

Abstract
Cascading speciation is predicted to occur when multiple interacting species diverge in parallel as a result of divergence in one species promoting adaptive differentiation in other species. However, there are few examples where ecological interactions among taxa have been shown to result in speciation that cascades across multiple trophic levels. Here, we test for cascading speciation occurring among the western pine beetle (Dendroctonus brevicomis), its primary host tree (Pinus ponderosa), and the beetle's fungal mutualists (Ceratocystiopsis brevicomi and Entomocorticium sp. B). We assembled genomes for the beetle and a fungal symbiont and then generated reduced representation genomic data (RADseq) from range-wide samples of these three interacting species. Combined with published data for the host tree, we present clear evidence that the tree, the beetle, and the fungal symbionts are all genetically structured into at least two distinct groups that have strongly codiverged with geographical isolation. We then combine our genomic results with diverse population and laboratory-based data to show evidence for reproductive isolation at each level of the cascade and for coevolution of both antagonistic and mutualistic species interactions within this complex network.
Keywords: symbiosis, ectosymbiosis, mutualism, coevolution, diversification
1. Introduction
Speciation, the splitting of one lineage into two, is the fundamental process that underlies the evolution of biodiversity. While our understanding of speciation has increased dramatically over the last few decades [1], most research has focused on understanding the evolution of reproductive isolation between pairs of diverging taxa. Far fewer investigations have looked at how interacting complexes of species may promote or constrain processes that lead to diversification [2–4]. Recently, the idea that increases in species diversity itself may promote speciation by creating new ecological niches has gained renewed attention [5,6]. So-called examples of ‘sequential’ or ‘cascading’ divergence and speciation have primarily been described in systems where a herbivorous insect shifts to a new host plant, diverges, and becomes reproductively isolated from its progenitor. In turn, parasitoids specialized on the herbivore also diverge and become reproductively isolated [7,8]. The end result of this dynamic process of codifferentiation is speciation that cascades across multiple trophic levels.
Cascading speciation among multiple interacting species probably extends beyond antagonistic plant–herbivore–parasitoid systems. Plant–herbivore–microbe systems are ubiquitous multi-trophic interactions where cascades could be common. Many plant-feeding insects are involved in endosymbiotic relationships with bacteria that are sequestered within their body and vertically transmitted from mother to offspring. These intimate interactions often play critical roles in adaptation to specific ecological niches [9–11] and it is easy to envision how speciation in the host insect could result in speciation in the endosymbiont [12]. Both theoretical and empirical studies have demonstrated insect and endosymbiont cospeciation [13,14], including multiple endosymbiotic taxa associated with a single host insect [15]. However, many insect–microbe associations are ectosymbioses with bacteria and/or fungi [9] which tend to have looser associations, with the symbiont more likely to be lost, swapped or added over evolutionary time. Further, how adaptation to diverging host plants might result in divergence in the insect–ectosymbiont system has not been fully explored. The extent to which adaptation and coevolution (i.e. reciprocal adaptation between interacting species) results in stable long-term ectosymbioses that may drive cascading speciation remains largely unknown.
The ponderosa pine–western pine beetle–fungi symbiosis affords the opportunity to study the phenomena of cascading speciation among interacting species across different trophic levels. The host tree, ponderosa pine (Pinus ponderosa), is broadly distributed across the western USA (figure 1a). Within this species there exist two genetically distinct subspecies [17] that formed in isolated southern glacial refugia [18–21] during the Pleistocene [22]. As the climate warmed, the two subspecies expanded their distributions northwards, forming a narrow zone of secondary contact [18,22]. The western pine beetle (Dendroctonus brevicomis) is one of the most significant pests of ponderosa pine [23] (figure 1b). Except in a very small portion of the beetle's range where it also reproduces in the closely related Pinus coulteri, the western pine beetle only attacks and reproduces in ponderosa pine [24]. Therefore, any host-associated adaptation and differentiation in the western pine beetle is likely to be driven by interactions with this tree species. The ability of the beetle to recognize and colonize host trees is intimately linked to host tree chemistry [25]. Once a host tree is located, the beetle stages a pheromone-mediated attack wherein hundreds to thousands of individuals overcome the tree's physical and chemical defences. After exhausting these defences, the beetle reproduces within the tree, first feeding in the phloem and then later moving into the outer bark (figure 1b). There are a number of phenotypic differences within and between the two subspecies of ponderosa pine (summarized in [19]), including extensive variation in their defensive chemicals [26]. Likewise, previous studies have identified substantial levels of mitochondrial DNA sequence divergence (approx. 7–9%) between beetles broadly associated with the two ponderosa pine lineages, suggesting parallel plant–herbivore divergence in this system [27,28].
Figure 1.

The ponderosa pine–western pine beetle–fungal symbiont interaction. (a) Ponderosa pine is an iconic tree widely distributed across the western USA. The western pine beetle kills ponderosa pine by constructing tunnels and reproducing in the phloem and bark. After successful tree colonization and inoculation of fungal symbionts, developing larvae leave the phloem and tunnel into the nutrient-poor outer bark where they feed heavily on symbiotic fungi that provide critical nutrients to the developing insect [16]. (b) The beetle–fungal symbiosis is maintained via an exoskeletal structure in the female (mycangium; its location highlighted with an ellipse) that harbours glands and excretes unknown substances thought to nurture and promote specificity [16]. After pupation, adult beetles incorporate spores into the mycangium for transport to the next host tree. (c) Shown is a scanning electron microscope image (see the electronic supplemental material, figure S1 for additional images) of symbiotic fungi and spores lining the pupal chamber. (d) Schematic of the complexity of the tree–beetle–fungi interaction. Arrow widths scaled to represent the strength of the interaction.
The beetle is involved in an obligate mutualism with two species of symbiotic fungi [16,29–31]. The beetle supplements its nutrient-poor diet through feeding on fungi inoculated into the tree during the initial stages of attack, while the fungi benefit from the interaction by gaining transportation and access to host trees (figure 1c). This symbiosis is maintained by vertical transmission of the symbiont from mother to offspring via specialized structures in the adult integument called mycangia (figure 1b). One species, Ceratocystiopsis brevicomi, is from an order of Ascomycetes (Ophiostomatales) commonly associated with bark beetles as well as mites [29]. The other, Entomocorticium sp. B, is in a group of mostly uncharacterized Basidiomycetes that all appear to be facultative or obligate symbionts of bark beetles [30]. Both fungi (hereafter ascomycete or basidiomycete) show morphological adaptations that aid in beetle feeding and dispersal [32]. The basidiomycete appears to be the more frequent and beneficial partner, although both fungi are found in most if not all populations [33]. The ability of either fungus to colonize host tree tissues is also intimately linked to host tree chemistry [34,35].
The ponderosa pine–western pine beetle–fungal symbiosis presents a network of antagonistic and mutualistic species interactions across three taxonomic kingdoms and three trophic levels (figure 1d). Population genetic structure of the symbionts in relation to genetic structure in the host beetles (or trees) has not been clearly resolved and shared geographical isolation may have played a role in promoting differentiation [27]. Fungal-swapping experiments have shown that beetles from the northwestern portion of the range failed to incorporate a basidiomycete isolated from southeastern portion of the range into their mycangia, suggesting coevolution [16]. Here we combine extensive population genomic data of the beetle, basidiomycete and ascomycete with published genetic data for the tree to resolve the evolutionary history of these interacting species. Integration of these novel genomic data with diverse population and laboratory-based experiments reveal a species interaction network consistent with cascading speciation driven by interspecific interactions.
2. Results
(a). Beetle and basidiomycete reference genome assemblies
It can be difficult to fully isolate symbionts from hosts. This issue is particularly worrisome for genome partitioning-based analyses that rely on de novo assemblies (i.e. RADseq and related approaches) where contamination from one or more of the species could confound patterns of codifferentiation. To overcome this issue we first generated whole-genome assemblies for the beetle and the basidiomycete (the ascomycete genome was recently published [36]) using Illumina sequencing and the Allpaths-lg assembler [37]. Our assembled beetle genome had a scaffold N50 of 5 kb and consisted of 35 469 scaffolds, with the maximum scaffold size of 540 064 bp. The total length of the assembly was 130 Mb (electronic supplementary material, table S1). Sequencing of the basidiomycete, resulted in a 36 Mb assembly with a scaffold N50 of 54 kb, a total of 1248 scaffolds, and a maximum scaffold size of 302 230 bp (electronic supplementary material, table S1). However, this assembly represents only approximately 50% of the estimated genome size (electronic supplementary material, table S1), suggesting the basidiomycete genome contains many repetitive elements that were collapsed during assembly. Completeness of the beetle and ascomycete genome assemblies, estimated by identifying single copy orthologues [38], was moderately high and comparable to closely related species (electronic supplementary material, table S2).
(b). Codivergence of the tree, beetle and fungal symbionts
To characterize the genetic structure of the four interacting species, we obtained range-wide genetic data for the tree, beetle, and the two fungal symbionts. For the tree, we reanalysed microsatellite data from a previously published analysis (n = 428 trees, seven simple sequence repeats (SSRs) [19]). We generated genome-wide reduced representation (RADseq) data for the beetle (n = 156) and three closely related species (Dendroctonus approximatus, Dendroctonus adjunctus and Dendroctonus frontalis), the ascomycete symbiont (n = 22) and an outgroup (Ceratocystiopsis ranaculosus) and the basidiomycete symbiont (n = 36, including a reference isolate of E. sp. B (B1037)) and four outgroups (E. sp. A, C, G and H); electronic supplementary material, table S3; figure 2a). We sequenced the beetles to a mean coverage of 13× (min = 4.4×, max = 40.1×), the ascomycete isolates to a mean coverage of 53× (min = 28.6×, max = 76.7×) and the basidiomycete isolates to a mean coverage of 39× (min = 10.3×, max = 78.5×). We then mapped the RADseq data to the de novo assembled or published [36] reference genomes for each species. On average, 87%, 95% and 80% of cleaned RADseq reads mapped to the basidiomycete, ascomycete, and beetle genome assemblies, respectively. This fairly high mapping rate indicated that our genome assemblies and RADseq data were both of high quality. After filtering and excluding outgroups (see the electronic supplementary material), we identified 65 624, 6874 and 6424 single nucleotide variants (SNVs) in the ascomycete, basidiomycete and beetle, respectively.
Figure 2.

Geographical distribution and codifferentiation in the tree–beetle–fungi system. (a) Ponderosa pine comprised two subspecies (var. ponderosa and var. scopulorum) thought to have formed in isolation in southern refugia during the Pleistocene. The distribution of the beetle currently follows its primary host tree, except where absent in the central and northern portion of the P. ponderosa var. scopulorum range. The northern range limits of the beetle (and fungi) in the var. scopulorum range is broadly coincident with a shift in tree defensive monoterpenes [26]. Tree, beetle and fungal collection locations are shown, and when present at a location, are represented in the pie chart. (b) Structure and Admixture results for the tree, beetle and two symbiotic fungi and the posterior probability of assignment for each individual (vertical bar) to the optimal number of genetic clusters (K) for each species.
To test for concordant genetic structure among the four species we first performed Structure [39] and Admixture [40] analyses independently for the tree, beetle and the two fungal mutualists. The optimal number of genetic clusters (K) was two in three of the four taxa (tree, beetle and ascomycete; figure 2b). These clusters split individuals into groups consistent with previously described host tree subspecies boundaries (hereafter referred to as East and West groups; figure 2a). For the basidiomycete, we found support for three groups with clear clustering of individuals into East and West groups with individuals from the West group being further partitioned into two subgroups. The West basidiomycetes were unambiguously assigned into two broadly distributed groups that co-occurred at two localities (SB and CQ).
Next, we used principal component analyses (PCA) to further visualize fine-scale genetic structure. Consistent with Structure and Admixture results, PCA clearly resolved East and West groups for the tree, beetle, ascomycete and basidiomycete (figure 3). The second principal component (PC2) for the West tree (figure 3a; green ellipse), beetle (figure 3b; green ellipse) and ascomycete (figure 3c; green ellipse) showed some evidence of a south-to-north cline in genetic variation with the most southern population (SB) showing strong differentiation (figure 3). For the basidiomycete (figure 3d), PC2 split the two clusters of individuals identified in previous Structure analyses.
Figure 3.

Fine-scale population structuring and relationships among sites. Results of PCA for the (a) tree, (b) beetle and symbiotic, (c) ascomycete and (d) basidiomycete. Highlighted in green are West sites, and in blue are East sites. Colours correspond to those used in figure 2b. Dense clusters of points are highlighted in boxes and expanded in the bottom right.
To directly test for codivergence between the tree, beetle and ascomycete (here excluding the basidiomycete, see below), we tested for correlations among the three genetic distance matrices using a framework [41] that has been used to detect correlated genetic structure in other bark beetle–fungus symbioses [42,43]. This analysis was restricted to nine sites (six West and three East) where we had genetic data for the tree, beetle and ascomycete. Using this approach, we strongly rejected the global null hypothesis of incongruence among the distance matrices (W = 0.82, Friedman's χ2 = 86.44, p = 0.001). A posteriori permutation tests of the influence of individual distance matrices on overall concordance indicated the strongest correlation was between beetle and ascomycete (r = 0.78, p = 0.001), followed by beetle and tree (r = 0.76, p = 0.010), and then tree and ascomycete (r = 0.66, p = 0.007).
Our population genomic analyses revealed geographical structuring within species and strongly correlated East–West genetic differentiation for all species with the strongest associations occurring between beetle and ascomycete. Next, we estimated rooted maximum-likelihood trees for the beetle and the two fungi to better resolve the evolutionary history of codivergence (figure 4). The East and West populations partitioned into reciprocally monophyletic groups defining the deepest phylogenetic split within both the beetle and the basidiomycete. Finer scale geographical structuring within East and West was largely absent within the beetle. Consistent with our population genetic analyses, three phylogenetically distinct basidiomycete lineages (previously identified as haplotypes A, B and C [33], and hereafter designated as B1, B2 and B3, respectively) were identified. We found very strong support for more recent ancestry between the two overlapping and broadly distributed West lineages (figure 4). The pattern of reciprocal East–West monophyly was somewhat more ambiguous for the ascomycete. We found strong bootstrap support (figure 4) for East ascomycetes being nested within the West group (i.e. paraphyletic) and being more closely related to the most southern West population (SB). However, the only available outgroup for this analysis is quite distant, resulting in low internode certainty values for this relationship (figure 4c) and many genomic partitions exhibited topologies consistent with East/West reciprocal monophyly. Regardless, all East isolates are found in one well-supported clade (figure 4c).
Figure 4.

Phylogenetic relationships among individuals. Rooted maximum-likelihood trees (RAxML, GTR+ gamma) for the (a) basidiomycete, (b) beetle and (c) ascomycete. For the beetle, only the most closely related outgroup (D. approximatus) is shown (see the electronic supplementary material, figure S4). Bootstrap support and the associated internode certainty (BS/IC/ICA) are shown at important locations in each tree. A total of 315 655, 2 901 196 and 2 926 074 bp of genomic sequence (both variant and invariant positions) was used to infer the relationships among the basidiomycete, beetle and ascomycete individuals, respectively. An important location of the ascomycete tree (c) is expanded to the right.
(c). Population genetics of East and West lineages
Given the presence of genetically differentiated East and West groups in the tree, beetle and fungal mutualists, we sought to characterize the levels of genetic diversity, differentiation and level of DNA sequence divergence. Of particular interest were the three distinct basidiomycete lineages. The presence of more than one West lineage was suggested in a previous study [33], but genome-wide resolution of two distinct lineages with no clear geographical association was a surprise. Further, both lineages were found to co-occur. For example, beetles collected at SB and CQ harboured one or the other basidiomycete lineage (figure 3d). To characterize genetic differentiation, we calculated Weir & Cockerham's Fst [44] and found universally high levels of differentiation approaching complete isolation between the three basidiomycete lineages (B1 versus B2, Fst = 0.92; B1 versus B3, Fst = 0.96; B2 versus B3, Fst = 0.97). The three lineages also showed exceptionally low levels of nucleotide diversity (table 1) with the vast majority of SNVs corresponding to fixed differences between lineages. Nonetheless, overall levels of absolute sequence divergence were low for these taxa (B1 and B2, Dxy = 0.17%; B1 and B3, Dxy = 0.33%; B2 and B3, Dxy = 0.32%). Thus, the western pine beetle harbours three distinct lineages of the basidiomycete characterized by low levels of divergence and diversity.
Table 1.
Mean nucleotide diversity and Tajima's D estimates (+1 s.d.) for East and West beetles (5 kb nonoverlapping windows) and their symbiotic fungi (2.5 kb nonoverlapping windows).
| organism | region (East or West) | number of individuals | nucleotide diversity π (%) | Tajima's D |
|---|---|---|---|---|
| basidiomycete (E. sp. B) | West (B1) | 17 | 0.015 (0.014) | −0.532 (0.783) |
| West (B2) | 8 | 0.011 (0.015) | −0.239 (0.671) | |
| East (B3) | 11 | 0.011 (0.013) | −0.390 (0.730) | |
| ascomycete (C. brevicomi) | West | 18 | 0.373 (0.154) | −1.249 (0.393) |
| East | 4 | 0.100 (0.116) | a | |
| beetle (D. brevicomis) | West | 14 | 0.805 (0.166) | −1.064 (0.747) |
| East | 14 | 0.577 (0.165) | −0.866 (0.797) |
aToo few individuals to estimate.
We found moderate differentiation between East and West ascomycetes (Fst = 0.33) and low levels of absolute divergence (Dxy = 0.43%). Further, we found that nucleotide diversity for ascomycete was substantially higher in West than in East, with an excess of rare alleles in West ascomycetes (Tajima's D = −1.249; table 1) consistent with a recent population expansion. Absolute (Dxy = 1.37%) and relative divergence (Fst = 0.51) was high between East and West beetles. We also found evidence of reduced diversity in East compared to West beetles (table 1), mirroring patterns of ascomycete genetic diversity. The lower nucleotide diversity in East for both beetle and ascomycete suggest that similar forces (e.g. a bottleneck or founder event) may have historically reduced diversity in both species.
(d). Fungal symbiont recombination
Fungal mutualists that are vertically transmitted from parent to offspring are often assumed to reproduce asexually [9]. Contrary to this expectation, we found clear evidence of recombination in the ascomycete (pairwise homoplasy index (PHI), p < 0.005, and four-gamete test). This result was not altogether surprising given previous work describing sexual structures [29]. For the basidiomycete, we also found evidence of recombination occurring within the three distinct lineages (PHI test p < 0.005, and four-gamete test) but no evidence of recombination occurring between the lineages or significant reticulation in phylogenetic networks (electronic supplementary material, figure S2). Taken together with the low genetic diversity observed, our results suggest the basidiomycete is highly selfing or primarily asexual, with infrequent bouts of recombination. Although sexual structures (basidia) have not been identified in the basidiomycete, a closely related species does produce basidia [45]. Infrequent bouts of recombination may be advantageous for a fungus in a tightly linked symbiosis to maintain favourable genotypic combinations that promote stability. Rare recombination events may help avoid the accumulation of deleterious mutations [46].
3. Discussion
The idea that that divergence in one organism can drive divergence across other tightly linked organisms is intuitive. However, clear demonstrations of this phenomenon across multiple trophic levels are rare and have been primarily restricted to plant–herbivore–parasitoid systems [4,7,8]. Our phylogenomic results reveal a striking history of codivergence between East and West populations of the tree, beetle, and their fungal symbionts. While codivergence may be an inevitable outcome of shared geographical isolation, a wealth of population and laboratory data in this system provide compelling evidence that adaptation and coevolution is also probably occurring between these interacting lineages. When considered in the context of genome-wide resolution of population histories, functional integration of these antagonistic and mutualistic interactions appears to have further cascaded into multiple instances of incompatible interactions within and across trophic levels (figure 5). Below we synthesize the available data for cascading speciation in this system and discuss how these data fit with general conditions observed in other systems (table 2).
Figure 5.
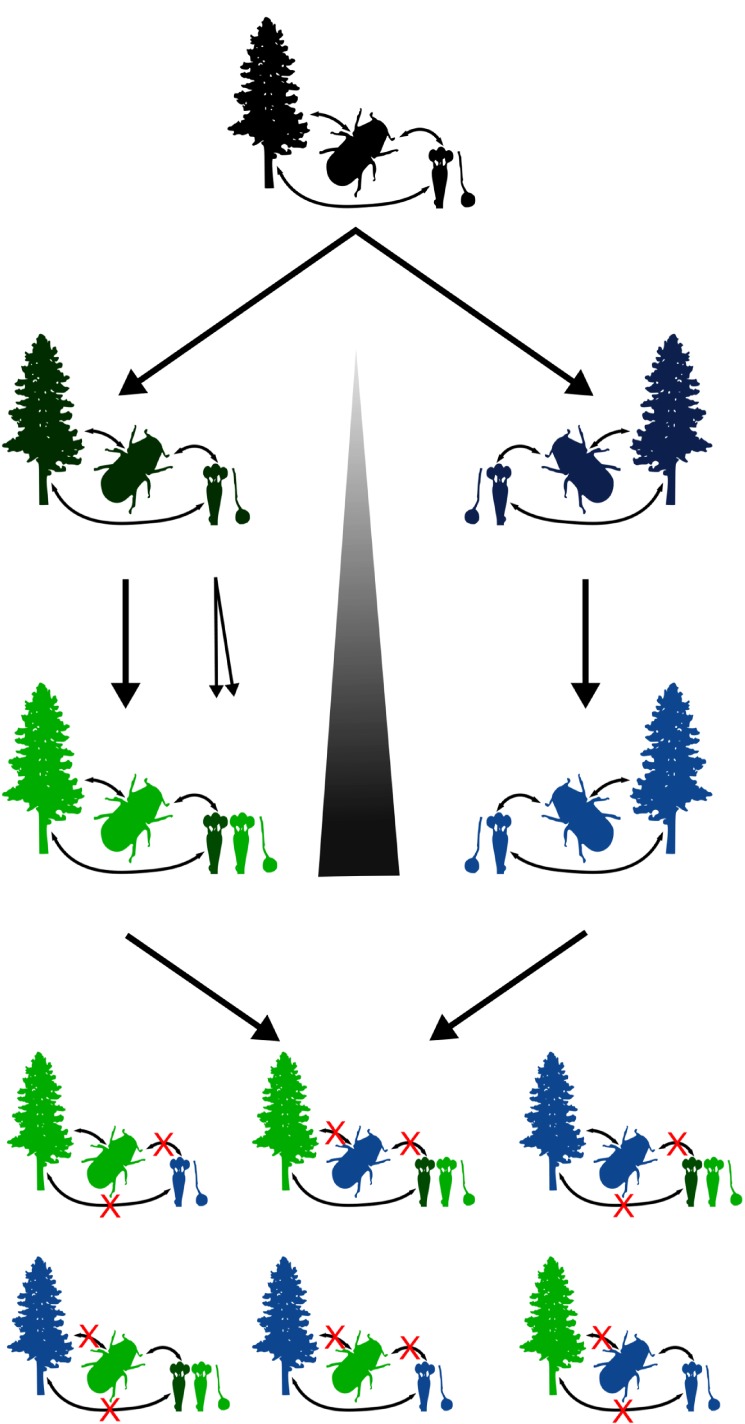
Model for cascading speciation in the ponderosa pine–western pine beetle–fungal symbiont system. Ancestors (black) were subdivided over time into geographically isolated populations. In isolation, strong antagonistic and mutualistic interactions occurred among partners and pushed species along unique evolutionary trajectories resulting in differentiated and unique tree–beetle–fungi systems (blue and green). An additional speciation event occurred in West basidiomycetes (light green and dark green). Levels of divergence among partners ultimately leads to heightened reproductive isolation because mismatched interspecific interactions break down the interaction network.
Table 2.
Conditions often present during cascading speciation as outlined in Abrahamson & Blair [4], Hood et al. [8] and Brodersen et al. [3], and available evidence for those conditions in the ponderosa pine–western pine beetle–symbiotic fungi system.
| condition | generalization for upward adaptive radiation cascade in a predator–prey system [3] | evidence in the ponderosa pine–western pine beetle–symbiotic fungi system |
|---|---|---|
| 1 | ecological speciation and ecotype formation in prey involves a shift to a new habitat or resource and genetic differentiation | [27,33] |
| 2 | species or ecotypes of prey as well as individual predators exhibit habitat preferences | [34,47] |
| 3 | predators have distinct preferences among alternative prey | unclear but likely [34,47] |
| 4 | predator–prey phenology match or overlap sufficiently | n.a. |
| 5 | significant predator trait × prey type interactions determine fitness variation among predators | [16] |
| 6 | mate choice among predators is mediated by habitat or is affected by feeding related behavioural, ecological or morphological traits | [47,48] |
| 7 | Populations of prey and predators at least partly overlap in their geographical ranges | n.a. |
| 8 | ecological diversification among predators is at least to some degree associated with genetic differentiation | ([19,27,33], present study) |
At the center of this system is the host tree. East and West ponderosa pine have evolved qualitative and quantitative differences in tree defensive chemistry. These differences are thoroughly characterized and have long been hypothesized to be the result of selection by bark beetles, and primarily by the western pine beetle [26,49]. For the beetle, host tree chemical profiles are important for host tree recognition, and particular components of the tree's defensive chemical cocktail are used by the beetle to make aggregation pheromones during tree colonization [50]. The direct link between the chemicals that underlie host defence and their co-option in pheromone-mediated beetle attacks is predicted to drive an evolutionary arms race between tree and beetle. Consistent with this, tree defensive chemicals that play a key role in host tree colonization [50] differ in quality and quantity between West and East trees [26]. For example, α-pinene and myrcene both play roles in host tree attractiveness and α-pinene has been found to be more prevalent in East trees while myrcene more prevalent in West trees [26]. Myrcene is an important component of the beetle's pheromones [51] but is undetectable in some East trees [26]. Evolved differences in beetle response to tree chemistry are likely to exist as lures developed to monitor East beetles were found to perform poorly in the West until modified to better match West tree chemistry [47]. East and West beetles also appear to have evolved dramatic differences in the composition of their pheromones [48]. In summary, chemical defences appear to be coevolving with beetle detection and aggregation pheromones, and these differences would probably lead to difficulties finding suitable hosts and attracting mates and conspecifics for mass attack. Additional genomic analyses are needed to further clarify the evolutionary history of divergence between populations of ponderosa pine.
In an interesting twist, recent sampling has led to the discovery that the western pine beetle may exist in very low population densities in northern and central Mexico [52]. These rarely encountered beetles differ anatomically from East and West and also have a more broad host range [52]. The available evidence suggests these beetles probably constitute a third cryptic species in the western pine beetle system [52]. Studies of this putative third cryptic species and identification of its mycangial fungi will undoubtedly further our understanding of how host tree use can influence beetle–fungal divergence.
The obligate mutualism between the beetle and the two species of symbiotic fungi probably are the strongest drivers of cascading speciation in this system. Mycangial incorporation and vertical transmission of fungi is essential for beetle development and survival. Using fungal-swapping experiments, we previously demonstrated that West beetles were incapable of incorporating the East lineage of the basidiomycete into their mycangia [16]. These experiments indicate rapid coevolution between beetle and fungal genotypes. This specificity has the potential to act as a strong barrier to reproductive isolation; even if East and West beetles are able to successfully attack West and East trees, respectively, genotypic mismatches between the beetle and fungal symbionts could result in a loss of symbiont prior to dispersal, thereby dooming subsequent beetle offspring [16]. Our understanding of the beetle–ascomycete interaction is less clear, but given the tight association [33], and the importance of closely related ascomycetes in other beetle symbioses [53], it is highly likely that there is coevolution of this interaction as well.
How differences in tree defensive chemistry could promote divergence in the symbiotic fungi has not been directly explored. However, fungal symbionts must contend with host tree defences much like the beetle and there is evidence that differences in tree defensive chemistry can impact the growth of the basidiomycete [34]. Little is known about how the ascomycete responds to tree defences. Recent evidence has challenged the idea that vertically transmitted fungal symbionts are exclusively asexual [54] and indeed, recombination has been found to occur in the few bark beetle symbionts thus far investigated [43,55]. We found clear genetic evidence of recombination in these systems, indicating that both fungal partners can reproduce sexually. However, the lack of recombination between basidiomycete B1 and B2, the two less divergent and broadly co-occurring West lineages, argues that all three basidiomycete lineages are completely reproductively isolated from one another. The presence of two co-occurring, reproductively isolated basidiomycetes in the absence of clear population structure suggest that diversification in the basidiomycete may evolve rapidly, perhaps in the absence of allopatry. It is unclear if both West lineages can occur with a single beetle host.
An important distinction between cospeciation and the special case of cascading speciation is that the former can be owing to a purely ‘passive’ process of genetic differentiation (i.e. mutation and drift) of co-isolated species, while the latter is a direct result of interspecific adaptation driving speciation [2,8,56]. The most convincing evidence for cascading speciation has been from systems where the interacting organisms are in sympatry [7,8] because many of the conditions facilitating speciation are more easily identified (outlined in table 2). However, sympatry is not a requirement for cascading speciation [8,56]. One prediction of purely ‘passive’ cospeciation owing to shared geographical isolation is that although reproductive isolation may evolve solely between species A and B, interspecific interactions and adaptation to a separate trophic level would not necessarily lead to increased reproductive isolation between A and B. Therefore, evidence of adaptation or coevolution with a separate trophic level that increases the extent of reproductive isolation between A and B, even if aided by geographical isolation, supports the existence of cascading speciation because adaptation itself is helping to promote diversification. Therefore, the tightly linked nature of this tree–beetle–fungi system, along with evidence of adaptation/coevolution, and our genetic data indicating codivergence at all levels of the system, strongly supports the hypothesis of cascading speciation and emphasizes how geographical isolation and adaptation/coevolution can promote diversification across multiple trophic levels.
4. Material and methods
(a). Beetle/fungal collections, DNA extractions and RADseq
Beetles and fungi were collected in the summer of 2011 (electronic supplementary material, table S3). We isolated Ceratocystiopsis from single spores and Entomocorticium from hyphal tips and all fungal collections were from Bracewell and Six [33]. DNA was extracted from beetles using an Omega E.Z.N.A tissue DNA kit (Prod. no. D3396-01, Omega Bio-Tek, Norcross, GA, USA). For fungi, tissue was bead-beat (Geno/Grinder 2000, Spex Certiprep, Metuchen, NJ) prior to DNA extraction with a Qiagen DNeasy Plant Mini Kit (Cat. no. 69104). Our RADseq methods followed Etter et al. [57], with slight modifications (see the electronic supplementary material). Libraries from each species were pooled and Illumina sequenced. RADseq read mapping, filtering and SNV identification are described in the electronic supplementary material.
(b). Genome sequencing and assembly
Draft genomes were assembled using Allpaths-lg [37] and Illumina overlapping and mate pair libraries for beetles from the OL population and a basidiomycete from SierraII (SierraII_2, accession CBS 137838). Genome assembly completeness was determined using BUSCO v. 1.22 [38]. Additional details on genome sequencing and assembly are available in the electronic supplementary material.
(c). Ponderosa pine population genetic data
We downloaded microsatellite data for seven SSRs from Potter et al. [19] and analysed 13 sites in close geographical proximity (mean = 28.9 K, min = 3 K, max = 66 K) to our collection locations.
(d). Genetic analyses
To identify population genetic structure we used Structure v. 2.3.2.1 [39] and Admixture v. 1.23 [40]. Structure was used on the fungal and tree data because it can handle both haploid and diploid data. Owing to computation limitations, Structure analyses were run on 3000 SNVs pulled at random from the two fungal datasets. For the tree and fungal analyses, the burnin was set at 20 000 with 50 000 Markov chain Monte Carlo replicates after burn-in. We tested K values from one to eight with eight replicates per K. The best K for each species was determined by visualizing the likelihoods in Structure Harvester [58] and based on the Evanno method [59]. Results for different runs for each species were averaged using Clumpp 1.1.2 [60]. For the beetle SNV data, we used Admixture v. 1.23 [40] on 6424 SNVs. We tested K values of one through to 19 with 10-fold cross-validation. The K value with the lowest cross-validation standard error was considered the best K [40]. PCA were done using Eigensoft [61]. To perform PCA on the tree microsatellite data, we used GenAlEx [62] and the covariance matrix of the allele frequencies with data standardization.
To test for congruence among the genetic distance data matrices of the four species, we used CADM [41] in the R package ape [63]. Genetic distance matrices were generated for the tree, beetle and C. brevicomi datasets using GenAlEx [62]. Analyses were done using nine sites where we had data for all three species and included three East sites (MTC, FL, RO) and six West sites (SB, OL, CQ, LA, LF, DA). In CADM, 1000 permutations were used to compute a posteriori tests and we used the Holm correction (default) to account for the multiple testing of distance matrices.
To infer phylogenetic relationships, we used supertree and supermatrix approaches to analyse the resulting concatenated (above) and subdivided alignments (20 kb for beetle and 10 kb for fungi). For the supertree approach, a best fitting nucleotide substitution model was selected for each alignment using Modelfinder with free rate heterogeneity implemented in IQ-Tree 1.6.1 [64,65]. Maximum-likelihood phylogenies [66] with 2000 UltraFast (UF) bootstrap replicates [67] were generated for each locus. We used the –minsup flag in IQ-Tree to collapse poorly supported nodes in bootstrap consensus trees for each loci (less than 95% UF bootstrap support). Internode certainty (IC) values were then calculated for an extended majority rule consensus tree constructed from the maximum-likelihood phylogeny for each subsequence using only the well-supported nodes with RAxML 8.2.9 [68,69]. For the supermatrix analysis, a maximum-likelihood phylogeny with 2000 UF bootstrap replicates was estimated using IQ-Tree 1.6.1. IC values were calculated for the supermatrix topology using the well-supported bootstrap trees from the supertree analysis.
For C. brevicomi and E. sp. B, we calculated Weir and Cockerham's weighted Fst [44] using VCFtools and nucleotide diversity (π), absolute sequence divergence (Dxy), and Tajima's D using DNAsp v. 5.10.1 [70]. For the beetle the above statistics were estimated using PopGenome [71]. SplitsTree4 [72] and the PHI test and the four-gamete test [73] as implemented in PopGenome were used to test for recombination.
Supplementary Material
Acknowledgements
We are grateful to Joseph Dysthe for help with data collection. We also thank John McCutcheon, the Good lab, and the Advanced Evolutionary Genetics and Genomics (AEGG) community at the University of Montana for helpful discussions. We thank all who donated their time to collect beetles (electronic supplementary material, table S3).
Data accessibility
Sequence that support the findings of this study have been deposited at the NCBI Sequencing Read Archive, BioProject ID PRJNA306782, PRJNA306780, PRJNA306779.
Authors' contributions
R.B., J.G. and D.S. conceived of the study. R.B. collected samples and generated sequence data. R.B. and D.V. assembled draft genomes. R.B. analysed the RADseq and microsatellite data. D.V. and R.B. conducted phylogenetic analyses. R.B., J.G. and D.S. wrote the manuscript with comments from D.V.
Competing interests
The authors declare no competing interests.
Funding
This project was supported by the McIntire-Stennis Cooperative Forestry Program and by Agriculture and Food Research Initiative Competitive grant no. 2013-67011-21113 from the USDA National Institute of Food and Agriculture. D.V. was supported by grants from NSF GRFP and NSF GROW travel award no. DGE-1313190. Genomic data were generated using instrumentation and services provided by the Vincent J. Coates Genomics Sequencing Laboratory at the University of California Berkeley, supported by the National Institutes of Health S10 Instrumentation grants S10RR029668 and S10RR027303 and the University of Montana Genomics Core, supported by a grant from the M.J. Murdock Charitable Trust.
References
- 1.Coyne JA, Orr HA. 2004. Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 2.Forister ML, Feldman CR. 2011. Phylogenetic cascades and the origins of tropical diversity. Biotropica 43, 270–278. ( 10.1111/j.1744-7429.2010.00702.x) [DOI] [Google Scholar]
- 3.Brodersen J, Post DM, Seehausen O. 2018. Upward adaptive radiation cascades: predator diversification induced by prey diversification. Trends Ecol. Evol. 33, 59–70. ( 10.1016/j.tree.2017.09.016) [DOI] [PubMed] [Google Scholar]
- 4.Abrahamson WG, Blair CP. 2008. Sequential radiation through host-race formation: herbivore diversity leads to diversity in natural enemies. In Specialization, speciation, and radiation: the evolutionary biology of herbivorous insects (ed. Tilmon KJ.), pp. 182–202. Berkeley, CA: University of California Press. [Google Scholar]
- 5.Emerson BC, Kolm N. 2005. Species diversity can drive speciation. Nature 434, 1015–1017. ( 10.1038/nature03450) [DOI] [PubMed] [Google Scholar]
- 6.Janz N, Nylin S, Wahlberg N. 2006. Diversity begets diversity: host expansions and the diversification of plant-feeding insects. BMC Evol. Biol. 6, 4 ( 10.1186/1471-2148-6-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbes AA, Powell THQ, Stelinski LL, Smith JJ, Feder JL. 2009. Sequential sympatric speciation across trophic levels. Science 323, 776–779. ( 10.1126/science.1166981) [DOI] [PubMed] [Google Scholar]
- 8.Hood GR, Forbes AA, Powell THQ, Egan SP, Hamerlinck G, Smith JJ, Feder JL. 2015. Sequential divergence and the multiplicative origin of community diversity. Proc. Natl Acad. Sci. USA 112, E5980–E5989. ( 10.1073/pnas.1424717112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller UG, Gerardo NM, Aanen DK, Six DL, Schultz TR. 2005. The evolution of agriculture in insects. Annu. Rev. Ecol. Evol. S 36, 563–595. ( 10.1146/annurev.ecolsys.36.102003.152626) [DOI] [Google Scholar]
- 10.Moran NA. 2007. Symbiosis as an adaptive process and source of phenotypic complexity. Proc. Natl Acad. Sci. USA 104, 8627–8633. ( 10.1073/pnas.0611659104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner SM, Martinez AJ, Ruan YM, Kim KL, Lenhart PA, Dehnel AC, Oliver KM, White JA. 2015. Facultative endosymbionts mediate dietary breadth in a polyphagous herbivore. Funct. Ecol. 29, 1402–1410. ( 10.1111/1365-2435.12459) [DOI] [Google Scholar]
- 12.Simon JC, Carre S, Boutin M, Prunier-Leterme N, Sabater-Munoz B, Latorre A, Bournoville R. 2003. Host-based divergence in populations of the pea aphid: insights from nuclear markers and the prevalence of facultative symbionts. Proc. R. Soc. Lond. B 270, 1703–1712. ( 10.1098/rspb.2003.2430) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark MA, Moran NA, Baumann P, Wernegreen JJ. 2000. Cospeciation between bacterial endosymbionts (Buchnera) and a recent radiation of aphids (Uroleucon) and pitfalls of testing for phylogenetic congruence. Evolution 54, 517–525. ( 10.1111/j.0014-3820.2000.tb00054.x) [DOI] [PubMed] [Google Scholar]
- 14.Conord C, Despres L, Vallier A, Balmand S, Miquel C, Zundel S, Lemperiere G, Heddi A. 2008. Long-term evolutionary stability of bacterial endosymbiosis in Curculionoidea: additional evidence of symbiont replacement in the Dryophthoridae family. Mol. Biol. Evol. 25, 859–868. ( 10.1093/molbev/msn027) [DOI] [PubMed] [Google Scholar]
- 15.Noda S, et al. 2007. Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 16, 1257–1266. ( 10.1111/j.1365-294X.2006.03219.x) [DOI] [PubMed] [Google Scholar]
- 16.Bracewell RR, Six DL. 2015. Experimental evidence of bark beetle adaptation to a fungal symbiont. Ecol. Evol. 5, 5109–5119. ( 10.1002/ece3.1772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conkle MT, Critchfield WB. 1988. Genetic variation and hybridization of ponderosa pine. In Ponderosa pine: the species and its management (eds Baumgartner DM, Lotan JE), pp. 27–43. Pullman, WA: Washington State University Press. [Google Scholar]
- 18.Betancourt JL, Van Devender TR, Martin PS. 1990. Packrat middens: the last 40,000 years of biotic change. Tucson, AZ: University of Arizona Press. [DOI] [PubMed] [Google Scholar]
- 19.Potter K, Hipkins V, Mahalovich M, Means R. 2015. Nuclear genetic variation across the range of ponderosa pine (Pinus ponderosa): phylogeographic, taxonomic and conservation implications. Tree Genet. Genomes 11, 1–23. ( 10.1007/s11295-015-0865-y) [DOI] [Google Scholar]
- 20.Potter KM, Hipkins VD, Mahalovich MF, Means RE. 2013. Mitochondrial DNA haplotype distribution patterns in Pinus ponderosa (Pinaceae): range-wide evolutionary history and implications for conservation. Am. J. Bot. 100, 1562–1579. ( 10.3732/ajb.1300039) [DOI] [PubMed] [Google Scholar]
- 21.Roberts DR, Hamann A. 2015. Glacial refugia and modern genetic diversity of 22 western North American tree species. Proc. R. Soc. B 282, 20142903 ( 10.1098/rspb.2014.2903) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latta RG, Mitton JB. 1999. Historical separation and present gene flow through a zone of secondary contact in ponderosa pine. Evolution 53, 769–776. ( 10.1111/j.1558-5646.1999.tb05371.x) [DOI] [PubMed] [Google Scholar]
- 23.Miller JM, Keen FP. 1960. Biology and control of the western pine beetle: a summary of the first 50 years of research. Washington, DC: U.S. Department of Agriculture. [Google Scholar]
- 24.Wood SL. 1982. The bark and ambrosia beetles of North and Central America (Coleoptera: Scolytidae). Great Basin Naturalist 6, 1–1359. [Google Scholar]
- 25.Byers JA. 1995. Host-tree chemistry affecting colonization in bark beetles. In Chemical ecology of insects 2 (eds Cardé RT, Bell WJ), pp. 154–213. Berlin, Germany: Springer. [Google Scholar]
- 26.Smith RH. 1977. Monoterpenes of ponderosa pine xylem resin in western United States. USDA Forest Service Technical Bulletin 1532. Washington, DC, USA.
- 27.Kelley ST, Mitton JB, Paine TD. 1999. Strong differentiation in mitochondrial DNA of Dendroctonus brevicomis (Coleoptera : Scolytidae) on different subspecies of ponderosa pine. Ann. Entomol. Soc. Am. 92, 193–197. ( 10.1093/aesa/92.2.193) [DOI] [Google Scholar]
- 28.Kelley ST, Farrell BD. 1998. Is specialization a dead end? The phylogeny of host use in Dendroctonus bark beetles (Scolytidae). Evolution 52, 1731–1743. ( 10.1111/j.1558-5646.1998.tb02253.x) [DOI] [PubMed] [Google Scholar]
- 29.Hsiau PTW, Harrington TC. 1997. Ceratocystiopsis brevicomi sp. nov., a mycangial fungus from Dendroctonus brevicomis (Coleoptera: Scolytidae). Mycologia 89, 661–669. ( 10.2307/3761004) [DOI] [Google Scholar]
- 30.Hsiau PTW, Harrington TC. 2003. Phylogenetics and adaptations of basidiomycetous fungi fed upon by bark beetles (Coleoptera : Scolytidae). Symbiosis 34, 111–131. [Google Scholar]
- 31.Paine TD, Birch MC. 1983. Acquisition and maintenance of mycangial fungi by Dendroctonus brevicomis Leconte (Coleoptera, Scolytidae). Environ. Entomol. 12, 1384–1386. ( 10.1093/ee/12.5.1384) [DOI] [Google Scholar]
- 32.Harrington TC. 2005. Ecology and evolution of mycophagous bark beetles and their fungal partners. In Ecological and evolutionary advances in insect-fungal associations (eds Vega FE, Blackwell M), pp. 257–291. Oxford, UK: Oxford University Press. [Google Scholar]
- 33.Bracewell RR, Six DL. 2014. Broadscale specificity in a bark beetle-fungal symbiosis: a spatio-temporal analysis of the mycangial fungi of the western pine beetle. Microb. Ecol. 68, 859–870. ( 10.1007/s00248-014-0449-7) [DOI] [PubMed] [Google Scholar]
- 34.Davis TS, Hofstetter RW. 2012. Plant secondary chemistry mediates the performance of a nutritional symbiont associated with a tree-killing herbivore. Ecology 93, 421–429. ( 10.1890/11-0231.1) [DOI] [PubMed] [Google Scholar]
- 35.Paine TD, Raffa KF, Harrington TC. 1997. Interactions among scolytid bark beetles, their associated fungi, and live host conifers. Annu. Rev. Entomol. 42, 179–206. ( 10.1146/annurev.ento.42.1.179) [DOI] [PubMed] [Google Scholar]
- 36.Vanderpool D, Bracewell RR, McCutcheon JP. 2017. Know your farmer: ancient origins and multiple independent domestications of ambrosia beetle fungal cultivars. Mol. Ecol 27, 2077–2094. ( 10.1111/mec.14394) [DOI] [PubMed] [Google Scholar]
- 37.Gnerre S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518. ( 10.1073/pnas.1017351108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. ( 10.1093/bioinformatics/btv351) [DOI] [PubMed] [Google Scholar]
- 39.Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. ( 10.1101/gr.094052.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legendre P, Lapointe FJ. 2004. Assessing congruence among distance matrices: single-malt Scotch whiskies revisited. Aust. NZ J. Stat. 46, 615–629. ( 10.1111/j.1467-842X.2004.00357.x) [DOI] [Google Scholar]
- 42.Tsui CKM, Farfan L, Roe AD, Rice AV, Cooke JEK, El-Kassaby YA, Hamelin RC. 2014. Population structure of mountain pine beetle symbiont Leptographium longiclavatum and the implication on the multipartite beetle-fungi relationships. PLoS ONE 9, e105455 ( 10.1371/journal.pone.0105455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roe AD, Rice AV, Coltman DW, Cooke JEK, Sperling FAH. 2011. Comparative phylogeography, genetic differentiation and contrasting reproductive modes in three fungal symbionts of a multipartite bark beetle symbiosis. Mol. Ecol. 20, 584–600. ( 10.1111/j.1365-294X.2010.04953.x) [DOI] [PubMed] [Google Scholar]
- 44.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. [DOI] [PubMed] [Google Scholar]
- 45.Whitney HS, Bandoni RJ, Oberwinkler F. 1987. Entomocorticium dendroctoni gen. et sp. nov. (Basidiomycotina), a possible nutritional symbiote of the mountain pine beetle in lodgepole pine in British Columbia. Can. J. Bot. 65, 95–102. ( 10.1139/b87-013) [DOI] [Google Scholar]
- 46.Schultz ST, Lynch M. 1997. Mutation and extinction: The role of variable mutational effects, synergistic epistasis, beneficial mutations, and degree of outcrossing. Evolution 51, 1363–1371. ( 10.1111/j.1558-5646.1997.tb01459.x) [DOI] [PubMed] [Google Scholar]
- 47.Hofstetter RW, Chen Z, Gaylord ML, McMillin JD, Wagner MR. 2008. Synergistic effects of alpha-pinene and exo-brevicomin on pine bark beetles and associated insects in Arizona. J. Appl. Entomol. 132, 387–397. ( 10.1111/j.1439-0418.2007.01263.x) [DOI] [Google Scholar]
- 48.Pureswaran DS, Hofstetter RW, Sullivan BT, Grady AM, Brownie C. 2016. Western pine beetle populations in Arizona and California differ in the composition of their aggregation pheromones. J. Chem. Ecol. 42, 404–413. ( 10.1007/s10886-016-0696-9) [DOI] [PubMed] [Google Scholar]
- 49.Sturgeon KB. 1979. Monoterpene variation in ponderosa pine xylem resin related to western pine beetle predation. Evolution 33, 803–814. ( 10.1111/j.1558-5646.1979.tb04736.x) [DOI] [PubMed] [Google Scholar]
- 50.Byers J. 1982. Male-specific conversion of the host plant compound, myrcene, to the pheromone, (+)-ipsdienol, in the bark beetle, Dendroctonus brevicomis. J. Chem. Ecol. 8, 363–371. ( 10.1007/BF00987784) [DOI] [PubMed] [Google Scholar]
- 51.Bedard WD, Tilden PE, Wood DL, Silverstein RM, Brownlee RG, Rodin JO. 1969. Western pine beetle: field response to its sex pheromone and a synergistic host terpene, myrcene. Science 164, 1284–1285. ( 10.1126/science.164.3885.1284) [DOI] [PubMed] [Google Scholar]
- 52.Valerio-Mendoza O, Armendáriz-Toledano F, Cuéllar-Rodríguez G, Negrón JF, Zúñiga G. 2017. The current status of the distribution range of the western pine beetle, Dendroctonus brevicomis (Curculionidae: Solytinae) in Northern Mexico. J. Insect. Sci. 17, 92 ( 10.1093/jisesa/iex070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Six DL. 2012. Ecological and evolutionary determinants of bark beetle-fungus symbioses. Insects 3, 339–366. ( 10.3390/insects3010339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mikheyev AS, Mueller UG, Abbot P. 2006. Cryptic sex and many-to-one colevolution in the fungus-growing ant symbiosis. Proc. Natl Acad. Sci. USA 103, 10 702–10 706. ( 10.1073/pnas.0601441103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsui CKM, Roe AD, El-Kassaby YA, Rice AV, Alamouti SM, Sperling FAH, Cooke JEK, Bohlmann J, Hamelin RC. 2012. Population structure and migration pattern of a conifer pathogen, Grosmannia clavigera, as influenced by its symbiont, the mountain pine beetle. Mol. Ecol. 21, 71–86. ( 10.1111/j.1365-294X.2011.05366.x) [DOI] [PubMed] [Google Scholar]
- 56.Feder JL, Forbes AA. 2010. Sequential speciation and the diversity of parasitic insects. Ecol. Entomol. 35, 67–76. ( 10.1111/j.1365-2311.2009.01144.x) [DOI] [Google Scholar]
- 57.Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA. 2011. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol. Biol. 772, 157–178. ( 10.1007/978-1-61779-228-1_9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Earl DA, Vonholdt BM. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. ( 10.1007/s12686-011-9548-7) [DOI] [Google Scholar]
- 59.Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. ( 10.1111/j.1365-294X.2005.02553.x) [DOI] [PubMed] [Google Scholar]
- 60.Jakobsson M, Rosenberg NA. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. ( 10.1093/bioinformatics/btm233) [DOI] [PubMed] [Google Scholar]
- 61.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. ( 10.1038/ng1847) [DOI] [PubMed] [Google Scholar]
- 62.Peakall R, Smouse PE. 2012. GenAlEx6.5: genetic analysis in Excel. Population genetic software for teaching and research: an update. Bioinformatics 28, 2537–2539. ( 10.1093/bioinformatics/bts460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. ( 10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 64.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. ( 10.1093/molbev/msu300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. ( 10.1038/nmeth.4285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Felsenstein J. 1981. Evolutionary trees from DNA sequences: a maximum-likelihood approach. J. Mol. Evol. 17, 368–376. ( 10.1007/BF01734359) [DOI] [PubMed] [Google Scholar]
- 67.Hoang DT, Chernomor O, von Haeseler A, Minh B, Vinh LS. 2017. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. ( 10.1093/molbev/msx281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salichos L, Rokas A. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497, 327–331. ( 10.1038/nature12130) [DOI] [PubMed] [Google Scholar]
- 69.Salichos L, Stamatakis A, Rokas A. 2014. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 31, 1261–1271. ( 10.1093/molbev/msu061) [DOI] [PubMed] [Google Scholar]
- 70.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. ( 10.1093/bioinformatics/btp187) [DOI] [PubMed] [Google Scholar]
- 71.Pfeifer B, Wittelsburger U, Ramos-Onsins SE, Lercher MJ. 2014. PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. ( 10.1093/molbev/msu136) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. ( 10.1093/molbev/msj030) [DOI] [PubMed] [Google Scholar]
- 73.Hudson RR, Kaplan NL. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111, 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence that support the findings of this study have been deposited at the NCBI Sequencing Read Archive, BioProject ID PRJNA306782, PRJNA306780, PRJNA306779.