

Abstract
Heat‐shock factor 1 (HSF1) is the master transcription factor that regulates the response to proteotoxic stress by controlling the transcription of many stress‐responsive genes including the heat‐shock proteins. Here, we show a novel molecular mechanism controlling the activation of HSF1. We demonstrate that transglutaminase type 2 (TG2), dependent on its protein disulphide isomerase activity, triggers the trimerization and activation of HSF1 regulating adaptation to stress and proteostasis impairment. In particular, we find that TG2 loss of function correlates with a defect in the nuclear translocation of HSF1 and in its DNA‐binding ability to the HSP70 promoter. We show that the inhibition of TG2 restores the unbalance in HSF1‐HSP70 pathway in cystic fibrosis (CF), a human disorder characterized by deregulation of proteostasis. The absence of TG2 leads to an increase of about 40% in CFTR function in a new experimental CF mouse model lacking TG2. Altogether, these results indicate that TG2 plays a key role in the regulation of cellular proteostasis under stressful cellular conditions through the modulation of the heat‐shock response.
Keywords: Cystic fibrosis, HSF1, HSP70, proteostasis, TG2
Subject Categories: Molecular Biology of Disease; Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control
Introduction
Transglutaminase type 2 (TG2) is a multifunctional, ubiquitously expressed member of the TG family that catalyses post‐translational modifications of proteins through both Ca2+‐dependent and Ca2+‐independent reactions 1. Several unique features, including its ubiquitous expression, widespread localization, binding to and hydrolysis of guanine nucleotides, distinguish TG2 from the other transglutaminases 2. Indeed, in addition to its crosslinking activity, TG2 might also act as a GTP‐binding protein that mediates intracellular signalling by coupling the alpha‐1 beta‐adrenergic receptor to phospholipase C‐gamma 1 3, 4. Substantial evidence indicates that under physiological circumstances the enzyme may also act as protein disulphide isomerase (PDI). In the last few years, it has been established that TG2 switches its 3D structure from a nucleotide‐bound “closed” to the transamidation/PDI prone “open” conformation 5, 6. These 3D changes influence the interaction of TG2 with multiple substrates or binding partners that are essential to carry out its biological functions 7. The peculiar biochemistry of this enzyme as well as its capacity to interact with the main proteins involved in the regulation of proteostasis suggests that TG2 plays a key role in this process. For example, among the TG2 intracellular partners, many are molecular chaperones, such as the heat‐shock proteins HSP70, HSP27, HSP90 as well as co‐chaperone from the DNAjs and the BAG families 7, 8, 9, 10. Indeed, recent compelling evidence places TG2 within the regulation of main pathways controlling the proteome homeostasis as autophagy, proteasome and exosomes 11, 12, 13, 14. In keeping with this, there is a vast literature describing the deregulation of this enzyme during the pathogenesis of major human diseases (neurodegenerative, liver and respiratory disorders as well as cancer) 15, 16, 17 in which there is a deregulated proteostasis that can be improved by modulators of TG2 activity 18, 19. In spite of differences related to cell‐ and tissue‐specific disease context, these disorders share common features of unbalanced cell adaptation to either cell autonomous or environmental stress. Thus, TG2 can be viewed as a dynamic platform that aims at restoring cellular imbalances. However, it remains elusive through which mechanisms the TG2‐mediated capability of fighting stress is disrupted, making TG2 a harmful, instead of beneficial, player in disease pathogenesis. Several TG2 inhibitors can ameliorate disease phenotype, either in pre‐clinical or in clinical settings 20. One typical example is cysteamine, a small molecule with pleiotropic functions, among which the capability of inhibiting TG2 transamidating activity, with proved to have beneficial pre‐clinical or clinical effects in neurodegenerative disorders 15, 21, fibrosis 16 as well as in cystic fibrosis (CF) 17. Cysteamine is capable of controlling the consequences of TG2 overactivation in CF, improving the trafficking of the most common misfolded F508del CFTR mutant 17, 22, 23, 24, 25.
Here, we demonstrate that TG2 regulates proteostasis and the general cellular adaptation to stress. By combining in vivo approaches in transgenic mice with in vitro studies, we demonstrate that TG2, by acting as a PDI, is able to activate HSF1, the master transcriptional regulator of the stress‐responsive genes. We show that TG2 controls the heat‐shock response by modulating the expression of the stress‐inducible chaperone HSP70. Moreover, we report that cysteamine inhibits the PDI activity of TG2, restoring the proteostasis imbalance in CF, a finding that might explain its superior activity as a therapeutic agent.
Results
HSP70 expression is dependent on TG2 both in vivo and in vitro
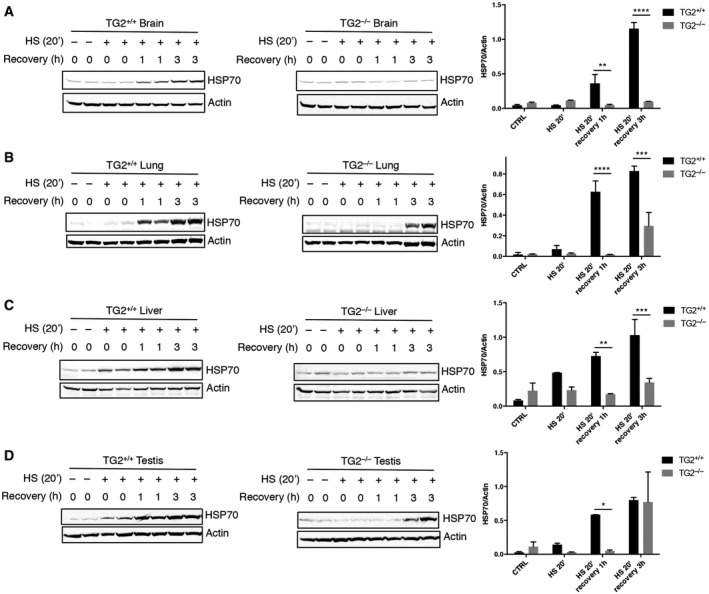
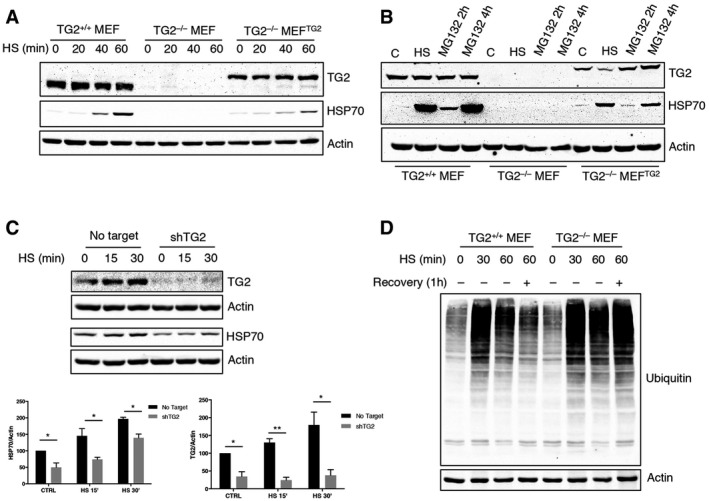
We have previously demonstrated that TG2 is involved in the assembly and clearance of ubiquitinated aggregates 12. In addition, the enzyme interacts with several molecular chaperones and proteins involved in the folding machinery 7 including the stress‐inducible form of HSP70 (HSPA1A). Based on these findings, we evaluated the hypothesis of whether TG2 might influence proteostasis by modulating the chaperone homeostasis. To this aim, we analysed the effect of TG2 ablation on HSP70 induction. First, mice that were either wild type or knockout for TG2 (TG2+/+ and TG2−/− mice) were exposed to hyperthermic stress at 42°C for 20 min to stimulate heat‐shock response and HSP70 induction. HSP70 protein expression was examined in different tissues (Fig 1) after heat shock (HS) or 1 and 3 h after recovery at room temperature. Immunoblotting revealed that mice lacking TG2 had a significant defect in HSP70 induction in all the examined organs (Fig 1A–D). We next confirmed this result in vitro, using mouse embryonic fibroblasts (MEFs) from TG2+/+ and TG2−/− mice. To validate the in vivo data, TG2+/+ and TG2−/− MEFs, as well as TG2−/− MEFs stably reconstituted with the unmutated TG2 (TG2−/− MEFTG2), were either exposed to heat shock for 20, 40, 60 min (Fig 2A) or treated with the proteasome inhibitor MG132 (Fig 2B). Both treatments are known to cause the accumulation of misfolded proteins and the consequent induction of HSP70 26, 27, 28. As shown in Fig 2A and B, in the absence of TG2, no induction of HSP70 occurred after HS or proteasome inhibition. Interestingly, the reintroduction of human TG2 into TG2−/− MEF was sufficient to rescue HSP70 induction. Similarly, shRNA‐mediated silencing of TG2 in TG2+/+ MEF significantly reduced the induction of HSP70 by heat shock (Fig 2C). Commensurate with a key role for TG2 and HSP70 in the recovery from stress, TG2+/+ MEF exhibited a transient increase in protein ubiquitination post‐HS, suggesting successful clearance of damaged proteins, while TG2−/− MEF showed a stable elevation of protein ubiquitination (Fig 2D). Moreover, in contrast to TG2+/+ MEF, the lack of HSP70 induction in TG2−/− MEF correlated to HS‐dependent apoptosis as highlighted by the activation of caspase 3 (Fig EV1).
Figure 1. The ablation of TG2 leads to a defect in the induction of HSP70 protein expression in mice tissues after exposure to a proteotoxic stress (HS).
-
A–DWestern blot and densitometric analysis of HSP70 protein levels in brain (A), lung (B), liver (C) and testis (D) tissues from TG2+/+ and TG2−/− mice after exposure to heat shock at 42°C for 20 min. HSP70 expression was also monitored after 1 and 3 h of recovery at 37°C. Actin was used as loading control.
Figure 2. HSP70 induction is dependent on TG2 both after heat shock and after proteasome inhibition.
-
A, BWestern blot analysis of HSP70 and TG2 protein levels in TG2+/+ MEF, TG2−/− MEF and TG2−/− MEFTG2 after heat shock for 20, 40 and 60 min (A) or proteasome inhibition with MG132 for 2 and 4 h (B). Actin was used as loading control.
-
CWestern blot and densitometric analysis of HSP70 and TG2 in TG2+/+ MEF silenced for TG2 (shTG2) after exposure to heat shock. Actin was used as loading control.
-
DWestern blot analysis of ubiquitinated proteins after exposure of TG2+/+ and TG2−/− MEF to a heat shock for 30 and 60 min or 60 min followed by 1 h of recovery to 37°C. Actin was used as loading control.
Figure EV1. Long‐term exposure to HS fails to induce HSP70 in TG2−/− MEF, leading to cell death.
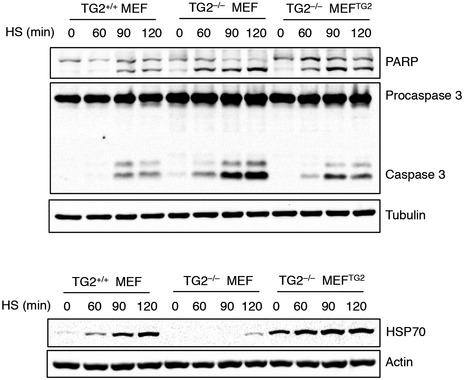
Western blot analysis of cleaved caspase 3 and PARP, as markers for apoptotic cell death induction, and HSP70 in protein extracts of TG2+/+ and TG2−/− MEFs as well as TG2−/− MEFTG2 after heat shock for 60, 90 and 120 min. Tubulin and actin were used as loading control.
TG2 mediates HSF1 activation
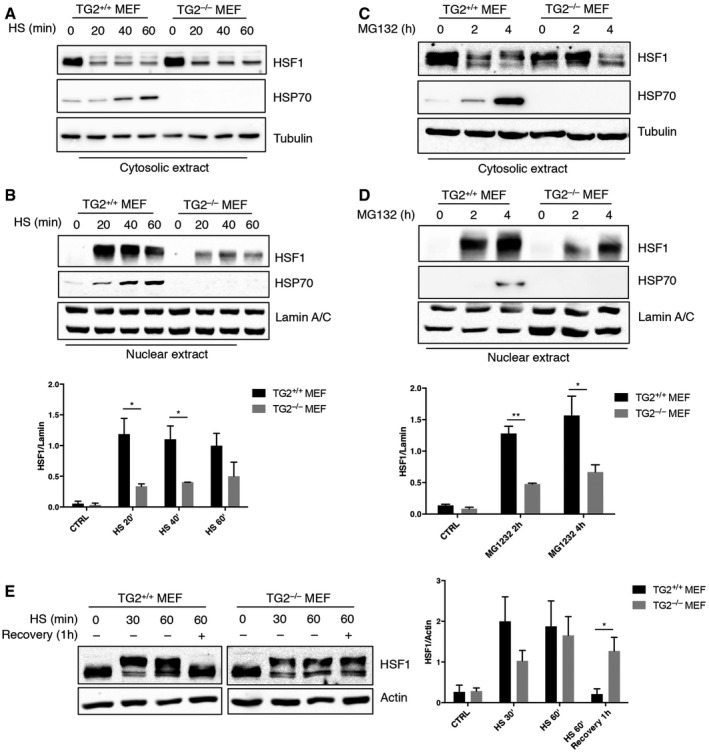
In order to define the molecular mechanism regulating the TG2‐dependent induction of HSP70, we focused our attention on the heat‐shock factor 1 (HSF1), the main transcription factor involved in HSP70 expression. Activation of HSF1, associated with hyperphosphorylation, requires a multi‐step process that includes the translocation into the nucleus, the transition from a monomeric to a trimeric form and the binding to target gene promoters 29, 30, 31. To verify whether the lack of HSP70 induction, in absence of TG2, was due to a defect in the HSF1 activation, we first analysed the nuclear translocation of the transcriptional factor after HS (Fig 3A and B) and proteasome inhibition with MG132 (Fig 3C and D), two cellular stresses known to activate the transcriptional response mediated by HSF1. As expected 32, 33, 34, in TG2+/+ MEF HSF1 was only present in the cytosol in basal conditions and rapidly translocated into the nucleus after heat shock or MG132 addition leading to HSP70 expression (Fig 3A–D). By contrast, the lack of HSP70 induction in TG2−/− MEF was correlated to a significant decrease in the nuclear translocation of HSF1 (Fig 3B and D). Interestingly, the amount of hyperphosphorylated HSF1, shortly after heat shock, was reduced in TG2−/− MEF as compared to TG2+/+ MEF (Fig 3E). Moreover, while HSF1 hyperphosphorylation was transient in TG2+/+ MEF, it persisted also during the recovery phase in TG2−/− MEF. These results indicate that TG2 is required for the optimal activation of HSF1.
Figure 3. TG2 protein levels affect HSF1 nuclear translocation and phosphorylation.
-
A–DWestern blot analysis of HSF1 nuclear translocation and subsequent HSP70 expression in cytosolic and nuclear fractions of TG2+/+ and TG2−/− MEF treated with heat shock for 20, 40 and 60 min (A, B) and MG132 for 2 and 4 h (C, D). Tubulin was used as loading control for cytosolic extract. Lamin A/C was used as loading control for nuclear extract. Densitometric analysis of nuclear HSF1 amount after HS and MG132 in TG2+/+ and TG2−/− MEF.
-
EWestern blot analysis of HSF1 expression in whole‐cell lysates of TG2+/+ and TG2−/− MEF after 30 and 60 min of heat shock or 60 min followed by 1 h of recovery to 37°C. The hyperphosphorylation‐dependent shift of HSF1 can be detected after HS. Actin was used as loading control. Densitometric analysis of the HSF1 hyperphosphorylated form after HS in TG2+/+ and TG2−/− MEF.
To characterize how TG2 could modulate HSF1 activation, we first verified whether the two proteins could interact. Co‐immunoprecipitation analyses revealed that TG2 interacts with HSF1 just after HS (Fig 4A and B). This result was also confirmed by immunofluorescence microscopy showing the co‐localization of TG2 and HSF1 in the nucleus after HS (Fig 4C and D).
Figure 4. TG2 interacts with HSF1 in the nuclear compartment of cells exposed to heat shock.
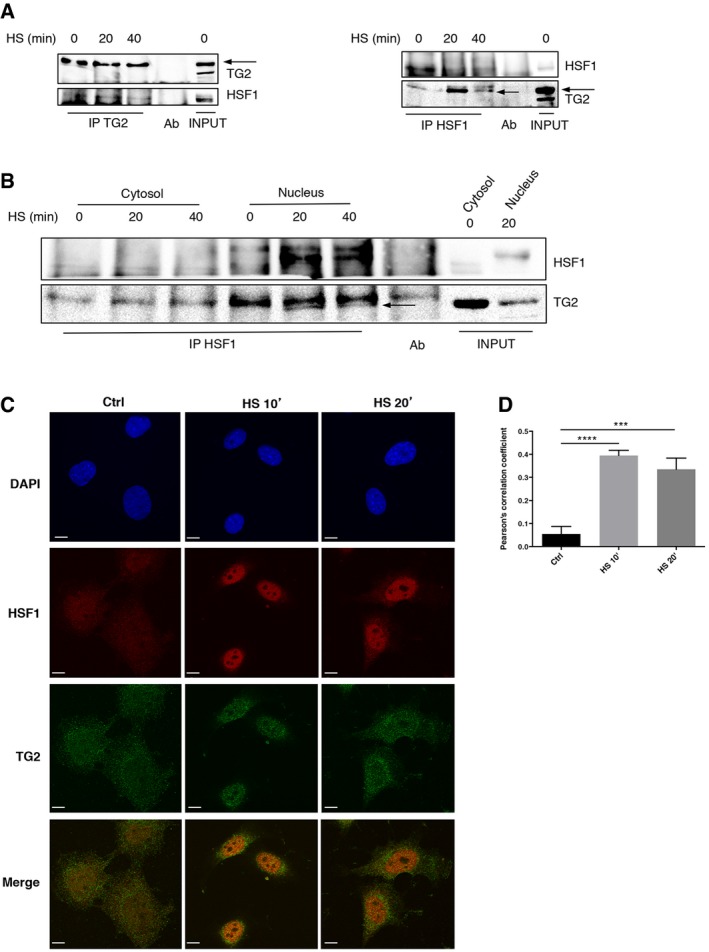
- TG2−/− MEFTG2 cell lysates were subjected to immunoprecipitation for TG2 (left) and HSF1 (right) upon heat shock for 20 and 40 min. Immuno‐ and co‐immunoprecipitated proteins were separated by SDS–PAGE and blotted using the indicated antibodies. Whole‐cell lysates (INPUT) were used as protein control.
- Nuclear and cytosolic extracts, obtained from TG2−/− MEFTG2 exposed to heat shock for 20 and 40 min, were subjected to immunoprecipitation of HSF1. Immuno‐ and co‐immunoprecipitated proteins were separated by SDS–PAGE and blotted using the indicated antibodies. Whole‐cell lysates (INPUT) were used as protein control. The arrows indicate the TG2‐specific band.
- Immunofluorescence analysis of TG2 (green) and HSF1 (red) co‐localization in TG2+/+ MEF after exposure to thermal stress for 10 and 20 min. Scale bar 10 μm.
- Histogram showing Pearson's correlation coefficient to evaluate co‐localization of TG2 and HSF1.
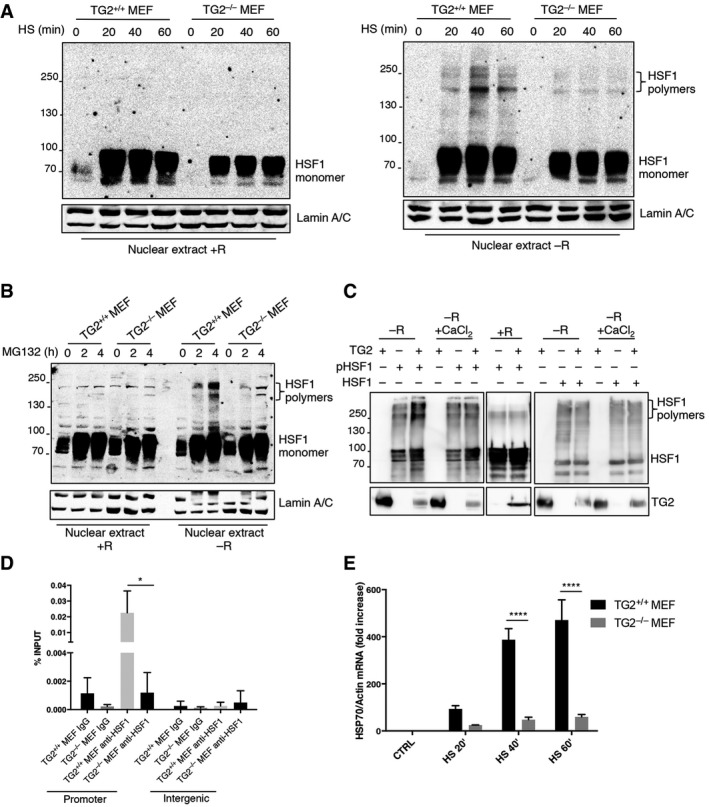
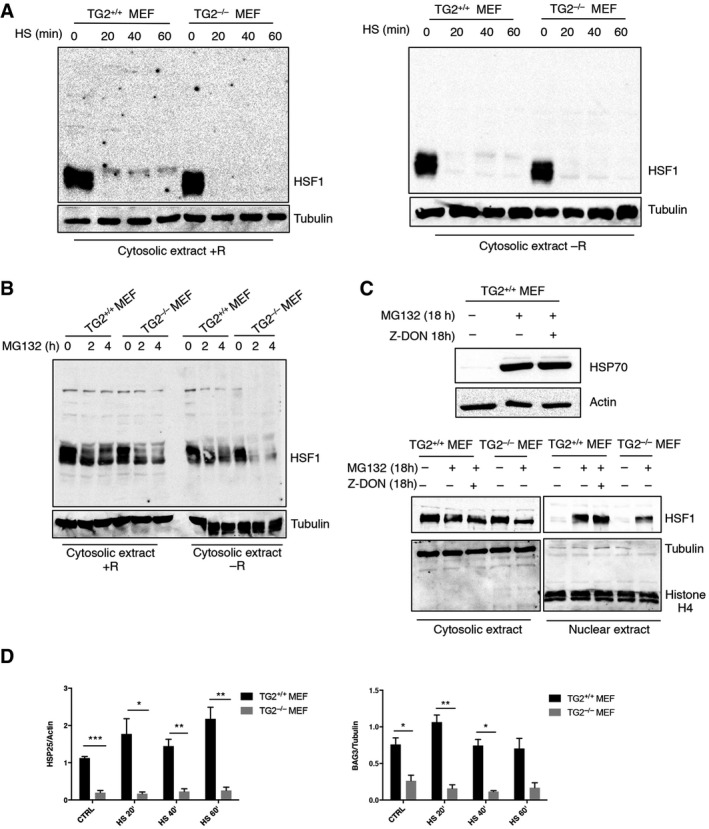
To bind DNA and transactivate target genes, HSF1 must trimerize following the formation of disulphide bonds between cysteine residues 35, 36. The formation of three intermolecular S‐S bonds between two cysteine residues (Cys36 and Cys103) is essential for HSF1 trimerization and DNA binding. However, so far the mechanism by which Cys36 and Cys103 form this intermolecular S‐S bond was elusive. Considering that TG2 interacts with HSF1 after HS, we asked whether the enzyme, through its PDI activity, would trigger HSF1 trimerization leading to its activation. To this aim, we analysed the trimerization of HSF1 in TG2+/+ and TG2−/− MEF after heat shock (Fig 5A) or proteasome inhibition (Fig 5B) using or not reducing agents. Interestingly, heat shock and proteasome inhibition induced the trimerization of HSF1 (molecular weight 240 kDa) only in TG2+/+ MEF, without reducing condition (Fig 5A and B), and this reaction was confined to the nuclear pool of HSF1 (Fig EV2A and B). Next, we performed an in vitro assay using recombinant TG2 and HSF1 proteins (Fig 5C) to show that TG2 effectively induced HSF1 polymerization. HSF1 polymerized only if it was phosphorylated and in the absence of calcium, which is known to interfere with the redox‐sensitive cysteines of TG2 and consequently with its PDI activity 37. Importantly, the regulation of HSF1‐HSP70 pathway by TG2 occurs through its PDI activity and not the transamidating one. In fact, the treatment with Z‐DON, a specific inhibitor of the transamidating activity of TG2, affects neither HSP70 induction nor HSF1 nuclear translocation after proteasome inhibition (Fig EV2C). These data suggest that TG2 PDI activity induces the trimerization of HSF1, which in turn facilitates transactivation of the HSP70 gene by binding to heat‐shock elements (HSEs) in the promoter. To confirm this hypothesis, we analysed the DNA‐binding ability of HSF1 to the HSP70 promoter in the presence and absence of TG2 by chromatin immunoprecipitation (ChIP) assay. We found that HSF1 was recruited to the HSP70 promoter after heat shock in TG2+/+ but not in TG2−/− MEF (Fig 5D). Accordingly, quantification of HSP70 mRNA indicated that the HSP70 gene was efficiently transcribed only in TG2+/+ MEF in response to HS (Fig 5E). Altogether, these results reveal that TG2 promotes HSF1 trimerization in heat‐stressed cells, thereby stimulating HSF1‐mediated transactivation of the HSP70 gene. TG2 dependent activation of HSF1 also regulates the expression of other target genes such as BAG3 and HSP25 (Fig EV2D). Thus, these findings demonstrate a key role played by TG2 in the control of cellular stress homeostasis via the post‐translational modification of HSF1.
Figure 5. TG2 causes the trimerization and transcriptional activation of HSF1 by its PDI activity.
-
A, BWestern blot analysis of HSF1 in nuclear extracts of TG2+/+ and TG2−/− MEFs after heat shock for 20, 40, 60 min (A) and proteasome inhibition treating with MG132 for 2 and 4 h (B). The analysis was performed in presence (+R) and absence (−R) of reducing agents to detect the formation of disulphide bonds. Lamin A/C was used as loading control.
-
CRecombinant HSF1 or the phosphorylated isoform was incubated with recombinant TG2, in the presence or absence of calcium (CaCl2) at 30°C for 1 h. The analysis was performed in presence (+R) and absence (−R) of reducing agents to detect the formation of disulphide bonds. The products of the in vitro assay were resolved by SDS–PAGE and blotted using the indicated antibodies.
-
DChIP‐enriched DNA was prepared from TG2+/+ and TG2−/− MEF treated with heat shock at 42°C for 10 min. ChIP‐qPCR analysis was performed in the distal HSE (promoter) of HSP70 promoter and in intergenic regions, as negative control, using HSF1 antibody. ChIP signal of HSF1 was normalized as % of input. The P‐value was determined by one‐way ANOVA test followed by Tukey's post‐test.
-
EHSP70 mRNA levels, quantified by qPCR, in TG2+/+ and TG2−/− MEF treated with heat shock at 42°C for 20, 40 and 60 min.
Figure EV2. TG2‐dependent HSF1 trimerization occurs in the nucleus and not in the cytosolic compartment.
-
A, BWestern blot analysis of HSF1 in cytosolic extracts of TG2+/+ and TG2−/− MEF after heat shock for 20, 40 and 60 min (A) and proteasome inhibition by treating with MG132 for 2 and 4 h (B). The analysis was performed in presence (+R) and absence (−R) of reducing agents to detect the formation of disulphide bonds. Tubulin was used as loading control.
-
CHSF1 nuclear translocation and HSP70 induction do not depend on TG2 transamidating activity. Western blot analysis of HSP70 expression (upper) and HSF1 nuclear translocation (lower) in TG2+/+ and TG2−/− MEF after proteasome inhibition with MG132 for 18 h in presence or not of Z‐DON, a specific inhibitor of TG2 transamidating activity. Tubulin was used as loading control for cytosolic extract. Histone H4 was used as loading control for nuclear extract. Actin was used as loading control for whole protein extract.
-
DDensitometric analysis of HSP25 and BAG3 protein in TG2+/+ and TG2−/− MEF after heat shock for 20, 40 and 60 min. Tubulin and actin were used as loading control.
TG2 ablation in CF mice restores F508del CFTR function
One sign of perturbed proteostasis that accompanies CF is the overexpression of HSP70 protein that associates with mutated CFTR 38, 39. Moreover, active trimeric HSF1 and several of its target chaperones are increased in CF cells and favour the degradation of mutated CFTR protein 40, 41. In contrast to wild‐type CFTR, F508del CFTR is unable to dissociate from HSP70 and consequently rapidly degraded by the proteasome 38, 39.
To translate our findings in vivo and elucidate the molecular mechanisms by which TG2 regulates CF pathogenesis, we developed a new experimental mouse model carrying the F508 CFTR deletion on a TG2 null background. We used the mouse model to test the effects of TG2 ablation on the capacity of F508del CFTR mice of counteracting luminal challenges, either in the intestine or in the airways. Mice homozygous for F508del mutations often died within the first week of life and the majority of the survivors succumbed to the intestinal obstruction just after weaning (between 21 and 28 days), if they were kept on standard chow 24. We first characterized the new mouse model in terms of survival immediately after birth, and we found that CF mice on TG2 null background (CFTRF508del/TG2−/−) mice not only were born at the expected Mendelian frequency (Fig EV3) but, fed with a standard diet, survived after weaning (Fig 6A). Subsequently, we analysed the sensitivity of these mice to the infection with Pseudomonas aeruginosa, a persistent bacteria that commonly infects and kills CF patients as a result of chronic lung inflammation. To this aim, mice were infected by intratracheal instillation of the P. aeruginosa PAO1 strain and either sacrificed 1 day later, to surgically collect the lungs and measure bacterial clearance, or kept in an isolator to monitor their survival. WT (CFTRwt/TG2+/+) and CF mice on TG2 null background (CFTRF508del/TG2−/−) indistinguishable survived in response to PAO1 infection, while CFTR homozygous ones (CFTRF508del/TG2+/+) succumbed to infection (Fig 6B). Moreover, CFTRF508del/TG2−/− mice cleared P. aerguinosa more efficiently from their lungs than CFTRF508del/TG2+/+ animals (Fig 6C). As a surrogate of inflammation, we measured the RNA levels of TNF‐α in different tissue. TNF‐α mRNA was reduced in the lungs and intestines from CFTRF508del/TG2−/− mice as compared to mice with the CFTRF508del/TG2+/+ genotype, indicating that the increased inflammation, usually observed in CF mice, was reduced when TG2 was absent (Fig 6D). These results suggest that TG2 ablation improves the overall conditions of CF mice. Finally, we measured CFTR‐dependent chloride secretion ex vivo in segment of the ileum mounted in Ussing chambers. This CFTR activity was defined as a forskolin‐induced increase in short‐circuit current (Isc) that was partially reverted by a selective CFTR inhibitor (CFTRinh172, Fig 6E), in line with previous publications 17. While CFTRF508del/TG2+/+ mice were devoid of CFTR function, CFTRF508del/TG2−/− mice exhibited approximately 40% of the CFTR activity found in wild‐type controls, thus indicating that the CFTR function was partially restored (Fig 6E).
Figure EV3. Mice strains crossed to generate CFTRF 508del/TG2−/− mice.
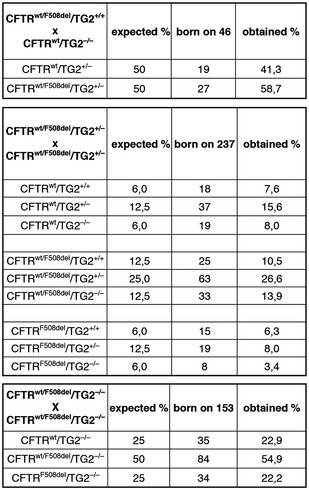
C57Bl/6 TG2−/− transgenic mice have been crossed with 129/FVB CFTRwt/F508del transgenic ones. The wild‐type littermates have been also produced. The presence of TG2 and F508del CFTR has been determined by PCR. The new mouse model has been characterized in terms of survival and number of littermates.
Figure 6. F508del CFTR function is restored in CF mice ablating TG2.
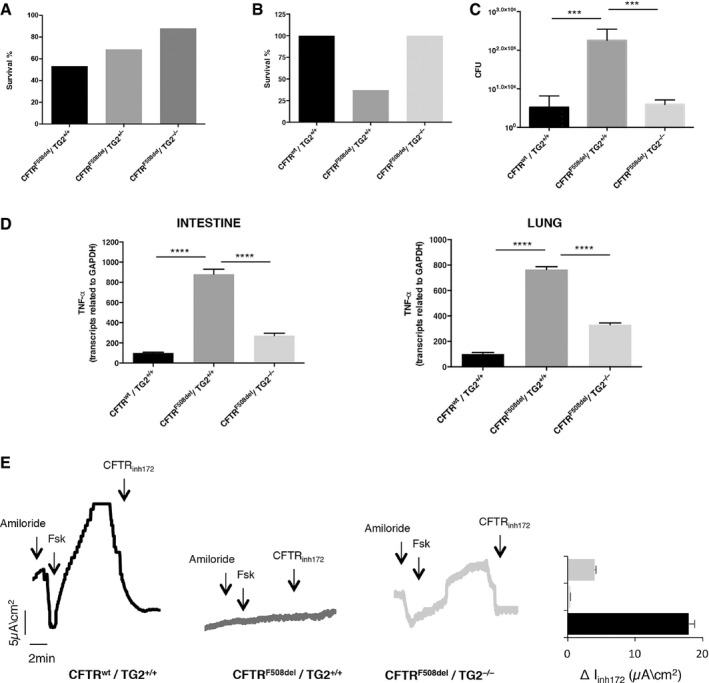
- Survival rate of CFTRF508del/TG2−/− (n = 5/42) and CFTRF508del/TG2+/− (n = 6/19) mice compared to the CFTRF508del/TG2+/+ (n = 7/15) ones fed with a standard diet.
- Survival rate of CFTRF508del/TG2−/− mice (n = 8/8) compared to the CFTRF508del/TG2+/+ ones (n = 3/8) and the wild‐type littermates (CFTRwt/TG2+/+)(n = 9/9) after infection with PAO1 strain.
- Number of colony‐forming units (CFU) of CFTRF508del/TG2−/− mice compared to the CFTRF508del/TG2+/+ ones and the wild‐type littermates (CFTRwt/TG2+/+) after 24 h of infection with PAO1 strain of Pseudomonas Aeruginosa.
- TNF‐α transcription levels, quantified by qPCR, in intestine (left) and lung (right) homogenates from 10‐week‐old CFTRF508del/TG2−/− (n = 3) and CFTRF508del/TG2+/+ (n = 3) mice.
- Representative traces of CFTR‐dependent Cl‐ secretion measured by forskolin (Fsk)‐induced increase of chloride current (Isc (μA/cm2)) in small intestines mounted in Ussing chambers; quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in tissue samples.
TG2 is involved in CF pathogenesis by regulating HSP70‐HSF1 pathway
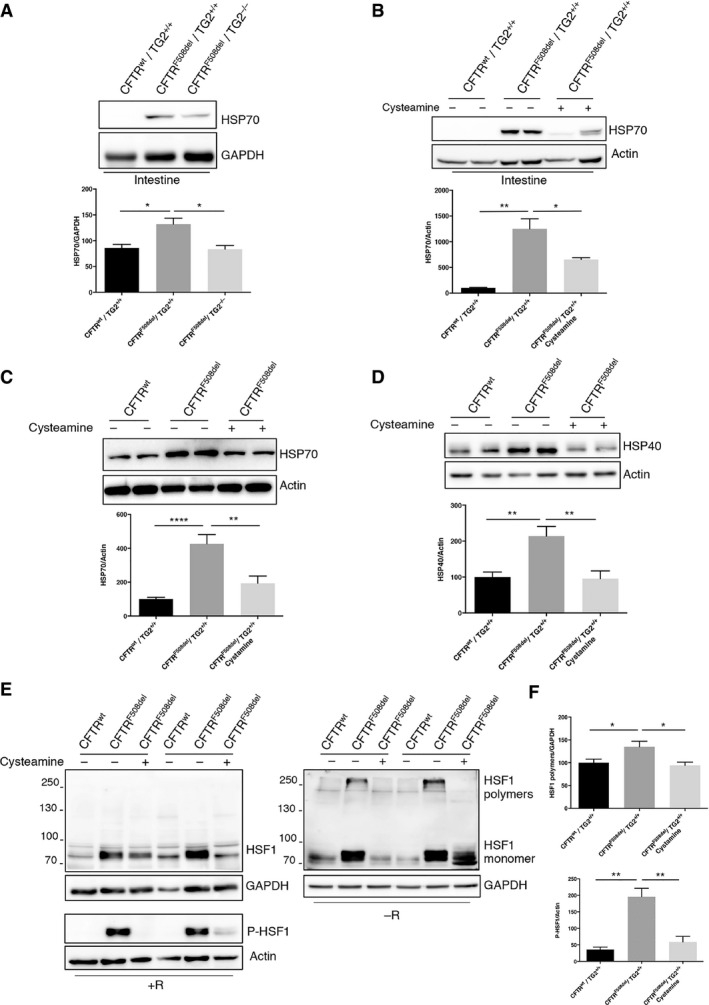
The above‐reported findings prompted us to investigate whether the effects of TG2 on HSF1‐mediated HSP70 transactivation may contribute to CF pathogenesis in the mouse model. Intestinal HSP70 expression was increased in CFTRF508del/TG2+/+ mice over WT (CFTRwt/TG2+/+) controls, and this HSP70 overexpression was reduced in CFTRF508del/TG2−/− mice (Fig 7A). Recently, it has been demonstrated that the administration of the TG2 inhibitor cysteamine restores CFTR function both in CF mice and patients bearing misfolded CFTR mutant proteins either in homozygous or in compound heterozygous form 17, 23, 24. By a computerized docking analysis, we found that cysteamine was able to interact with TG2 close to its active site, thus inhibiting the transamidating and the PDI activities (Fig EV4A). In particular, the protonated amino group of cysteamine interacts with the triad His 335, Asp 358 and Glu 396 of TG2, thus being tightly bound to the enzyme. Moreover, this interaction drives the correct orientation of cysteamine with Cys 371, forming a covalent disulphide bridge. Notably, residue 371 is one of the cysteines involved in oxidative inactivation of TG2, through the formation of disulphide bonds 37. These data indicate that the mechanism through which cysteamine inhibits TG2 could involve both the physical blockage of the active site and oxidative inactivation of the enzyme (Fig EV4A). Prompted by this evidence, we decided to verify whether the TG2 inhibition by cysteamine could reduce HSP70 protein levels leading to its positive effects. We therefore analysed HSP70 expression in the intestine of CF mice (CFTRF508del/TG2+/+) after 5 days of oral administration with cysteamine (Fig 7B). Interestingly, cysteamine led to a significant reduction in HSP70 protein levels that returned to the levels observed in the wild‐type mice (CFTRwt/TG2+/+). Next, to verify whether the TG2‐HSF1‐HSP70 axis was also detectable in CF patients homozygous for the F508del CFTR mutation, we examined HSP70 expression in primary nasal epithelial cells. Ex vivo treatment of freshly brushed patient cells with cysteamine significantly reduced the HSP70 protein levels observed in CF cells (Fig 7C). Cysteamine also decreased protein expression of HSP40 (Fig 7D) and HSP27 (Fig EV4B), others HSF1‐regulated chaperones involved in F508del CFTR handling 42, 43. Moreover, we verified whether the inhibition of TG2 by cysteamine could modulate HSP70 expression by interfering with its PDI activity. Notably, formation of HSF1 trimers in the nasal epithelial cells from CF patients was increased with respect to normal controls; however, cysteamine treatment abolished this increase (Fig 7E and F). According to this, phosphorylation of HSF1 at S326, a hallmark for HSF1 activation, occurred mainly in CF patients and largely decreased with cysteamine (Fig 7E and F). We also analysed freshly brushed nasal epithelial cells from two F508del CFTR homozygous patients who underwent a phase II clinical trial (EudraCT 2013‐001258‐82) with cysteamine bitartrate 17. Both patients, who showed functional rescue of mutant CFTR protein after 4 weeks of in vivo therapy 17, also manifested reduced HSP70 expression together with a decrease in TG2 protein (Fig EV4C). Altogether, our data indicate a pivotal role of TG2 in favouring F508del CFTR degradation through regulation of HSP70‐HSF1 pathway. They also suggest that cysteamine can improve the rescue of a functional F508del CFTR by modulating the PDI activity of TG2 in the epithelial cells. Thus, cysteamine can exert pleiotropic effects, as it is capable of either avoiding TG2‐mediated Ca2+‐dependent crosslinking of substrate proteins as well as of interfering with the activity of PDIs in CF epithelial cells.
Figure 7. TG2 regulates HSF1 activity and HSP70 expression in CF .
-
AWestern blot analysis and densitometric analysis of HSP70 protein levels in the intestine tissue lysates of CFTRF508del/TG2−/− mice (n = 4) compared to the CFTRF508del/TG2+/+ ones (n = 4) and the wild‐type littermates (CFTRwt/TG2+/+) (n = 4). GAPDH was used as loading control.
-
BWestern blot analysis and densitometric analysis of HSP70 protein levels in the intestine tissue lysates of CFTRF508del/TG2+/+ mice administrated with the TG2 inhibitor cysteamine (n = 3), compared to the untreated CFTRF508del/TG2+/+ ones (n = 3) and the wild‐type littermates (CFTRwt/TG2+/+) (n = 3). Actin was used as loading control.
-
C, DWestern blot and densitometric analysis of HSP70 and HSP40 protein levels in nasal epithelial cells, from CF patients (n = 6), cultured ex vivo with cysteamine for 18 h.
-
EWestern blot analysis of HSF1 and phosphorylated HSF1 at Ser326 in nasal epithelial cells, from CF patients (n = 6), cultured ex vivo with cysteamine for 18 h. The analysis was performed in presence (+R) and absence (−R) of reducing agents to detect the formation of disulphide bonds. GAPDH and Actin were used as loading control.
-
FDensitometric analysis of HSF1 polymers formation and HSF1 phosphorylation in nasal epithelial cells, from CF patients, cultured ex vivo with cysteamine.
Figure EV4. Cysteamine inhibition of TG2's PDI activity rescues HSP27 and HSP70 protein levels.
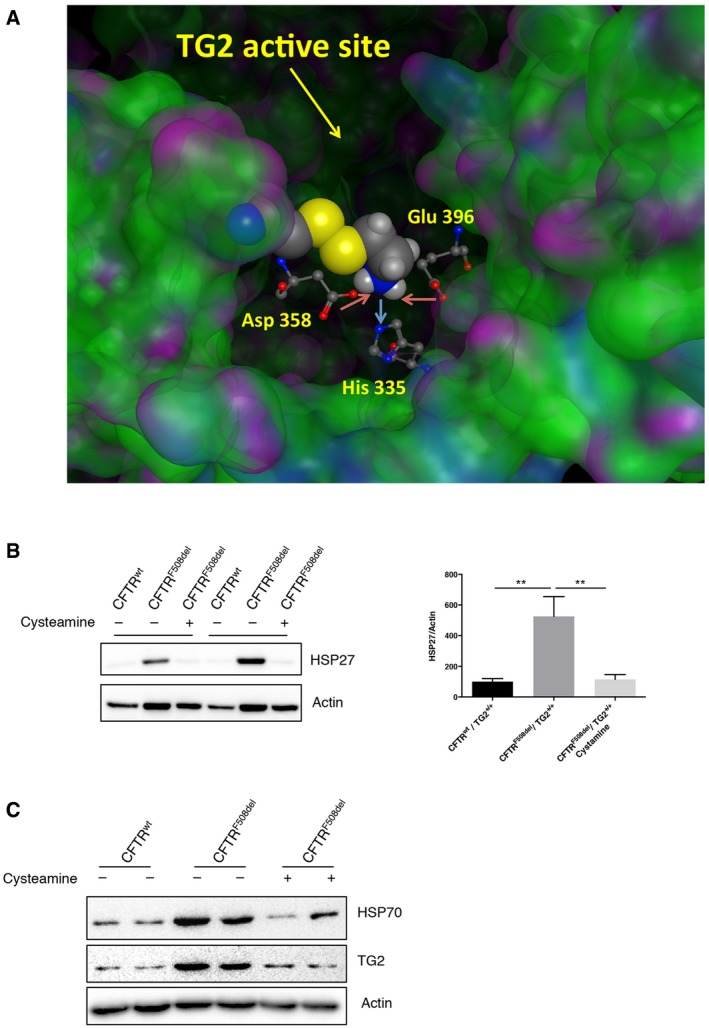
- Docking analysis of cysteamine binding to TG2.
- Western blot and densitometric analysis of HSP27 protein levels in nasal epithelial cells, from CF patients (n = 6), cultured ex vivo with cysteamine for 18 h.
- Western blot of HSP70 in brushed nasal epithelial cells freshly collected from F508del CFTR homozygous patients (n = 2) underwent a phase II clinical trial with cysteamine bitartrate. Actin was used as loading control.
Discussion
One of the mechanisms through which eukaryotic cells adapt to intracellular and environmental stress is the heat‐shock response. The master transcription factor regulating this pathway is HSF1 that, by controlling a large set of target genes, allows the stressed cell to survive. Under normal conditions, monomeric HSF1 is localized in the cytoplasm bound to some HSPs (HSP70 and 90). Upon stress, HSF1 is released from these binding partners and trimerizes, forming a complex that translocates into the nucleus where it binds to DNA and activates target genes 30, 31. Our study demonstrates that TG2 plays a key upstream role in the regulation of proteostasis by catalysing the trimerization of HSF1. Here, we provide the first evidence that the HSF1 trimerization is mediated by the PDI activity of TG2 (Fig EV5). The formation of three intermolecular S‐S bonds between two cysteine residues (Cys36 and Cys103) is essential for HSF1 trimerization and DNA binding 35. However, so far the mechanism by which Cys36 and Cys103 form this intermolecular S‐S bond was elusive. Here, we show that the PDI activity of TG2 catalyses HSF1 trimerization and its absence/inhibition impairs translocation of the HSF1 trimeric complex in the nucleus. In keeping with these evidences, mice lacking TG2 displayed a markedly impaired response to HS characterized by the absence of HSF1 trimer formation. In the absence of TG2, we also detected a marked difference in the HSF1 phosphorylation profile, largely proposed as a requirement for HSF‐driven transcription 44, even if recent evidences show that the phosphorylation status of HSF1 does not affect the subcellular localization and DNA‐binding activity of HSF1 45. Taken together, these findings demonstrate the key role played by TG2 in the control of cellular stress homeostasis via the post‐translational modification of HSF1. Another aspect of the present study is that it clarifies and extends prior observations on the role of TG2 in degenerative disorders, such as the Huntington disease (HD), in which TG2 inhibitors such as cysteamine reversed transcriptional deregulation due to the expression of mutated huntingtin 46, 47. These studies showed that the treatment of HD cells with TG2 inhibitors resulted in a broad normalization of several hundred genes that are deregulated by the expression of the mutated huntingtin. Interestingly, among these TG2‐sensitive genes, many are known to be also controlled by HSF1 47. It is important to note that, so far, no model was proposed to explain how the TG2 inhibition regulates transcription under proteomic stress, but based on the present study it is possible to suggest that TG2 might exert its transcriptional function through modulation of HSF1 activity. In agreement with this novel function of TG2, it was previously described that stressed cells lacking TG2 are defective in the clearance of ubiquitinated protein aggregates thus predisposing them to death 12, 48. Indeed, the peculiar biochemistry of TG2, as well its capacity to interact with the major proteins involved in the regulation of proteostasis 18, 48 in various intracellular compartments, confers to TG2 a unique ability to act as a guardian of the proteome under stressful conditions. In keeping with this assumption, recent evidence has highlighted a role for TG2 in the regulation of the main pathways controlling proteome homeostasis including autophagy, proteasomes and exosomes 4, 11, 13, 14. Likewise, HSF1 and the HSPs have been shown to play a key regulatory role in dictating the fate of their abnormal client proteins versus the proteasome and/or autophagy 31. TG2 function/regulation has been implicated in the pathogenesis of several major human diseases, such as cancer and metastasis formation, cystic fibrosis, coeliac disease and neurodegenerative disorders 49. Here, we focused our attention on CF, showing that the ablation/inhibition of TG2 significantly ameliorated the typical CF symptoms in a double‐transgenic mouse model in which the F508del CFTR mutation was backcrossed into a TG2 null background. Interestingly, CFTRF508del TG2−/− mice exhibited a partial recovery (about 40%) of the CFTR functionality and exhibited a marked amelioration of CF symptoms, including reduced inflammatory and pulmonary inflammation as well as improved clearance of, and resistance to, Pseudomonas aeroginosa. Importantly, restoring even < 30% of CFTR function in vivo is believed to confer a clinical benefit to CF patients 24. In fact, rescuing approximately 20% of function of WT CFTR prevents the CF‐associated intestinal manifestations in newborn F508del CFTR homo‐zygous pigs 24. Thus, even a partial rescue of CFTR function, as that elicited by the ablation of TG2, may explain the amelioration of the CF phenotype. The improvements observed in the absence of TG2 were paralleled by reduction in the HSF1 trimerization, resulting in a drastic reduction in HSP70 levels. It is important to note that similar findings were also detected in epithelial cells obtained from the nasal brushing of CF patients treated with the TG2 inhibitor cysteamine, which has been demonstrated to be clinically efficient in CF. Thus, systemic cysteamine treatment switches off inflammation, rescues F508del CFTR protein expression and restores its function at the PM, both in the lungs from CFTRF508del TG2+/+ mice and in F508del CFTR bronchial epithelial cells from CF patients 24. Taken together, these data highlight a key upstream novel role played by TG2 and HSF1 as general regulators of proteostasis in the CF pathogenesis. Indeed, the results of this study uncover a new and unexpected paradigm: reduction in HSPs levels, in the course of the CF disease, results in decreased degradation of the CFTR, thus leading to a positive outcome. Indeed, immature F508del CFTR fails to mature, does not transit to the late secretory pathway 50 and is trapped in the ER where interacts with the cytosolic chaperones, such as HSP70, as well as with the ER chaperone calnexin 38, 42, 51, 52. Ultimately, F508del CFTR, retained in the ER, is ubiquitinated and retrotranslocated to the cytosol where it undergoes proteasomal degradation by the ER‐associated degradation (ERAD) pathway 53, 54, 55, 56. In this regard, in the last years it has become clear that HSF1 is able to regulate not only cytosolic but also ER and mitochondria proteins 57, 58, 59, 60, suggesting a possible interplay between HSF1 activation and ERAD pathway, involved in CFTR degradation. In addition, recent findings show that HSF1 is also able to regulate gene transcription for the maintenance of proteostasis capacity in unstressed conditions 61. In fact, accumulating evidences show that HSF1 is activated in the nucleus as a consequence of diverse specific physiological cellular requirements and directs transcriptional programmes distinct from the heat‐shock response.
Figure EV5. Scheme of TG2 dependent trimerization of HSF1.
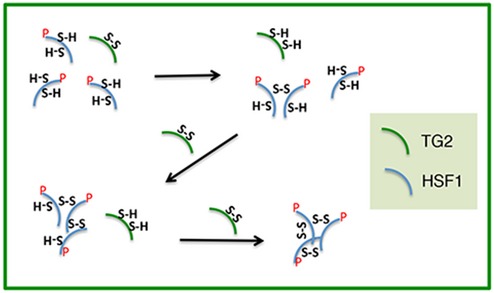
It is showed the hypothetical mechanism of action of TG2's PDI activity on HSF1.
In conclusion, our findings may open new perspectives in drug discovery programmes aimed at the identification of new inhibitors of PDIs to circumvent CFTR defect. Given the fact that cysteamine can inhibit the PDI activity of TG2 in a direct fashion, by a direct molecular interaction with the enzyme and its critical cysteines, it appears that most, if not all, of the cysteamine effects on CF must be considered as “on‐target”. It remains to be determined whether yet‐to‐be‐developed specific TG2 PDI inhibitors may replace cysteamine advantageously for the treatment of CF or other diseases linked to TG2 deregulation.
Materials and Methods
Cell lines and drug treatments
TG2+/+ and TG2−/− MEFs (murine embryonic fibroblasts) were obtained by spontaneous immortalization of fibroblasts derived from C57BL/6 mice embryos either wild type or knockout for TG2. TG2−/− MEFTG2 stably reconstituted with the unmutated TG2 was obtained as previously described 4. To silence TG2, TG2+/+ MEF was transfected with pLKO.1 plasmid (Sigma) containing an shRNA insert specific for TG2 (shTG2) or an shRNA insert that does not target any known genes from any species (no target shRNA). Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, and the cells were selected for puromycin (Sigma) resistance using the antibiotic added to the culture medium (2 μg/ml). MEF cells were cultured in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% foetal bovine serum, 100 μg/ml streptomycin and 100 units/ml penicillin, at 37°C and 5% CO2 in a humidified atmosphere. To induce heat shock, cells were placed in a water bath at 42°C followed, where indicated, by recovery to 37°C. To block proteasome activity, cells were incubated in full medium in the presence of 5 μM MG132 (Z‐Leu‐Leu‐Leu‐al, Sigma‐Aldrich) for the indicated time. TG2 transamidating activity was inhibited incubating the cells with 40 μM Z‐DON (Zedira). Nasal epithelial cells were collected by nasal brushing from two F508del CFTR homozygous CF patients enrolled in a previous open‐label phase II clinical trial (EudraCT number #2013‐001258‐82 approved by Local Ethics Committee, Protocol reference #85/13) at the Regional Cystic Fibrosis Care Center, University of Naples Federico II A. All patients gave written informed consent at the time of the clinical study. The patients were treated as described 17, 24, and nasal cells were collected previously and after 8 weeks of treatment with cysteamine. After nostril washing to remove mucus, cytological brushes (Robimpex, MO11157) were used to scrape the mid part of the inferior turbinate from both nasal nostrils. Brushes with cells were immediately transferred into RPMI 1640 medium (Invitrogen) containing 1% penicillin–streptomycin (Lonza), in 15‐ml sterilized tubes. The tubes were incubated at 37°C for 2 h on a thermoshaker, to remove all cells from brushes, the brushes were then removed, and the cells were centrifuged at 800 × g for 20 min. The supernatant fractions were discarded and the cell pellet treated with 150 μl of trypsin–versene (EDTA) solution (Lonza) for 4 min at 37°C to disaggregate possible cell clusters. Trypsin solution was inactivated by adding 3 ml of serum‐free bronchial epithelial cell growth medium BEGM (Clonetics, Lonza). After centrifugation at 800 × g for 10 min, cells were plated with 10 ml of BEGM medium. Non‐specific epithelial cells were removed during the daily cell washing and medium changes. Counterstaining with specific antibody was used to exclude the presence of lymphocytes, inflammatory cells and mucipar differentiation. For ex vivo treatments, the cells were incubated with or without cysteamine (250 μM) for 18 h.
Generation of CFTRF508del/TG2−/− mice
In order to obtain TG2−/− mice carrying F508del mutation in CFTR, C57Bl/6 knockout mice for TG2 62 (obtained from Gerry Melino, Department of Experimental Medicine and Biochemical Sciences, University of Rome “Tor Vergata”, Rome, Italy) were crossed with 129/FVB mice heterozygous for F508del mutation (obtained from Bob Scholte, Erasmus Medical Centre Rotterdam, the Netherlands, CF coordinated action programme EU FP6 LSHM‐CT‐2005‐018932). TG2 genotype was assessed by PCR using primers:
5′‐TCCTGACCTGAGTCCTCGTC‐3′
5′‐TACTCCAGCTTCTCGTTCTG‐3′
5′‐ACGAGACTAGTGAGACGTGC‐3′, following the conditions 95°C for 5 min, then 35 cycles at 94°C for 1 min + 56°C for 45 s + 72°C for 1 min and then 72°C for 5 min, as previously described 62. To assess the presence of F508del mutation, DNA was digested with SspI restriction enzyme (Thermo Scientific) at 37°C and PCR was carried out with primers:
5′‐GGACGCAAAGAAAGGGATAAG‐3′
5′‐CACAACACTGACACAAGTAGC‐3′, following the conditions 95°C for 5 min, then 33 cycles at 95°C for 1 min + 52°C for 1 min + 72°C for 1 min and then 72°C for 7 min.
After weaning, mice were fed with a standard diet and survival rate was evaluated in the 4 weeks following.
Animal treatments
To induce an hyperthermic stress in mice, an empty mouse cage and a tray filled with water were placed in an oven at least 2 h at 42°C. These conditions should provide a relative humidity of 75%, which favours efficient HS 63, 64. Eight‐week‐old mice were placed in the cage for 20 min at 42°C, after which they were transferred to a clean cage at room temperature. Cysteamine administration was performed in CFTRF508del mice via gavage with cysteamine (60 mg/kg in 100 ml saline/day) or 100 ml saline/day for 5 consecutive day/week for 5 weeks. P. aeruginosa (PAO1) infection was performed in 6‐week‐old mice by intratracheal injection of 5 × 106 cell/mice for 24 h. At the end of the treatment, mice were anesthetized with avertin (tribromoethanol, 250 mg/kg, Sigma) and killed and lungs were collected for analysis. The whole lung of each mouse was homogenized in 1 ml of sterile 0.9% saline and 100 μl of appropriately serial diluted lung homogenates samples was plated in agar Petri dish in the presence of carbenicillin (100 μg/ml, Sigma) overnight at 37°C. Colony factor unit was enumerated 24 h later 65, 66. All the procedures in mice were approved by the Local Ethics Committee for Animal Welfare (IACUC No. 713) and were carried out in strict respect of European and National regulations.
Western blot analysis
Tissues were lysed in 50 mM Tris–HCl pH 7.5, 50 mM NaCl, 320 mM sucrose, 1% Triton X‐100, 10% glycerol supplemented with protease and phosphatase inhibitor cocktail. Cells were rinsed in ice‐cold phosphate‐buffered saline (PBS) and collected in lysis buffer containing 20 mM Tris–HCl pH 7.4, 150 mM NaCl and 1% Triton X‐100 with protease inhibitor cocktail. Nuclear and cytosolic extracts were obtained using the NE‐PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific). Protein concentrations were determined by the Bradford assay, using bovine serum albumin as a standard. Aliquots of total protein extracts from cells and tissues after different treatments were resolved on SDS–polyacrylamide gel and transferred to a nitrocellulose membrane. Blots were blocked in 5% non‐fat dry milk in T‐PBS (PBS + 0.05% Tween‐20) for 1 h at room temperature and then incubated overnight with the described antibodies. The membranes were incubated with HRP‐conjugated secondary antibody for 1 h at room temperature, and the signal was detected by Immun‐Star WesternC Kit (Bio‐Rad Laboratories).
Immunoprecipitation
Cells were lysed in a buffer containing 150 mM NaCl, 50 mM Tris–HCl pH 7.5, 2 mM EDTA, 2% NP‐40 and freshly added protease inhibitor cocktail. An amount of 1.5 mg of proteins from cell lysates were subjected to immunoprecipitation using 2 μg of specific antibodies in combination with 80 μl of Protein G PLUS‐Agarose beads (Santa Cruz), overnight at 4°C. LDS Sample Buffer 4× (Life Technologies) containing 2.86 M 2‐mercaptoethanol (Sigma‐Aldrich) was added to beads, and samples were boiled at 95°C for 10 min. Supernatants were analysed by Western blot.
Fluorescence microscopy
TG2+/+ MEF, grown on coverslips, were subjected to heat shock, then washed with PBS and fixed in 4% paraformaldehyde for 10 min at room temperature. Fixed cells were permeabilized with 0.1% Triton X‐100 in PBS for 10 min, blocked with 3% BSA in PBS for 20 min and incubated with anti‐TG2 and anti‐HSF1 primary antibodies for 1 h. After washing, cells were incubated with Alexa Fluor 488‐conjugated and Alexa Fluor 594‐conjugated secondary antibodies. The coverslips were mounted on microscope slides, sealed with an antifade solution and examined under a Leica TCS SP5 confocal microscope equipped with an ×40 (NA 1.25) or ×63 (NA 1.4) oil‐immersion objective (Deerfield, IL, USA). Quantification of co‐localization was performed on six sets of images acquired with the same optical settings and excluding the consideration of autofluorescence. To determine fluorescent signal co‐localization between different channels, the Coloc module of ImageJ was used. A median filter was applied to the images to reduce the background noise before threshold analysis. The degree of channels co‐localization was analysed by considering the Pearson's coefficient.
In vitro assay of HSF1 polymerization
Recombinant His‐TG2 (0.25 μg, Zedira) was incubated either with recombinant His‐HSF1 (0.5 μg, Enzo) or with the phosphorylated His‐HSF1 recombinant protein (0.5 μg, Enzo) in reaction buffer including 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% glycerol in presence or not of 5 mM CaCl2 for 1 h at 30°C. The reaction products were than denaturated with LDS Sample Buffer 4× containing (+R) or not (−R) 2‐mercaptoethanol, heated 10 min at 70°C and directly analysed by Western blotting.
Chromatin Immunoprecipitation assay
Chromatin Immunoprecipitation was performed as previously described 67. TG2+/+ and TG2−/− MEFs, exposed to heat shock, were crosslinked in 1% (vol/vol) formaldehyde for 10 min at room temperature and quenched with 125 mM glycine for 5 min. Then, cells were washed twice with PBS, scraped and pelleted by centrifugation at 1,300 × g for 10 min and stored at 80°C until further use. Cells were lysed with a mild lysis buffer, and nuclei were enriched by centrifugation at 14,000 × g for 10 min. Nuclei were lysed with a nuclear lysis buffer and chromatin was sheared by sonication (Diagenode, Belgium, 30‐s pulses, 30‐s rests) on ice. Successful fragmentation was confirmed on a 0.8% agarose gel electrophoresis by comparing to unsheared chromatin. Immunoprecipitation was only done when sheared DNA had a size between 100 and 1,000 bp. Sheared chromatin was diluted in ChIP dilution buffer, and 100 μg was immunoprecipitated with magnetic protein G beads and 3 μg of primary antibody (α‐HSF‐1, Millipore ABE1044) overnight at 4°C by head‐to‐head rotation. The precipitated material was washed subsequently with a low salt buffer, high salt buffer and Tris–EDTA buffer for 5 min each with head‐to‐head rotation. The precipitated material was also treated with RNAse R at 37°C for 15 min and with 0.5 mg/ml Proteinase K at 50°C for 1 h to remove all contaminating RNA molecules and proteins. Eluted DNA was purified and subjected to qPCR analysis with primers directed against the HSP70.3 promoter region (mHSP70.3 dHSE Fwd 5′‐ACCCTCCCCCTCAGGAATC‐3′; mHSP70.3 dHSE Rev 5′‐TGTCCAGAACTCTCCAGAGGTTT‐3′). A primer set amplifying an HSP70.3 intergenic region (mHSP70.3 Intergenic Region Fwd 5′‐GTGGCGCATGCCTTTGAT‐3′; mHSP70.3 Intergenic Region Rev 5′‐CTTTGTAGAACAGGCTGACCTTGA‐3′) was used as control for the ChIP.
Ussing chamber
CFTR‐dependent chloride secretion was measured ex vivo in segment of mice ileum mounted in Ussing chambers. Chambers were obtained from Physiologic Instruments (model P2300, San Diego, CA, USA). Chamber solution was buffered by bubbling with a mixture of 95% O2 and 5% CO2. Tissues were short‐circuited using Ag/AgCl agar electrodes. Short‐circuit current and resistance were acquired or calculated using the VCC‐600 transepithelial clamp from Physiologic Instruments and the Acquire &Analyze2∙3 software for data acquisition (Physiologic Instruments), as previously described 17. A basolateral‐to‐apical chloride gradient was established by replacing NaCl with Na‐gluconate in the apical (luminal) compartment to create a driving force for CFTR‐dependent Cl− secretion. CFTR channels present at the apical surface of the epithelium (lumen side of the tissue) were activated. Stimulations with forskolin, CFTR inhibitor 172 and amiloride were performed as described 17.
RNA isolation and qPCR
Total RNA from MEF cells was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions and then treated with Dnase I to remove contaminant DNA. Total RNA from mouse lung and intestine homogenates was extracted with the Pure Link RNA Mini Kit (Ambion, Life Technologies). 2 μg of RNA was retro‐transcribed using AMV RT reverse transcriptase (Promega) and used in quantitative RT–PCR (qPCR) experiment, using SYBR green Supermix (BIO‐RAD) following manufacturer's instructions. Thermocycling consisted of an initial polymerase activation step at 98°C for 5 min, and amplification was performed with 35 cycles of 95°C for 15 s, 68°C for 10 s and 72°C for 20 s with data acquisition at this stage and the reaction finished by the built‐in melt curve. The relative amounts of mRNA were calculated by using the comparative Ctmethod. HSP70 primers: 5′‐ATGGACAAGGCGCAGATCC‐3′, 5′‐CTCCGACTTGTCCCCCAT‐3′. Actin primers: 5′‐GGCTGTATTCCCCTCCATCG‐3′, 5′‐CCAGTTGGTAACAATGCCATGT‐3′. TNFα primers: 5′‐CCACCACGCT CTTCTGTCTA‐3′ and 5′‐AGGGTCTGGGCCATAGAACT‐3′.
Computerized docking analyses
TG2 crystal structure was retrieved from the protein data bank (PDB code: 3S3J) and processed in order to remove unwanted ligands and water molecules. Hydrogen atoms were added to the protein structure using standard geometries. To minimize contacts between hydrogens, the structure was subjected to Amber99 force‐field minimization until the rms (root‐mean‐square) of conjugate gradient was <0.1 kcal/mol/Å (1Å = 0.1 nm) keeping the heavy atoms fixed at their crystallographic positions 68. After protein preparation, a site finder approach was used to retrieve information about all the possible interaction sites suitable with cysteamine size and chemical properties. Only one binding site, defined by His 335, Asp 358, Cys 371, Phe 392 and Glu 396, resulted to be compatible and was then selected for the docking analysis. The docking procedure was performed using AUTODOCK software 69.
Statistical analysis
GraphPad was used for statistical analysis. ImageJ64 software was used for densitometric analysis. Statistical significance was determined using the Student's t‐test or one‐way ANOVA test. P‐value smaller than 0.05 (P < 0.05) was considered to be significant.
Author contributions
FR designed and performed most of the experiments. VRV and SE processed the human samples from CF patients. MD and MGF produced MEF cells and along with LO who helped with the experiments. EF, RM and NAB performed the experiments on CF mice. VP and CS planned and performed the CHIP assay. GC performed the bioinformatics analysis. LF performed the immunofluorescence analysis. GMF and GK analysed all data. VR provided the human samples from CF patients. MP and LM conceived the project and designed the experiments. MP, FR and LM wrote the manuscript. All authors read and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors would like to thank Prof. Gerry Melino from the Department of Experimental Medicine and Surgery, University of Rome “Tor Vergata”, Rome, Italy, to have kindly provided TG2−/− mice. The authors would like to thank Dr. E Romano from the Centre of Advanced Microscopy, Department of Biology, University of Rome Tor Vergata, for her skilful assistance in the use of the facility. This work was supported in part by grants from AIRC (IG2015 n. 17404 to G.M.F., IG2014 n. 15244 to M.P. and IG18790 to C.S.), grant from Telethon (GGP14095 to C.S.), the Italian Ministry of University and Research (FIRB Accordi di Programma 2011), the Italian Ministry of Health (Ricerca Corrente and Ricerca Finalizzata RF2010 2305199), Fondazione Fibrosi Cistica (FFC#8/2015 to M.P., FFC#2/2016 to G.C.), (E‐Rare Rescue CFTR pre‐clinic), F.R. was supported by “Fondazione Umberto Veronesi” and AIRC fellowships. The authors also acknowledge the support of the grant from the Russian Government Programme for the Recruitment of the Leading Scientists into the Russian Institutions of Higher Education 14.W03.31.0029 to MP.
EMBO Reports (2018) 19: e45067
References
- 1. Fesus L, Piacentini M (2002) Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci 27: 534–539 [DOI] [PubMed] [Google Scholar]
- 2. Lorand L, Graham RM (2003) Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4: 140–156 [DOI] [PubMed] [Google Scholar]
- 3. Nurminskaya MV, Belkin AM (2012) Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 294: 1–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossin F, D'Eletto M, Macdonald D, Farrace MG, Piacentini M (2012) TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 42: 1793–1802 [DOI] [PubMed] [Google Scholar]
- 5. Mastroberardino PG, Farrace MG, Viti I, Pavone F, Fimia GM, Melino G, Rodolfo C, Piacentini M (2006) “Tissue” transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim Biophys Acta 1757: 1357–1365 [DOI] [PubMed] [Google Scholar]
- 6. Gundemir S, Colak G, Tucholski J, Johnson GVW (2012) Transglutaminase 2: a molecular swiss army knife. Biochim Biophys Acta 1823: 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altuntas S, Rossin F, Marsella C, D'Eletto M, Diaz‐Hidalgo L, Farrace MG, Campanella M, Antonioli M, Fimia GM, Piacentini M (2015) The transglutaminase type 2 and pyruvate kinase isoenzyme M2 interplay in autophagy regulation. Oncotarget 6: 44941–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caccamo D, Condello S, Ferlazzo N, Currò M, Griffin M, Ientile R (2013) Transglutaminase 2 interaction with small heat shock proteins mediate cell survival upon excitotoxic stress. Amino Acids 44: 151–159 [DOI] [PubMed] [Google Scholar]
- 9. Ergulen E, Bécsi B, Csomós I, Fésüs L, Kanchan K (2016) Identification of DNAJA1 as a novel interacting partner and substrate of human transglutaminase 2. Biochem J 473: 3889–3901 [DOI] [PubMed] [Google Scholar]
- 10. Min B, Park H, Lee S, Li Y, Choi JM, Lee JY, Kim J, Choi YD, Kwon YG, Lee HW et al (2016) CHIP‐mediated degradation of transglutaminase 2 negatively regulates tumor growth and angiogenesis in renal cancer. Oncogene 35: 3718–3728 [DOI] [PubMed] [Google Scholar]
- 11. D'Eletto M, Farrace MG, Falasca L, Reali V, Oliverio S, Melino G, Griffin M, Fimia GM, Piacentini M (2009) Transglutaminase 2 is involved in autophagosome maturation. Autophagy 5: 1145–1154 [DOI] [PubMed] [Google Scholar]
- 12. D'Eletto M, Farrace MG, Rossin F, Strappazzon F, Giacomo GD, Cecconi F, Melino G, Sepe S, Moreno S, Fimia GM et al (2012) Type 2 transglutaminase is involved in the autophagy‐dependent clearance of ubiquitinated proteins. Cell Death Differ 19: 1228–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rossin F, D'Eletto M, Falasca L, Sepe S, Cocco S, Fimia GM, Campanella M, Mastroberardino PG, Farrace MG, Piacentini M (2015) Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ 22: 408–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diaz‐Hidalgo L, Altuntas S, Rossin F, D'Eletto M, Marsella C, Farrace MG, Falasca L, Antonioli M, Fimia GM, Piacentini M (2016) Transglutaminase type 2‐dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim Biophys Acta 1863: 2084–2092 [DOI] [PubMed] [Google Scholar]
- 15. Van Raamsdonk JM, Pearson J, Bailey CD, Rogers DA, Johnson GV, Hayden MR, Leavitt BR (2005) Cystamine treatment is neuroprotective in the YAC128 mouse model of Huntington disease. J Neurochem 95: 210–220 [DOI] [PubMed] [Google Scholar]
- 16. Dohil R, Meyer L, Schmeltzer S, Cabrera BL, Lavine JE, Phillips SA (2012) The effect of cysteamine bitartrate on adiponectin multimerization in non‐alcoholic fatty liver disease and healthy subjects. J Pediatr 161: 639–645 [DOI] [PubMed] [Google Scholar]
- 17. Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A et al (2016) A novel treatment of cystic fibrosis acting on‐target: cysteamine plus epigallocatechin gallate for the autophagy‐dependent rescue of class II‐mutated CFTR. Cell Death Differ 23: 1380–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Altuntas S, D'Eletto M, Rossin F, Hidalgo LD, Farrace MG, Falasca L, Piredda L, Cocco S, Mastroberardino PG, Piacentini M et al (2014) Type 2 transglutaminase, mitochondria and Huntington's disease: menage a trois. Mitochondrion 19: 97–104 [DOI] [PubMed] [Google Scholar]
- 19. Piacentini M, D'Eletto M, Farrace MG, Rodolfo C, Del Nonno F, Ippolito G, Falasca L (2014) Characterization of distinct sub‐cellular location of transglutaminase type II: changes in intracellular distribution in physiological and pathological states. Cell Tissue Res 358: 793–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siegel M, Khosla C (2007) Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther 115: 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bousquet M, Gibrat C, Ouellet M, Rouillard C, Calon F, Cicchetti F (2010) Cystamine metabolism and brain transport properties: clinical implications for neurodegenerative diseases. J Neurochem 114: 1651–1658 [DOI] [PubMed] [Google Scholar]
- 22. Anderson MP, Sheppard DN, Berger HA, Welsh MJ (1992) Chloride channels in the apical membrane of normal and cystic fibrosis airway and intestinal epithelia. Am J Physiol 263: L1–L14 [DOI] [PubMed] [Google Scholar]
- 23. Luciani A, Villella VR, Esposito S, Brunetti‐Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello‐Mantovani M et al (2010) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS‐mediated autophagy inhibition. Nat Cell Biol 12: 863–875 [DOI] [PubMed] [Google Scholar]
- 24. De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, Gregorio FD, Salvadori L, Grassia R, Leone CA, Rosa GD et al (2014) Restoration of CFTR function in patients with cystic fibrosis carrying the F508del‐CFTR mutation. Autophagy 10: 2053–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Esposito S, Tosco A, Villella VR, Raia V, Kroemer G, Maiuri L (2016) Manipulating proteostasis to repair the F508del‐CFTR defect in cystic fibrosis. Mol Cell Pediatr 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xia W, Voellmy R (1997) Hyperphosphorylation of heat shock transcription factor 1 is correlated with transcriptional competence and slow dissociation of active factor trimers. J Biol Chem 272: 4094–4102 [DOI] [PubMed] [Google Scholar]
- 27. Du ZX, Zhang HY, Meng X, Gao YY, Zou RL, Liu BQ, Guan Y, Wang HQ (2009) Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol 218: 631–637 [DOI] [PubMed] [Google Scholar]
- 28. Qi W, White MC, Choi W, Guo C, Dinney C, McConkey DJ, Siefker‐Radtke A (2013) Inhibition of inducible heat shock protein‐70 (hsp72) enhances bortezomib‐induced cell death in human bladder cancer cells. PLoS One 8: e69509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12: 3788–3796 [DOI] [PubMed] [Google Scholar]
- 30. Åkerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calderwood SK, Xie Y, Wang X, Khaleque MA, Chou SD, Murshid A, Prince T, Zhang Y (2010) Signal transduction pathways leading to heat shock transcription. Sign Transduct Insights 2: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baler R, Dahl G, Voellmy R (1993) Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol 13: 2486–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pirkkala L, Nykänen P, Sistonen L (2001) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15: 1118–1131 [DOI] [PubMed] [Google Scholar]
- 34. Jin YH, Ahn SG, Kim SA (2015) BAG3 affects the nucleocytoplasmic shuttling of HSF1 upon heat stress. Biochem Biophys Res Commun 464: 561–567 [DOI] [PubMed] [Google Scholar]
- 35. Ahn SG, Thiele DJ (2003) Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev 17: 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu M, Kim HE, Li CR, Kim S, Kwak IJ, Lee YJ, Kim SS, Moon JY, Kim CH, Kim DK et al (2008) Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry 47: 6007–6015 [DOI] [PubMed] [Google Scholar]
- 37. Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM (2010) Redox regulation of transglutaminase 2 activity. J Biol Chem 285: 25402–25409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang Y, Janich S, Cohn J, Wilson JM (1993) The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp7O and degraded in a pre‐Golgi non lysosomal compartment. Proc Natl Acad Sci USA 90: 9480–9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kopito RR (1999) Biosynthesis and degradation of CFTR. Physiol Rev 79: 167–173 [DOI] [PubMed] [Google Scholar]
- 40. Roberts RG (2014) Living in constant crisis–when stress management becomes the problem. PLoS Biol 12: e1001999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roth DM, Hutt DM, Tong J, Bouchecareilh M, Wang N, Seeley T, Dekkers JF, Beekman JM, Garza D, Drew L et al (2014) Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol 12: e1001998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM (1999) The Hdj‐2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J 18: 1492–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahner A, Gong X, Schmidt BZ, Peters KW, Rabeh WM, Thibodeau PH, Lukacs L, Frizzell RA (2013) Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin‐like modifier‐dependent pathway. Mol Biol Cell 24: 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chou SD, Prince T, Gong J, Calderwood SK (2012) mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One 7: e39679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Budzyński MA, Puustinen MC, Joutsen J, Sistonen L (2015) Uncoupling stress‐inducible phosphorylation of heat shock factor 1 from its activation. Mol Cell Biol 35: 2530–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mastroberardino PG, Piacentini M (2010) Type 2 transglutaminase in Huntington's disease: a double‐edged sword with clinical potential. J Intern Med 268: 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McConoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, Mahishi L, Cooper AJ, Antonyak MA, Cerione RA et al (2010) Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med 2: 349–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rossin F, D'Eletto M, Farrace MG, Piacentini M (2014) Transglutaminase type 2: a multifunctional protein chaperone? Mol Cell Oncol 1: e968506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iismaa SE, Mearns BM, Lorand L, Graham RM (2009) Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89: 991–1023 [DOI] [PubMed] [Google Scholar]
- 50. Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S (1994) Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J 13: 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pind S, Riordan JR, Williams DB (1994) Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 269: 12784–12788 [PubMed] [Google Scholar]
- 52. Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR (1998) Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J 17: 6879–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83: 129–135 [DOI] [PubMed] [Google Scholar]
- 54. Knittler MR, Dirks S, Haas IG (1995) Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc Natl Acad Sci USA 92: 1764–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gelman MS, Kannegaard ES, Kopito RR (2002) A principal role for the proteasome in endoplasmic reticulum‐associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J Biol Chem 277: 11709–11714 [DOI] [PubMed] [Google Scholar]
- 57. Takemori Y, Sakaguchi A, Matsuda S, Mizukami Y, Sakurai H (2006) Stress‐induced transcription of the endoplasmic reticulum oxidoreductin gene ERO1 in the yeast Saccharomyces cerevisiae. Mol Genet Genomics 275: 89–96 [DOI] [PubMed] [Google Scholar]
- 58. Sakurai H, Ota A (2011) Regulation of chaperone gene expression by heat shock transcription factor in Saccharomyces cerevisiae: importance in normal cell growth, stress resistance, and longevity. FEBS Lett 585: 2744–2748 [DOI] [PubMed] [Google Scholar]
- 59. Heldens L, Hensen SM, Onnekink C, van Genesen ST, Dirks RP, Lubsen NH (2011) An atypical unfolded protein response in heat shocked cells. PLoS One 6: e23512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim E, Sakata K, Liao FF (2017) Bidirectional interplay of HSF1 degradation and UPR activation promotes tau hyperphosphorylation. PLoS Genet 13: e1006849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fujimoto M, Nakai A (2010) The heat shock factor family and adaptation to proteotoxic stress. FEBS J 277: 4112–4125 [DOI] [PubMed] [Google Scholar]
- 62. De Laurenzi V, Melino G (2001) Gene disruption of tissue transglutaminase. Mol Cell Biol 21: 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fujio N, Hatayama T, Kinoshita H, Yukioka M (1987) Induction of four heat‐shock proteins and their mRNAs in rat after whole‐body hyperthermia. J Biochem 101: 181–187 [DOI] [PubMed] [Google Scholar]
- 64. Nowak TS, Bond U, Schlesinger MJ (1990) Heat shock protects RNA levels in brain and other tissues after hyperthermia and transient ischemia. J Neurochem 54: 451–458 [DOI] [PubMed] [Google Scholar]
- 65. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F et al (2013) Depletion of the ubiquitin‐binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr DF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem 288: 2049–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D'Eletto M, Tosco A, De Gregorio F, Izzo V et al (2017) Cysteamine re‐establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis‐relevant F508del‐CFTR mutation. Cell Death Dis 8: e2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cappellari M, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Saarikettu J, Silvennoinen O, Sette C (2014) The transcriptional co‐activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 33: 3794–3802 [DOI] [PubMed] [Google Scholar]
- 68. Cozza G, Girardi C, Ranchio A, Lolli G, Sarno S, Orzeszko A, Kazimierczuk Z, Battistutta R, Ruzzene M, Pinna LA (2014) Cell‐permeable dual inhibitors of protein kinases CK2 and PIM‐1: structural features and pharmacological potential. Cell Mol Life Sci 71: 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 16: 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File