

Abstract
Background
Structural and dynamic studies of chromosomes tagged with green fluorescent protein (GFP) in yeast and cultured animal cells have revealed some surprises. Although this technology can be very powerful, only a few studies using this approach with developed multicellular systems have been reported for the study of chromatin behavior in situ.
Results
We established vectors and conditions to visualize tagged loci stably inserted in the Arabidopsis genome via GFP fused to a bacterial DNA-binding protein. Using this system, three-dimensional coordinates for tagged loci within nuclei from cells of a live plant can be directly determined with concomitant visualization of the position of the nucleolus. Chromosome polyploidization in epidermal cells at the elongation zone of the root in transgenic plants can be visualized in situ using this technique.
Conclusion
We have established that GFP fusion with DNA-binding proteins can be used in conjunction with concatameric binding-site arrays to track genomic loci in living Arabidopsis plants. It should now be feasible to study the mechanisms of organization and dynamics of chromatin in specific cell types during various times of plant development, taking advantage of the well developed genetic systems and resources available for Arabidopsis.
Background
The organization of chromatin within the eukaryotic nucleus has been a subject of great interest as it is likely to impact on important processes such as recombination [1] and epigenetic mechanisms of gene control [2]. Early observations by Rabl (cited by [3]) in plants suggested a preferential polarization of chromosomes, with their centromeres clustered at one end of the nuclear envelope (the apical side) and the telomeres at the opposite end (the basal side). More recently, evidence from fluorescence in situ hybridization (FISH) studies in different systems has suggested that various genomic loci can be found consistently in specific regions of the nucleus [4], and that subnuclear location can be correlated with gene activity [5]. Together with results from real-time measurements with living cells [6], these studies argue for a structured organization of chromosomes.
The present model we have of chromatin organization within the nucleus is a dynamic one, with 'territories' for each chromosome defined by their relative location to each other [7]. During the mitotic cycle, chromosomes are duplicated in the S phase and pulled to the two daughter cells during M phase. Chromosome territories are re-established after cell division, and once the cell is arrested in a long interphase, chromosome movement becomes restricted again [8]. Some nucleus-to-nucleus variations in the exact position of each locus relative to the rest of the genome can be expected [9]. As some random organization within each territory is possible [10], there is also likely to be time-dependent positional differences in the relative coordinates of each locus within a single nucleus [11]. These predicted nucleus-to-nucleus variations and the mobility of the established territories within the nuclei of various cell types in eukaryotes remain poorly defined in most systems.
So far, most in situ studies of gene positioning and changes in higher-order chromatin architecture have been carried out using FISH. This method can produce artifacts, however, because it requires the fixation and permeabilization of cells and denaturation of chromatin before hybridization with labeled single-strand probes [12]. It is also very difficult to perform kinetic studies with FISH to track the dynamics of chromatin movement in a single nucleus. Recent advances in the application of green fluorescent protein (GFP) as an in vivo tag of specific chromosomal regions promises to revolutionize our ability to observe chromatin-based processes in near real-time [13]. This technique, first established by Andrew Belmont and collaborators [14], entails the construction of a fusion protein between GFP and the DNA-binding domain (DBD) of a known heterologous transcription factor. The binding site for the DBD is multimerized into a concatameric array which is then inserted into the genome of animal or yeast cells. Expression of the GFP-DBD fusion protein results in fluorescent tagging of the concatamer in situ. This provides a 'beacon' that allows one to track the position of this region in the genome with high specificity and sensitivity. In yeast and animal cells, a single concatamer insert can be visualized with high resolution and fidelity, and the application of this technique has provided new insights on chromosome behavior [15,16,17].
These findings might, however, be true only for the few cell types and yeast strains studied so far, and may not be generally applicable to all eukaryotic cells. For example, even though chromosome territories showed only small movements during an observation period of several hours [6], they may show larger movements over longer time periods such as days. Also, the degree of nuclear order appears to depend on the particular cell type [18]. Earlier application of this technology in multicellular organisms involved extrachromosomal arrays of the lac operator created in C. elegans by co-injecting plasmids containing the operator array and GFP sequences along with other constructs [19]. The samples were fixed for immunological detection and the sizes as well as stability of the arrays studied remain undefined. Thus, application of that technique to study chromatin behavior may be rather limited. Very recently, Vazquez et al. [20] applied this technology in dissected Drosophila spermatocytes from animals that have integrated the lac operator arrays at cytogenetically defined genomic loci. Real-time monitoring of chromosome movement in this specialized cell type indicated that progression through G2 dramatically restricts the slower, long-range component of chromatin movement. This study provided the first direct evidence for cell-cycle control of chromatin movement and elegantly illustrated the power of this technology in transgenic organisms to facilitate the understanding of chromosome behavior in a developmental context.
The model plant Arabidopsis offers an excellent system in which to apply this new technology for the study of chromatin organization and dynamics. Aside from being easily transformable and having a well characterized genome [21], it is relatively easy to track cell types and individual cells. One can easily distinguish one cell from another, and individual cells can be tracked for a week or more [22]. In the present work, we have successfully adapted the 'GFP beacon' approach to create the necessary tools and transgenic lines for visualizing stably inserted concatameric sites in the genome in live Arabidopsis plants. This material and technology should open up new opportunities for characterizing the physical organization and movement of chromosomes in the nuclei of Arabidopsis.
Results and discussion
Vector construction and molecular characterization of transgenic Arabidopsis
To control accumulation of lac repressor (LacI) proteins in plant nuclei, a glucocorticoid-inducible gene expression system (pTA7002) was used for vector construction [23]. A gene encoding a plant-optimized GFP variant, mGFP5 [24], was placed in frame with a LacI-SV40 nuclear localization signal fusion (GFP-LacI/NLS) [25] and then subcloned into the vector pTA7002 under the control of the glucocorticoid receptor (GR)-responsive promoter near the left border. The resulting plasmid is designated as pEL700. At the other end of the pTA7002 vector, we inserted 256 copies of the lac operator sequence (LacOs, 10.1 kb) and designated this second vector pEL702 (Figure 1a). The entire plasmid of pEL702 encompasses about 25 kilobases (kb) and the size of the integrating region flanked by the right and left borders of the Ti plasmid is about 17 kb. The plasmid was designed such that the DNA between the right and left borders can be transferred into plant nuclei via Agrobacterium [26]. Thus, when stable transgenic Arabidopsis plants are treated with the synthetic glucocorticoid dexamethasone (Dex), the expressed fusion proteins would be able to localize to the integrated loci by association with LacOs. This inducible expression system allows controlled expression of a small amount of GFP-LacI/NLS protein for optimal signal-to-noise and to prevent aggregation of GFP fusion proteins in the nuclei as a result of high protein concentrations. As Belmont and co-workers [14,25] reported that the LacOs is unstable in a recombination-competent bacterial strain, we are concerned with the stability of LacOs in the Agrobacterium strain that is used to transform Arabidopsis. Thus, we examined the stability of LacOs in the Agrobacterium tumefaciens strain GV3101/MP90 by Southern blot analysis after transformation. We found that the LacOs may be unstable in Agrobacterium initially, as many colonies examined turned out to contain truncated arrays that are less than 10.1 kb, the size of the original LacOs in pEL702 (Figure 1b, lanes 1 and 2). We found, however, that colonies that do show the proper size array can be maintained for at least 2 days at 28°C in liquid culture without a significant reduction of the array size (Figure 1b, lane 3). Thus, we first monitored the size of LacOs in our Agrobacterium culture before transforming Arabidopsis with colonies carrying full-length or near full-length LacOs.
Figure 1.
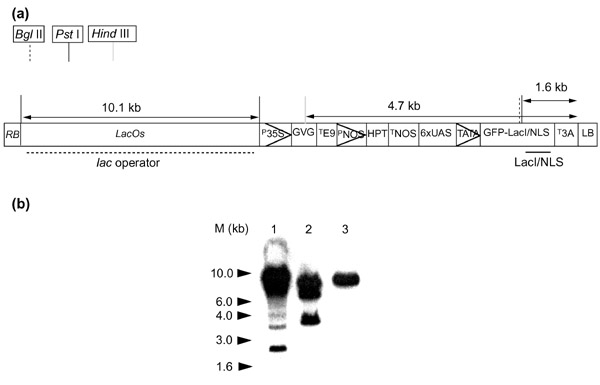
Stability of lac operator arrays in Agrobacteriumtumefaciens strain GV3101/MP90. (a) Schematic map of the construct, restriction sites and probe sites. RB, right border; p35S, cauliflower mosaic virus 35S promoter, LacOs, 256mer of lac operator arrays; GVG, glucocorticoid-binding domain-VP16 acidic activation domain-Gal4 DNA-binding domain; TE9, pea rbcs-E9 terminator; pNOS, nopaline synthase gene (NOS) promoter; HPT, hygromycin phosphotransferase; TNOS, NOS terminator; 6×UAS, 6×Gal4 UAS; TATA, TATA box; T3A, pea rbcs-3A terminator; LB, left border. (b) Three independent Agrobacterium colonies transformed with the binary vector, pEL702 (25 kb) which contains LacOs were cultured for 2 days in liquid culture. The vectors were extracted and subjected to Southern blot analysis after PstI digestion, which releases the LacOs from the vector. The blotted membrane was hybridized with random primer-labeled 40× lac operator arrays (thick dashed line in (a)). Kb, kilobase pairs; M, 1 kb size marker.
We observed low transformation efficiency of Arabidopsis with Agrobacterium carrying pEL702 under our transformation conditions (<10% of that with pEL700, data not shown). We speculate that a large T-DNA region (approximately 17 kb) and/or the presence of a large number of tandem repeats of LacOs in the T-DNA region may contribute to the low efficiency of gene transfer. After we obtained several independent lines that grew on selection plates containing hygromycin, we used Southern blot analysis to determine the size of the LacOs and the relative copy number of the inserted T-DNAs in the transformed lines. PstI digestion of genomic DNA from the transgenic lines, which should release the full-length LacOs (10.1 kb) from the integrated T-DNA inserts, was hybridized with a probe consisting of 40 copies of the lac operator sequence. Thus, in this analysis, each fragment should correspond to the size of an integrated lac operator array.
We detected several bands of different apparent sizes on the Southern blot (Figure 2), indicating that arrays with different numbers of lac operator sequences were integrated into the genome in each of the lines examined. Figure 2 shows data from analysis of two independently transformed lines. One line is designated EL702C and showed hybridizing bands with apparent sizes of approximately 10, 9 and 7 kb. The second is designated EL702E and DNA from this line showed a single band of approximately 4.5 kb that hybridized with the lac operator probe. Southern analysis with homozygous plants from the fifth-generation progenies of both lines also showed similar signal(s) (data not shown). These results indicated that the LacOs could be truncated before or during integration into the Arabidopsis genome, but the integrated genes were subsequently stable in the plant genome. For simplicity, we will refer to the integration site as a tagged locus.
Figure 2.
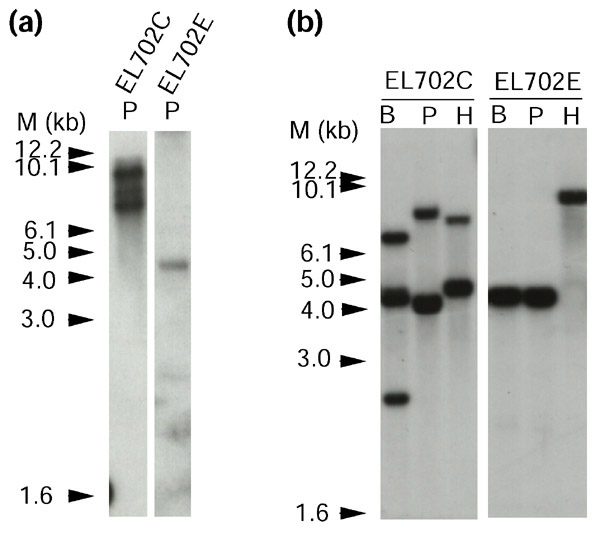
Southern blot analysis of EL702 transgenic Arabidopsis. Transformed Arabidopsis lines were characterized by Southern blot analysis and two independent lines identified. Line EL702C contains three copies of the inserts with near full-length LacOs (10.1 kb, approximately 250mer) incorporated into the genome. Line EL702E contains a single copy of LacOs with about half the number of binding sites (approximately 4.5 kb, approximately 120mer) incorporated into the genome. (a,b) Genomic DNA (20 μg) was digested with restriction enzymes and blotted onto nitrocellulose membranes after electrophoresis in a 0.7% agarose gel. The membranes were hybridized with (a) the lac operator and (b) the LacI/NLS probes described in Figure 1a. M, 1 kb size marker. B, BglII; P, PstI; H, HindIII.
The copy numbers of insertions in these lines were estimated with restriction digests of genomic DNA using BglII and HindIII in addition to PstI, followed by Southern blot analysis using the LacI/NLS fragment as a probe (Figures 1,2). Using this probe, we should detect fragments generated by enzymatic cleavage upstream of the LacI/NLS sequence in the inserts as well as restriction sites present at variable distances in the genomic DNA neighboring the left border of the insertions. Thus, each fragment detected in the Southern blot probably corresponds to a distinct copy of the transferred DNA that has integrated into the genome. Clustered integrations will, however, be more difficult to analyze in cases where a large number of insertions are present in the particular transgenic line and digestions with multiple restriction enzymes will be necessary to ascertain the structure of the insertion loci. Our results from digestions with three different restriction enzymes suggest that there are three insertions in the EL702C line and a single insertion in the EL702E line per haploid genome, consistent with the data obtained with the lac operator probe (Figure 2).
Dexamethasone-inducible expression of GFP fusion protein
Western blot analysis with anti-GFP polyclonal antibodies (Clonetech) was used to test inducibility of GFP-LacI/NLS in transgenic plants. We analyzed homozygous transgenic plants from two independent lines, EL702C and EL702E, as well as a line from our control vector, EL700S. This latter was transformed with pEL700, which is identical to pEL702 except for the absence of LacOs. As pEL700 does not contain the LacOs, the transgenic line EL700S can be used as a negative control for specific association of GFP-LacI/NLS with the LacOs in our experimental system. In this line, we expect the accumulation of GFP-LacI/NLS proteins in the nuclei after induction but specific binding of the fusion protein to chromatin should be absent. We treated transgenic seedlings with an excess concentration of Dex (30 μM) in 10 ml water to achieve uniform induction. Samples were collected at 0, 6, 12, 24, 48, 72 and 96 hours after the treatment and proteins were extracted in sodium dodecyl sulfate (SDS)-containing buffer for immunological detection of GFP fusion proteins.
We found that boiling the plant material in the extraction buffer resulted in loss of all GFP signals on our western blots. This appears to be a result of aggregation of GFP-LacI/NLS fusion proteins with some insoluble materials in plant cells as we do not encounter this problem with recombinant GFP-LacI/NLS produced in bacteria (data not shown). To circumvent this problem, we omitted the heating step in our protein sample preparation and anti-GFP cross-reacting proteins can be readily detected in our assay. Western blot signals were first detected 6 hours after the addition of Dex and increased over time in all three lines (Figure 3). After 48 hours, the detection levels did not change significantly and comparison between the three lines revealed that the Dex-inducible system in each line performed similarly with and without truncated LacOs, and minimal leakiness of the promoter was observed before induction. Although the predicted size of GFP-LacI/NLS fusion protein is 67 kDa, we detected several signals between 70 to 30 kDa in addition to a major species of around 81 kDa on our blots for all lines over time after 24 hours. The cause of this size heterogeneity of our GFP fusion protein is unclear at present.
Figure 3.
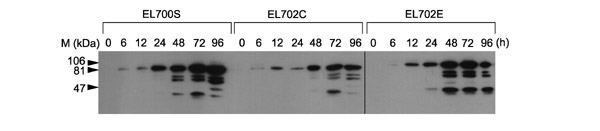
Kinetic study of the induced expression of mGFP5-LacI/NLS fusion proteins. Homozygous lines of EL700S (containing no LacOs), EL702C (containing three copies of LacOs) and EL702E (containing a single copy of about half-size LacOs) were tested. Seedlings germinated on 0.5× MS agar plates for 10 days were transferred to Petri dishes containing 30 μM dexamethasone (Dex) in 10 ml water. Three seedlings from each line were analyzed at 0, 6, 12, 24, 48, 72 and 96 h. The figure shows a composite of three independent blots with anti-GFP antibodies. Time of induction after Dex addition is shown on top of the figure in hours (h). M, protein size markers.
In situ detection of the tagged loci in transgenic plants
We then examined nucleus labeling by GFP after Dex treatment in homozygous transgenic plants from lines EL702C and E (with LacOs), and line EL700S (without LacOs) by fluorescence microscopy. Each line was collected at the same time points as for the western blot analysis, and GFP fluorescence was observed with appropriate filters. All lines examined showed fluorescence in nuclei 6 hours after the treatment (Figure 4, rows A). The intensities of GFP fluorescence in nuclei were increased by 48 hours and stabilized after 48 hours in all lines. These responses were correlated with the results of the western blot analysis. Both western blot analysis and microscopy studies confirmed that the LacOs does not interfere with glucocorticoid-inducible expression.
Figure 4.
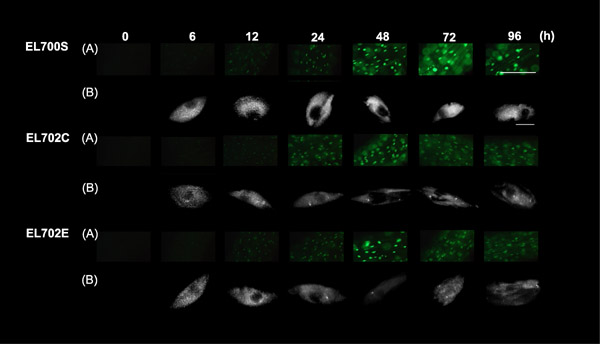
Kinetic study of mGFP5-LacI/NLS fluorescence in Dex-induced seedlings. Root epidermal cells of induced Arabidopsis seedlings were examined. Three seedlings from each homozygous line (EL700S, EL702C and EL702E) and three nuclei from each specimen were observed with fluorescence microscopy at 0, 6, 12, 24, 48, 72 and 96 h after Dex induction. One out of three sets of results is shown. (A) Fluorescence microscopic observation of epidermal cells. Scale bar = 100 μm. (B) Three-dimensional fluorescence microscopic observation of nuclei in A. Scale bar = 10 μm.
To investigate the response of the tagged loci, we observed each nucleus with three-dimensional fluorescence microscopy. If the system functions properly, we should be able to detect the tagged loci as bright spots above the GFP background signals in nuclei of the lines EL702C and E, and should not detect any spot in line EL700S because of the absence of LacOs. Bright spots were clearly observed above the diffused GFP fluorescence in nuclei observed 24 h after induction in lines EL702C and E, whereas significant fluorescence spots above the diffuse background signals were not observed in EL700S (Figure 4, rows B). The spots were detected with similar signal-to-background ratios until the end of the time course (96 h after induction) for lines EL702C and E, whereas line EL700S did not show any spots during this time course. The study also revealed that an array of about 120 copies of the lac operator sequence is sufficient to visualize LacOs in planta via GFP, as the size of the LacOs in the single inserted locus in line EL702E is approximately 4.5 kb (Figure 2).
Inhibition of chimeric GFP-LacI/NLS protein binding in situ to LacOs by IPTG
To verify if the spots in nuclei correspond to the tagged loci, we examined whether the detected subnuclear localization of the expressed GFP-LacI/NLS fusion protein is sensitive to isopropyl-β-D-thiogalactoside (IPTG). As the lac repressor variant that we used binds to the operator sites as dimers, we should be able to inhibit its binding to the LacOs of the tagged loci by adding IPTG [27]. If the binding is specific to the LacOs inserted in the genome, the observed fluorescence spots should be suppressed by the addition of IPTG.
We used homozygous plants from line EL702C, which carries three copies of the T-DNA insert, for this experiment. Forty microliters of solution containing 0.3 μM of Dex and 100 mM IPTG were dropped on each seedling on the culture plates. Guard cells in the stomata of cotyledons were observed 12 hours after induction in the presence or absence of IPTG and the results from observed nuclei were tabulated (Figure 5a,b). Without IPTG, we detected one spot in 15%, two spots in 65% and three spots in 20% of total observed nuclei (n = 25) among three seedlings. With IPTG, we detected no spots in 70% and one spot in 30% of total observed nuclei (n = 25) in three seedlings. We also tested protein accumulation levels in the specimens examined by western blot analysis. Similar levels of signals were detected with and without IPTG (Figure 5c), thus indicating that lac repressor binding is inhibited by IPTG without change in protein accumulation levels. This study shows that the observed fluorescence spots in lines EL702C and E indeed correspond to specific binding of the induced GFP-LacI/NLS fusion protein to the tagged loci. However, the relationship between the spot numbers and tagged loci is unclear at this point. For this purpose, we are currently determining the precise genomic location of the tagged loci and collecting the statistics of spot numbers for homozygous and heterozygous plants of line EL702C.
Figure 5.
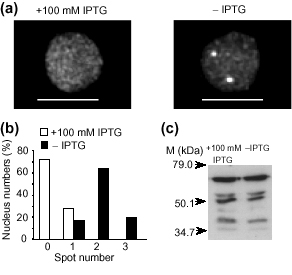
IPTG inhibition of mGFP5-LacI/NLS binding in nucleus. Homozygous plants of line EL702C were tested. Seedlings germinated on 0.5× MS agar plates for 10 days were transplanted onto 0.5× MS agar plates with and without 0.3 μM Dex. (a) Typical three-dimensional fluorescence images of nuclei from each treatment. (b) Graph of the observed number of spots and their percentages among total observed nuclei. (c) Levels of protein accumulation measured by western blot analysis using anti-GFP antibodies. M, protein size markers. Scale bar = 5 μm.
Detection and relational mapping of tagged loci configuration
As plants contain chloroplasts in leaf and stem cells and many autofluorescent compounds are localized in cell walls, a signal-to-background ratio for GFP detection is probably lower than in animal systems. Thus, higher levels of accumulation of GFP-LacI/NLS may be required to detect the loci in our system. Under this condition, we found that we could obtain spatial information for subnuclear structures with excess unbound GFP-LacI/NLS fusion proteins. The nuclear contour as well as the shape of the nucleolus, for which GFP-LacI/NLS has relatively low accessibility and which thus appears as an opaque object within the nucleus, can be captured along with the tagged loci positions by single channel excitation. A guard-cell nucleus from homozygous plants of line EL702C is shown (Figure 6). The data can be converted to a three-dimensional model shown for the detected spots (shown as red objects) and the nucleolus (shown as a blue object) within the nucleus (outline shown in green wire-frame) (Figure 6b). The distance between the three spots can also be easily determined with the existing program of our system (Figure 6c).
Figure 6.
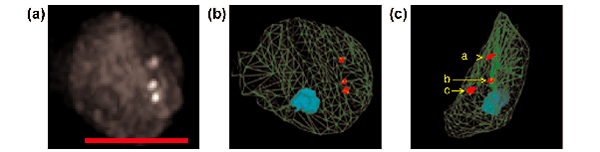
Measurement of the configuration of tagged loci in homozygous plants from line EL702C. (a) A stacked image from a guard cell nucleus that contains three GFP spots was created. Scale bar = 5 μm. (b) Outline of nucleus (green wire-frame), position of the nucleolus (blue solid) and GFP fluorescence spots (red solids) were extracted from the stacked images, and a three-dimensional model was created. (c) The three-dimensional model was used to rotate the image by 90° in the y-axis, and the distance between the three GFP spots (a, b and c) was measured. Distance dab = 0.88 μm, dbc= 1.13 μm, dac = 1.58 μm.
Comparison of tagged loci between guard cells and root epidermal cells
Plant organs are composed of ordered collections of various cell types that differ in their shape, size, function and DNA content. The positive correlation observed in most cells between cell volume and the degree of polyploidy indicates that the nuclear DNA content may have a key role in controlling cell growth [28]. In Arabidopsis, the ploidy level varies from 2C (guard cells, sepal and petal epidermal cells) to 64C (leaf trichomes) [29]. However, the mechanism of endocycle control in plants remains poorly understood. As an application of our chromatin-tagging system, we show here that cell-specific polyploidy can be observed directly with this technology without measuring nuclear DNA content.
We used the homozygous plants of line EL702E, which contain a single tagged locus, for this experiment. GFP signals observed in a cotyledon guard cell (Figure 7a) and an elongated root epidermal cell (Figure 7b) of a 10-day-old seedling were compared using three-dimensional projections of their nuclei after Dex induction. In the guard cell, where the shape of its nucleus is usually flat or spherical, we often detected one spot, which suggests that the tagged site is frequently paired with its homolog. In contrast, in the elongated root epidermal cells where the nucleus usually elongates at the two opposite sides, we often detected multiple fluorescence spots (five spots for the root nucleus shown in Figure 7), which indicates the polyploid nature of the observed nuclei. Upon further refinement of this technology, one may be able to study the dynamics and regulation of endoreduplication in plant cells that contribute to cell-specific ploidy differences.
Figure 7.
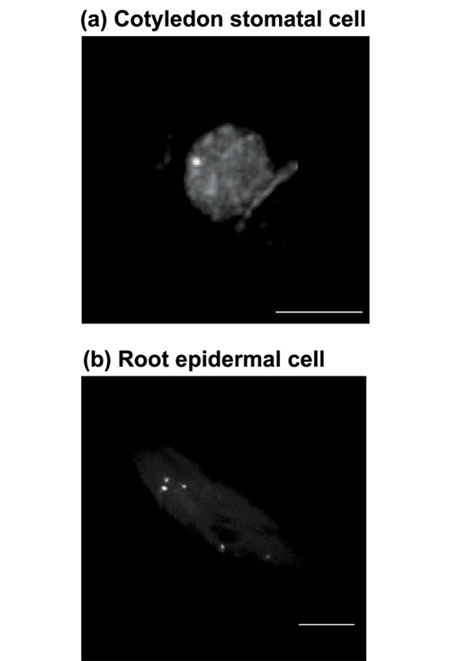
Detection of tagged loci in different cell types. Homozygous seedlings with a single tagged locus from the transgenic line EL702E were analyzed by DeltaVision restoration microscopy as described in the Materials and methods section and stacked images of nuclei were created from different cell types after induction with Dex. Representative images from (a) a guard cell nucleus and (b) an elongated epidermal cell from the seedlings are shown. Scale bar = 5 μm.
Conclusions
We have established transgenic plants that enable us to observe specific tagged genomic loci together with the nucleolar location in cells of a living plant without any fixation. Using this system, we have visualized GFP-tagged chromatin in different cell types in living Arabidopsis plants. GFP-tagged chromosome observation has been applied in several studies involving yeast, cultured animal cells and, more recently, transgenic Drosophila spermatocytes. These studies have revealed new principles of chromosome dynamics and organization. However, the relationship between chromatin structure and cell types and developmental stages is an important question that remains unanswered. The intact Arabidopsis seedlings that were used for microscopy under our conditions remain viable after transplanting to soil from the slides. Thus, it should be possible to monitor the same plants continuously for up to several weeks. In future, by marking individual cells with visual tags it should be possible to follow changes in nuclear shape as well as relative subnuclear position of the nucleolus and specific tagged genomic loci over long times-scales of the order of weeks. These types of studies may reveal new aspects of chromatin organization in relationship to developmental timing.
Materials and methods
Gene constructions
LacI/NLS (nuclear localization signal from SV40 large T-antigen) [25] was fused to a plant-optimized GFP sequence, mGFP5 [24], and designated mGFP5-LacI/NLS. mGFP5-LacI/NLS was inserted into a glucocorticoid-inducible expression vector pTA7002 [23] and the resulting vector designated pEL700. lac operator array, LacOs (256-mer, 10.1 kb) [25] was then inserted into pEL700 and the resulting vector designated pEL702.
Agrobacterium binary vector preparation and Southern blot analysis
After the Agrobacterium culture (2 ml) was collected by centrifugation in a microfuge, the pellet was resuspended in a solution of 50 mM glucose, 25 mM Tris and 10 mM ethylene aminotetraacetic acid (pH8.0). Cell lysis solution (0.2 M sodium hydroxide and 1.0% (w/v) sodium dodecyl sulfate (SDS)), phenol and neutralization solution (3 M sodium acetate (pH5.2)) were then added to the resuspended cells in order, and the solution was vortexed for a few seconds. After centrifugation, the upper-phase solution was transferred to a new tube, and the DNA precipitated with ethanol. The DNA was dissolved in water and digested with PstI, and the digested DNA was then separated in a 0.7% (w/v) agarose gel by electrophoresis. The DNA was transferred to a membrane (Zeta-probe GT genomic tested blotting membrane, Bio-Rad, CA) with 0.4 M sodium hydroxide after treatment with 0.25 N hydrochloric acid as suggested by the manufacturer. The membrane was prehybridized for 0.5 h at 65°C, hybridized for 1 h at 65°C with random primer-labeled lac operator concatamer (40 copies) probe in ExpressHyb hybridization solution (Clontech Laboratories, CA), and then washed at 60°C in washing solution (0.1% (w/v) SDS, 15 mM sodium chloride, 1.5 mM sodium citrate).
Transformation of Arabidopsis thaliana and Southern blot analysis
Arabidopsis thaliana (ecotype Columbia) plants were transformed by dipping the influorescence of flowering plants into transformation solution (5% (w/v) sucrose and 0.05% (v/v) Silwet L-77) with Agrobacterium tumefaciens strain GV3101/MP90 transformed with pEL700 or pEL702. To transform A. thaliana with the Agrobacterium carrying pEL702, independent Agrobacterium colonies were first grown for 12 h in several different culture tubes with 2 ml culture medium. One milliliter from each of the cultures was allocated to measure the size of the LacOs. The LacOs size in Agrobacterium was analyzed by Southern blots with a probe of 40 copies of the lac operator. The cultures in which full-length or near full-length LacOs was found to be maintained were then increased to 500 ml and incubated at 28°C for 12 h before use for Arabidopsis transformation. LacOs size and copy numbers were assayed by hybridization of two gene-specific probes separately to genomic DNA extracted from transgenic Arabidopsis. The first probe was a random primer-labeled 40 copies of the lac operator. The second was the random primer-labeled LacI/NLS. Membranes were prehybridized for 0.5 h at 65°C, hybridized for 1 h at 65°C in ExpressHyb hybridization solution and washed at 60°C in washing solution.
Western blot analysis
Samples for western blot analysis were ground in liquid nitrogen. Extraction buffer (10%(v/v) glycerol, 10% (v/v) β-mercaptoethanol, 5% (w/v) SDS) was added immediately to the samples. After 5 min incubation on ice, the mixture was centrifuged in a microfuge to remove insoluble material. Ten microliters of non-heated supernatant was used for western blot analysis and Coomassie Brilliant Blue (CBB) staining after SDS-polyacrylamide gel electrophoresis. Anti-GFP polyclonal antibodies (Clontech) were used at 1/5,000 dilution to detect mGFP5-LacI/NLS proteins. Protein amounts in each sample were qualitatively compared in CBB dye staining SDS-polyacrylamide gel and normalized before use for western blot analysis.
Growth of Arabidopsis thaliana
Surface-sterilized seeds were germinated on 0.5× MS agar plates (2.1 g/l Murashige and Skoog salts (Invitrogen, CA) and 0.7% bacto-agar, pH 6.0) containing 37 μg/l of hygromycin for 2-3 weeks in a plant growth chamber with 15 h light and 9 h dark at 22°C. Healthily growing plants were transplanted to the soil in small pots and grown until seeds were obtained in the plant growth chamber.
Sample preparation for microscopy
Surface-sterilized seeds were germinated on 0.5× MS agar plates. One- to two-week-old seedlings were transferred to fresh 0.5× MS agar plates and 40 μl of a 0.3 μM dexamethasone (Dex) solution was then dropped on each seedling. After 10-12 h, the seedlings were placed in between two coverslips with water. The coverslips were then placed on the microscope stage of our imaging set-up. When time course experiments were performed, 1- to 2-week-old seedlings were placed in Petri dishes (60 × 15 mm) and 10 ml water containing 30 μM Dex was added. The dishes were slowly shaken until seedlings were collected. Three seedlings from each time point were collected for western blot analyses.
Fluorescence microscopy
A DeltaVision restoration microscope system (Applied Precision, WA) equipped with a TE200 microscope (Nikon) was used to observe nuclei in induced seedlings. Forty images at 0.2 μm Z-steps were collected using a Nikon PlanApo 60×, 1.2 N.A. water-immersion objective lens. The exposure times were 0.3-3 sec. The filters used are exciter: 436 nm/10 nm, emitter: 470 nm/30 nm and JP4 beamsplitter (Chroma, VT). When larger areas of root epidermal tissues were observed, a TE200 microscope equipped with a Nikon PlanFluor 20×, 0.45 N.A. objective lens and a 3CCD camera (DEI-750, Optronics, CA) was used.
Image processing
The stacked images of nuclei were analyzed by softWoRx software (Applied Precision) on an Octane Workstation (Silicon Graphics, CA). The images were then processed by Adobe Photoshop 5.5 on a PowerMac G4 computer for the final images.
Acknowledgments
Acknowledgements
This work is supported by a grant from the Plant Genome Research Program of NSF (#9872636) and in part also by a grant from the Charles and Johanna Busch Memorial Fund at Rutgers University (#6-49142) to E.L. We would like to thank Jim Haseloff, Aaron Straight and Nam-Hai Chua for constructs. In addition, we gratefully acknowledge advice on fluorescence microscopy instrumentation by J. Sedat (UCSF) during the early phase of this work, and sharing of preprints and constructive suggestions by Andrew Belmont.
References
- Cobbe N, Heck MMS. SMCs in the world of chromosome biology - from prokaryotes to higher eukaryotes. J Struct Biol. 2000;129:123–143. doi: 10.1006/jsbi.2000.4255. [DOI] [PubMed] [Google Scholar]
- Cavalli G, Paro R. Epigenetic inheritance of active chromatin after removal of the main transactivator. Science. 1999;286:955–958. doi: 10.1126/science.286.5441.955. [DOI] [PubMed] [Google Scholar]
- Franklin AE, Cande WZ. Nuclear organization and chromosome segregation. Plant Cell. 1999;11:523–534. doi: 10.1105/tpc.11.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Dernburg AF, Harmon B, Agard DA, Sedat JW. Specific interactions of chromatin with the nuclear envelope: positional determination within the nucleus in Drosophila melanogaster. Mol Biol Cell. 1996;7:825–842. doi: 10.1091/mbc.7.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz A, Lampel S, Nickolenko JE, Bradl J, Benner A, Zirbel RM, Cremer T, Lichter P. Active and inactive genes localize preferentially in the periphery of chromosome territories. J Cell Biol. 1996;135:1195–1205. doi: 10.1083/jcb.135.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfleth H, Edelmann P, Zink D, Cremer T, Cremer C. Quantitative motion analysis of subchromosomal foci in living cells using four-dimensional microscopy. Biophys J. 1999;77:2871–2886. doi: 10.1016/S0006-3495(99)77119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink D, Cremer T, Saffrich R, Fischer R, Trendelenburg MF, Ansorge W, Stelzer EH. Structure and dynamics of human interphase chromosome territories in vivo . Hum Genet. 1998;102:241–251. doi: 10.1007/s004390050686. [DOI] [PubMed] [Google Scholar]
- Manders EM, Kimura H, Cook PR. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. J Cell Biol. 1999;144:813–821. doi: 10.1083/jcb.144.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcobia I, Dilao R, Parreira L. Spatial associations of centromeres in the nuclei of hematopoietic cells: evidence for cell-type-specific organizational patterns. Blood. 2000;95:1608–1615. [PubMed] [Google Scholar]
- Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Rykowski MC. Direct evidence for interphase chromosome movement during the mid-blastula transition in Drosophila. Curr Biol. 2000;10:285–288. doi: 10.1016/s0960-9822(00)00360-2. [DOI] [PubMed] [Google Scholar]
- Kozubek S, Lukasova E, Amrichova J, Kozubek M, Liskova A, Slotova J. Influence of cell fixation on chromatin topography. Analyt Biochem. 2000;282:29–38. doi: 10.1006/abio.2000.4538. [DOI] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- Belmont AS, Straight AF. In vivo visualization of chromosomes using lac operator-repressor binding. Trends Cell Biol. 1998;8:121–124. doi: 10.1016/s0962-8924(97)01211-7. [DOI] [PubMed] [Google Scholar]
- Tumbar T, Belmont AS. Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat Cell Biol. 2001;3:134–139. doi: 10.1038/35055033. [DOI] [PubMed] [Google Scholar]
- Tatebe H, Goshima G, Takeda K, Nakagawa T, Kinoshita K, Yanagida M. Fission yeast living mitosis visualized by GFP-tagged gene products. Micron. 2001;32:67–74. doi: 10.1016/s0968-4328(00)00023-8. [DOI] [PubMed] [Google Scholar]
- Aragon-Alcaide L, Strunnikov AV. Functional dissection of in vivo interchromosome association in Saccharomyces cerevisiae. Nat Cell Biol. 2000;2:812–818. doi: 10.1038/35041055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandley AC, Speed RM, Leitch AR. Different distributions of homologous chromosomes in adult human Sertoli cells and in lymphocytes signify nuclear differentiation. J Cell Sci. 1996;109:773–776. doi: 10.1242/jcs.109.4.773. [DOI] [PubMed] [Google Scholar]
- Carmi I, Kopczynski JB, Meyer BJ. The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature. 1998;396:168–173. doi: 10.1038/24164. [DOI] [PubMed] [Google Scholar]
- Vazquez J, Belmont AS, Sedat JW. Multiple regimes of constrained chromosome motion are regulated during interphase in the Drosophila nucleus. Curr Biol. 2001;11:1227–1239. doi: 10.1016/s0960-9822(01)00390-6. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Geisler M, Nadeau J, Sack FD. Oriented asymmetric divisions that generate the stomatal spacing pattern in Arabidopsis are disrupted by the too many mouths mutation. Plant Cell. 2000;12:2075–2086. doi: 10.1105/tpc.12.11.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Chua NH. A glucocorticoid-mediated transcriptional induction system in transgenic plants. Plant J. 1997;11:605–612. doi: 10.1046/j.1365-313x.1997.11030605.x. [DOI] [PubMed] [Google Scholar]
- Haseloff J, Siemering KR, Prasher DC, Hodge S. Removal of a cryptic intron and subcellular localization of Green Fluorescent Protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci USA. 1997;94:2122–2127. doi: 10.1073/pnas.94.6.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JS, Citovsky V. Agrobacterium-plant cell DNA transport: Have virulence proteins, will travel. Plant Cell. 1996;8:1699–1710. doi: 10.1105/tpc.8.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Matthews KS. Deletion of lactose repressor carboxyl-terminal domain affects tetramer formation. J Biol Chem. 1992;267:13843–13850. [PubMed] [Google Scholar]
- Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell. 2001;105:297–306. doi: 10.1016/s0092-8674(01)00334-8. [DOI] [PubMed] [Google Scholar]
- Melaragno JE, Mehrotra B, Coleman AW. Relationship between endopolyploidy and cell size in epidermal tissue of Arabidopsis. Plant Cell. 1993;5:1661–1668. doi: 10.1105/tpc.5.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]