature increased the proportion of OTUs assigned to Polaribacter sp. decreased 40-fold from 8.1 to 0.2% while the proportion of OTUs assigned to Staphylococcus sp. increased 12-fold from 0.5 to 5.9% (electronic supplementary material, figure S2a). In the intestines, as temperature increased, the proportion of family Marinilabiaceae decreased 15-fold from 0.96 to 0.06% (electronic supplementary material, figure S2b). In both the coelomic fluid and the faeces, the proportion of order Campylobacterales decreased as temperature increased—threefold, from 24.1 to 8.1%, in the coelomic fluid and ninefold from 14.3 to 1.6% in the faeces (electronic supplementary material, figure S2c,d). In the digested pellet, the proportion of family Marinilabiaceae decreased with increased temperature approximately fourfold from 46.5 to 9.9% while the proportion of Arcobacter sp. increased fivefold (from 1.9 to 10.1%). The proportions of order Clostridiales and Vibrio sp. in the digested pellet also increased 10-fold (from 1.2 to 12.2% and 1.9 to 19.0%, respectively) and the proportion of family Vibrionaceae increased 20-fold with temperature (from 0.5 to 11.2%; electronic supplementary material, figure S2e). Overall, however, due to the large number of taxa included in the analysis and small sample size, these shifts were not large enough to have a statistically significant effect on the overall microbial community structure.
(b). Predicted microbial functioning
PICRUSt analysis predictions at KEGG level two (general pathways of predicted functions) revealed membrane transport was the most common predicted microbial functional category, regardless of sample type or temperature treatment. Membrane transport comprised between 14.3% (feed) and 32.5% (seawater) of the predicted level two functional categories (figure 3). Other commonly predicted functional categories in both temperature treatments included carbohydrate (8.6–11.0%) and amino acid metabolism (5.5–11.4%), replication (4.2–8.7%), cell motility (4.0–12.5%), and lipid metabolism (2.1–5.8%). The top two sub-categories predicted at KEGG level three (more specific pathways of predicted functions) for all sample types and temperature treatments were transporters (8.6–20.9%) and ABC transporters (4.6–10.5%). Other abundant level three sub-categories were general function prediction only (3.3–4.4%), two-component systems (1.8–6.5%), bacterial motility proteins (1.5–6.1%), transcription factors (1.0–2.6%), and bacterial chemotaxis (0.9–4.7%). The NSTI for each sample type ranged from 0.092 (digested pellet) to 0.136 (feed; electronic supplementary material, table S2).
Figure 3.

The predicted microbial functions of samples collected from Lytechinus variegatus held at 26° and 30°C for 90 days. Samples taken were: (a) seawater, (b) intestines, (c) coelomic fluid, (d) digested pellet (sampled from inside the intestinal tract), and (e) faeces. Functions were categorized to level two (left column) and three (right column; 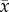 ± s.e.) using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) software. The top five functions contributing to dissimilarity (in order from largest contribution (left/top) to smallest contribution (right/bottom) as determined using Similarity Percentages (SIMPER) analysis; see electronic supplementary material, tables S3 and S4) for each sample type at levels two and three are shown.
± s.e.) using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) software. The top five functions contributing to dissimilarity (in order from largest contribution (left/top) to smallest contribution (right/bottom) as determined using Similarity Percentages (SIMPER) analysis; see electronic supplementary material, tables S3 and S4) for each sample type at levels two and three are shown.
Elevated seawater temperature had a significant effect on the predicted microbial functioning of every sample type (figure 3). This was consistent when categorized at both level two (Adonis; seawater [F1,2745 = 122.66, R2 = 0.043, p = 0.001], intestines [F1,393 = 2.25, R2 = 0.006, p = 0.05], coelomic fluid [F1,3499 = 11.635, R2 = 0.003, p = 0.001], digested pellet [F1,973 = 135, R2 = 0.122, p = 0.001], or faeces [F1,1311 = 9.55, R2 = 0.007, p = 0.001]) and level three (Adonis; seawater [F1,2745 = 97.448, R2 = 0.034, p = 0.001], intestines [F1,393 = 3.339, R2 = 008, p = 0.006], coelomic fluid [F1,3499 = 13.2, R2 = 0.004, p = 0.001], digested pellet [F1,973 = 104.31, R2 = 0.097, p = 0.001], or faeces [F1,1311 = 12.876, R2 = 0.010, p = 0.001]). The most abundant level two (membrane transport, carbohydrate and amino acid metabolism, and cell motility) and level three functions (transporters, ABC transporters, general function prediction only, bacterial motility, and two-component systems) contributed the most to the significant dissimilarity between samples collected from sea urchins held in the 26° and 30°C temperature treatments (electronic supplementary material, tables S3 and S4).
4. Discussion
Our results indicate that the microbial diversity of the intestines was low, indeed approximately 20-fold lower than the microbial diversity of the feed and the surrounding seawater at 26°C, and more than 30-fold lower than the microbial diversity of the feed and the surrounding seawater at 30°C (table 1). Similarly, the microbial diversity of other echinoderms—the intestines of the sea cucumber Apostichopus japonicus [30] and the subcuticle of the brittle star Amphipholis squamate [15]—is relatively low, dominated by Proteobacteria and Octadecabacter sp., respectively. In the sea cucumber, investigators attributed low microbial diversity to selective feeding [30]. However, as L. variegatus was not allowed to feed selectively in this experiment, the relatively low intestinal microbial diversity may indicate selection for microbes that facilitate digestion and nutrient intake in the intestines of L. variegatus.
Previous experiments have also demonstrated that the intestines of L. variegatus have a low microbial diversity dominated by the order Campylobacterales (family Campylobacteraceae) [10,11]. Hakim et al. [11] demonstrated that one oligotype of the order Campylobacterales comprised approximately 92% of the gut microbiota of L. variegatus maintained under laboratory conditions at 22°C and fed an artificial feed. This oligotype closely matched that of Arcobacter sp., Sulfuricurvum sp., and Arcobacter bivalviorum [11]. Similarly, the order Campylobacterales comprised approximately 93% of the microbiota of L. variegatus sampled in situ at Eagle Harbour, FL [10]. A BLAST search of the order Campylobacterales sequence isolated from this study revealed that it closely corresponded to Arcobacter sp. (Identity: 91%, E value: 1 × 10−87) and Arcobacter bivalviorum (Identity: 91%, E value: 1 × 10−87). Although this genus contains both pathogenic and non-pathogenic taxa [31], the prevalence of the order Campylobacterales in the intestines of visibly healthy sea urchins suggests that the near exclusive presence of this bacteria is involved with digestion and nutrient uptake. Most species in the order Campylobacterales are microaerophilic, requiring lower levels of oxygen and/or higher levels of carbon dioxide [32]. The gut of the sea urchin may possess conditions which are suitable for the order Campylobacterales, even under variable environmental conditions. Our study indicates that L. variegatus exposed to prolonged periods of near-future ocean temperature (30°C) maintained a consistent intestinal microbial diversity as well as the most abundant taxon groups observed at lower temperature both in the present study and previous studies.
Although the overall microbial community structure of the samples did not change significantly with increased seawater temperature, subtle shifts in microbial taxa may still be important. For example, in the digested pellet from the 30°C treatment, the proportion of OTUs assigned to Vibrionaceae, Vibrio sp. and Alteromonas sp. increased between 10- and 20-fold. These increases were expected, as Vibrio sp. and Alteromonas sp. are opportunistic species with fast growth rates and high thermal tolerances [33,34]. However, these taxa include pathogenic bacteria which have been associated with stress and disease in other marine invertebrates [32]. Additionally, elevated seawater temperatures may also increase the virulence of some pathogens, including Vibrio spp. [33]. Also in the digested pellet, the significant fourfold decrease in the proportion of OTUs assigned to the family Marinilabiaceae may impair the digestion of cellulose and other plant polysaccharides [35] at elevated seawater temperatures. Finally, the proportion of OTUs assigned to the class Actinobacteria decreased sixfold in the intestines of L. variegatus at 30°C. Many bacteria in the class Actinobacteria produce antibiotics and suppress pathogenic infections [32] and their reduction may further increase the incidence and severity of sea urchin diseases. Within this study, however, no signs of disease or mortality were observed at either temperature.
It was not possible to determine whether observed microbial shifts are a direct result of temperature on the microbes or indirect selection of different microbial abundances by sea urchins requiring different microbes in response to changing metabolic demands. Furthermore, in situ, elevated seawater temperature may have more complicated effects on microbial community structure than observed in the present laboratory-based study. However, we observed highly significant effects of increased temperature on predicted microbiome functioning across every sample type indicating that small changes in microbial community structure can have large effects on community function. PICRUSt provides a prediction rather than an actual measurement of functionality and further metagenomics analyses should be performed. However, the NSTI, which quantifies the availability of nearby genome representatives, was low across all sample types, suggesting that PICRUSt-predicted functions are relatively accurate [27]. In the present study, we did not observe higher predicted functional stability with higher microbial diversity, instead, we saw significant changes in microbial function in samples with high microbial diversity (e.g. seawater) as well as samples with low diversity (e.g. intestines).
The implications of our observed changes in predicted microbiome functionality are difficult to evaluate. The predicted functional changes may be a result of natural selection and could be advantageous to the sea urchin under near-future ocean conditions. Lytechinus variegatus held at 30°C for 90 days grew (in test diameter and height) at the same rate as sea urchins held at 26°C [36]. Although the feeding rate of L. variegatus held at 30°C decreased over the course of the experiment, the feed absorption efficiency of individuals maintained at 30°C increased over the same period by 6.5%, indicative of a potential compensatory response [36]. Intestinal microbes in L. variegatus may be acclimating and facilitating increased nutrient uptake in response to higher metabolic demands.
However, as 30°C represents the upper thermal limit of L. variegatus [8], changes in predicted microbiome functionality could also be destabilizing, indicating stress and an inability of the sea urchin to regulate its microbiome. In this case, there could be negative impacts on sea urchin fitness. Dedicating more energy to membrane transport at the cost of carbohydrate and amino acid metabolism may indicate a shift away from microbial mutualistic interactions and towards opportunistic pathogens [37]. In corals, destabilization of the ‘healthy’ microbial community is associated with increased rates of disease [37]. As sea urchins experience stress associated with ocean warming host immunity may also become compromised. In the sea urchin Heliocidaris erythrogramma, elevated seawater temperatures (+6°C) decreased coelomocyte concentrations and significantly affected the types of coelomocytes comprising the coelomic fluid [38].
Ocean acidification, occurring simultaneously with ocean warming, may have additive, synergistic or antagonistic effects on species-specific microbial communities [39,40]. For example, an experimental study found the microbial community structure associated with the spines of the sea urchin Echinometra sp. was unaffected by elevated seawater temperature or reduced pH, whereas these same stressors had an interactive effect on the microbial communities associated with the crustose coralline algae Hydrolithon onkodes and the foraminifera Marginopora vertebralis [40]. The results of the present study demonstrate the importance of determining both microbial community structure and functionality to more accurately evaluate fitness in sea urchins and other benthic marine invertebrates under near-future ocean conditions.
Supplementary Material
Acknowledgements
We thank Katie McArdle and Sarah Mays for assistance with sea urchin husbandry. We also thank Dr Peter Eipers for assistance with PCR.
Ethics
All applicable international, national and/or institutional guidelines for the care and use of animals were followed.
Data accessibility
All NextGen raw sequence data files are deposited in the NCBI Sequence Read Archive for public access (Accession no. SRP100630).
Authors' contributions
C.J.B., C.D.M., J.A.H., H.K. and J.B.M. conceived and designed the experiment. C.J.B. performed the experiment. C.D.M. and J.B.M. contributed materials and funding. C.J.B., C.D.M., J.A.H., H.K. and W.V.D.P. analysed the data. All authors drafted the manuscript and approve its publication.
Competing interests
We declare we have no competing interests.
Funding
The following are acknowledged for their support of the Microbiome Resource at the University of Alabama at Birmingham: Comprehensive Cancer Center (P30AR050948), Center for AIDS Research (5P30AI027767), Center for Clinical Translational Science (UL1TR001417), University Wide Institutional Core and Heflin Centre for Genomic Sciences, as well as an Endowed University Professorship in Polar and Marine Biology to J.B.M.
References
- 1.Stocker T. (ed.). 2014. Climate change 2013: the physical science basis: working group I contribution to the fifth assessment report of the intergovernmental panel on climate change. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 2.Pörtner HO, Bennett AF, Bozinovic F, Clarke A, Lardies MA, Lucassen M, Pelster B, Schiemer F, Stillman JH. 2006. Trade-offs in thermal adaptation: the need for a molecular to ecological integration. Physiol. Biochem. Zool. 79, 295–313. ( 10.1086/499986) [DOI] [PubMed] [Google Scholar]
- 3.Beddingfield SD, McClintock JB. 2000. Demographic characteristics of Lytechinus variegatus (Echinoidea: Echinodermata) from three habitats in a North Florida Bay, Gulf of Mexico. Mar. Ecol. 21, 17–40. ( 10.1046/j.1439-0485.2000.00688.x) [DOI] [Google Scholar]
- 4.Beddingfield SD, McClintock JB. 1998. Differential survivorship, reproduction, growth and nutrient allocation in the regular echinoid Lytechinus variegatus (Lamarck) fed natural diets. J. Exp. Mar. Biol. Ecol. 226, 195–215. ( 10.1016/S0022-0981(97)00247-5) [DOI] [Google Scholar]
- 5.Valentine J, Heck K Jr. 1999. Seagrass herbivory: evidence for the continued grazing of marine grasses. Mar. Ecol.: Prog. Ser. 176, 291–302. ( 10.3354/meps176291) [DOI] [Google Scholar]
- 6.Challener RC, Robbins LL, McClintock JB. 2016. Variability of the carbonate chemistry in a shallow, seagrass-dominated ecosystem: implications for ocean acidification experiments. Mar. Freshwater Res. 67, 163–172. ( 10.1071/mf14219) [DOI] [Google Scholar]
- 7.Biasutti M, Sobel AH, Camargo SJ, Creyts TT. 2012. Projected changes in the physical climate of the Gulf Coast and Caribbean. Clim. Change 112, 819–845. ( 10.1007/s10584-011-0254-y) [DOI] [Google Scholar]
- 8.Brothers CJ, McClintock JB. 2015. The effects of climate-induced elevated seawater temperature on the covering behavior, righting response, and Aristotle's lantern reflex of the sea urchin Lytechinus variegatus. J. Exp. Mar. Biol. Ecol. 467, 33–38. ( 10.1016/j.jembe.2015.02.019) [DOI] [Google Scholar]
- 9.Fong W, Mann KH. 1980. Role of gut flora in the transfer of amino acids through a marine food chain. Can. J. Fish. Aquat. Sci. 37, 88–96. ( 10.1139/f80-009) [DOI] [Google Scholar]
- 10.Hakim JA, Koo H, Kumar R, Lefkowitz EJ, Morrow CD, Powell ML, Watts SA, Bej AK. 2016. The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. 92, fiw146 ( 10.1093/femsec/fiw146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hakim JA, et al. 2015. An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 6, 343 ( 10.3389/fmicb.2015.01047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawrence JM, Lawrence AL, Watts SA. 2007. Feeding, digestion, and digestibility of sea urchins. In Developments in aquaculture and fisheries science, vol. 38 (ed. JM Lawrence), pp. 135–154. New York, NY: Elsevier. [Google Scholar]
- 13.Prim P, Lawrence JM. 1975. Utilization of marine plants and their constituents by bacteria isolated from the gut of echinoids (Echinodermata). Mar. Biol. 33, 167–173. ( 10.1007/bf00390722) [DOI] [Google Scholar]
- 14.Becker PT, Samadi S, Zbinden M, Hoyoux C, Compère P, De Ridder C. 2009. First insights into the gut microflora associated with an echinoid from wood falls environments. Cah. Biol. Mar. 50, 343. [Google Scholar]
- 15.Morrow KM, Tedford AR, Pankey MS, Lesser MP. 2018. A member of the Roseobacter clade, Octadecabacter sp., is the dominant symbiont in the brittle star Amphipholis squamata. FEMS Microbiol. Ecol. 94, 1967 ( 10.1093/femsec/fiy030) [DOI] [PubMed] [Google Scholar]
- 16.Galac MR, Bosch I, Janies DA. 2016. Bacterial communities of oceanic sea star (Asteroidea: Echinodermata) larvae. Mar. Biol. 163, 32 ( 10.1007/s00227-016-2938-3) [DOI] [Google Scholar]
- 17.Buckley KM, Ho EC, Hibino T, Schrankel CS, Schuh NW, Wang G, Rast JP. 2017. IL17 factors are early regulators in the gut epithelium during inflammatory response to Vibrio in the sea urchin larva. eLife 6, 860 ( 10.7554/eLife.23481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho EC, Buckley KM, Schrankel CS, Schuh NW, Hibino T, Solek CM, Bae K, Wang G, Rast JP. 2016. Perturbation of gut bacteria induces a coordinated cellular immune response in the purple sea urchin larva. Immunol. Cell Biol. 94, 861–874. ( 10.1038/icb.2016.51) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. ( 10.1111/1574-6976.12025) [DOI] [PubMed] [Google Scholar]
- 20.Dishaw LJ, Cannon JP, Litman GW, Parker W. 2014. Immune-directed support of rich microbial communities in the gut has ancient roots. Dev. Comp. Immunol. 47, 36–51. ( 10.1016/j.dci.2014.06.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith LC, Rast JP, Brockton V, Terwilliger DP, Nair SV, Buckley KM, Majeske AJ. 2006. The sea urchin immune system. Invertebr. Survival J. 3, 25–39. [Google Scholar]
- 22.Nakagawa S, et al. 2017. Microbiota in the coelomic fluid of two common coastal starfish species and characterization of an abundant Helicobacter-related taxon. Sci. Rep. 7, 4974 ( 10.1038/s41598-017-09355-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaneveld JR, et al. 2016. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 7, 11833 ( 10.1038/ncomms11833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108, 4516–4522. ( 10.1073/pnas.1000080107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. ( 10.1038/ismej.2012.8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar R, Eipers P, Little RB, Crowley M, Crossman DK, Lefkowitz EJ, Morrow CD. 2014. Getting started with microbiome analysis: sample acquisition to bioinformatics. Curr. Protoc. Hum. Genet. 215, 18.8.1–18.8.29. ( 10.1002/0471142905.hg1808s82). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langille MG, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. ( 10.1038/nbt.2676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. 2004. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, 277D–2280. ( 10.1093/nar/gkh063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Team RC. 2016. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; https://www.R-project.org. [Google Scholar]
- 30.Gao F, Li F, Tan J, Yan J, Sun H. 2014. Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS ONE 9, e100092 ( 10.1371/journal.pone.0100092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collado L, Figueras MJ. 2011. Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin. Microbiol. Rev. 24, 174–192. ( 10.1128/cmr.00034-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrity G, Staley JT, Boone DR, De Vos P, Goodfellow M, Rainey FA, Schleifer KH. 2006. Bergey's Manual® of systematic bacteriology: volume two: the proteobacteria. Berlin, Germany: Springer Science & Business Media. [Google Scholar]
- 33.Lokmer A, Wegner KM. 2015. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 3, 670–682. ( 10.1038/ismej.2014.160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourne DG, Morrow KM, Webster NS. 2016. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Ann. Rev. Microbiol. 70, 317–340. ( 10.1146/annurev-micro-102215-095440) [DOI] [PubMed] [Google Scholar]
- 35.van Kessel MA, Dutilh BE, Neveling K, Kwint MP, Veltman JA, Flik G, Jetten MS, Klaren PH, Op den Camp HJ. 2011. Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express 1, 41 ( 10.1186/2191-0855-1-41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brothers CJ, McClintock JB. 2018. Sea urchins exposed to near-future elevated seawater temperature alter resource allocation under low-quality food conditions. Mar. Bio. 165, 235 ( 10.1007/s00227-018-3301-7) [DOI] [Google Scholar]
- 37.Meron D, Atias E, Kruh LI, Elifantz H, Minz D, Fine M, Banin E. 2011. The impact of reduced pH on the microbial community of the coral Acropora eurystoma. ISME J. 5, 51–60. ( 10.1038/ismej.2010.102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brothers CJ, Harianto J, McClintock JB, Byrne M. 2016. Sea urchins in a high-CO2 world: the influence of acclimation on the immune response to ocean warming and acidification. Proc. R. Soc. B 283, 20161501 ( 10.1098/rspb.2016.1501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesser MP, Fiore C, Slattery M, Zaneveld J. 2016. Climate change stressors destabilize the microbiome of the Caribbean barrel sponge, Xestospongia muta. J. Exp. Mar. Biol. Ecol. 475, 11–18. ( 10.1016/j.jembe.2015.11.004) [DOI] [Google Scholar]
- 40.Webster NS, Negri AP, Botté ES, Laffy PW, Flores F, Noonan S, Schmidt C, Uthicke S. 2016. Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci. Rep. 6, 681 ( 10.1038/srep19324) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All NextGen raw sequence data files are deposited in the NCBI Sequence Read Archive for public access (Accession no. SRP100630).