

Abstract
Citrullination is a posttranslational modification of arginine catalyzed by five peptidylarginine deiminases (PADs) in humans. The loss of a positive charge may cause structural or functional alterations, and while the modification has been linked to several diseases, including rheumatoid arthritis (RA) and cancer, its physiological or pathophysiological roles remain largely unclear. In part, this is owing to limitations in available methodology to robustly enrich, detect, and localize the modification. As a result, only a few citrullination sites have been identified on human proteins with high confidence. In this study, we mined data from mass-spectrometry-based deep proteomic profiling of 30 human tissues to identify citrullination sites on endogenous proteins. Database searching of ∼70 million tandem mass spectra yielded ∼13,000 candidate spectra, which were further triaged by spectrum quality metrics and the detection of the specific neutral loss of isocyanic acid from citrullinated peptides to reduce false positives. Because citrullination is easily confused with deamidation, we synthetized ∼2,200 citrullinated and 1,300 deamidated peptides to build a library of reference spectra. This led to the validation of 375 citrullination sites on 209 human proteins. Further analysis showed that >80% of the identified modifications sites were new, and for 56% of the proteins, citrullination was detected for the first time. Sequence motif analysis revealed a strong preference for Asp and Gly, residues around the citrullination site. Interestingly, while the modification was detected in 26 human tissues with the highest levels found in the brain and lung, citrullination levels did not correlate well with protein expression of the PAD enzymes. Even though the current work represents the largest survey of protein citrullination to date, the modification was mostly detected on high abundant proteins, arguing that the development of specific enrichment methods would be required in order to study the full extent of cellular protein citrullination.
Keywords: Post-translational modifications*, Tissues*, Data evaluation, Omics, Tandem Mass Spectrometry, citrullination, human proteome, peptidylarginine deiminase, synthetic peptides
Citrullination is a protein posttranslational modification (PTM) of arginine specifically catalyzed by peptidylarginine deiminases (PADs). 1 This irreversible PTM leads to a small increase in mass of 0.9840 Da and the loss of a positive charge from the protein that may cause structural and/or functional alterations. Five PAD isoforms have been found in humans, PAD1–4 and the catalytically inactive PAD6 (1), which are believed to have distinct tissue specificities (2–6). PAD-catalyzed citrullination has been implicated in many cellular processes such as terminal epidermal differentiation, apoptosis, central nervous system stability, immune response, gene regulation, and embryonic development (7, 8). The modification has gained more attention recently because its pathological relevance has been established by the identification of autoantibodies recognizing citrullinated proteins in rheumatoid arthritis (RA) patients (9). Citrullinated fibrinogen alpha chain, filaggrin, Type II collagen, α-enolase, and vimentin have been found in the synovial fluid of RA patients (10), and these citrullinated antigens have become important biomarkers for monitoring disease progression (11, 12). Deregulation of PADs expression or activity and increased citrullinated protein levels have also been found in other diseases such as cancer, multiple sclerosis, and Alzheimer's disease, but the functional role of the modification remains largely unclear (reviewed in (7) and (8)). Furthermore, citrullination has recently been shown to participate in epigenetic regulation via the modification of histones (13). The interaction between coactivators and histone-modifying enzymes can also be regulated by citrullination, exemplified by the enhanced interaction between the coactivator GRIP1 and the histone acetyltransferase p300 when p300 was citrullinated (14). Moreover, citrullination of the splicing factor SFPQ has been shown to antagonize its methylation and thus regulate its association with mRNA (15).
Despite a lot of emerging biology, protein citrullination is far less well studied, let alone understood, compared to other PTMs. This can in part be attributed to a lack of robust biochemical enrichment and detection methods. The major current approach is immunodetection relying on the specificity and sensitivity of the antibodies available for this purpose. However, most of these reagents have narrow or poor specificity and fail to recognize the PTM globally in human proteomes (16, 17). In addition, the abundance of protein citrullination under physiological conditions is thought to be very low because, most of the time, the intracellular concentration of calcium (10–100 nm) may not be sufficient to activate the calcium-dependent PAD enzymes (7, 18, 19). As for other PTMs, mass spectrometric analysis is an attractive alternative as it does not require antibodies for detection. However, the mass increment of 0.9840 Da compared with the unmodified arginine is small and in fact identical to the frequently occurring deamidation of Asn/Gln residues (17), leading to serious ambiguity in database searching of data from complex proteome digests and calling a part of the citrullination literature into question. Biochemical enrichment is a potential way to improve the identification of low-abundance citrullinated peptides. Glyoxal derivatives have been reported to react specifically with the ureido group of citrulline under acidic conditions (20, 21), and biotinylated glyoxal derivatives have been used to enrich citrullinated proteins from the synovial fluid of RA patients and from cultured cells. This way, Tutturen et al. reported a 20-fold increase in the detection of citrullinated spectra, but the peptide identification rate was low due to the poor fragmentation efficiency of the glyoxal derivatives (22). Lewallen et al. identified more than 50 potential citrullinated proteins from human HEK293T cells when overexpressing PAD2. It is, however, unclear if these proteins would also be citrullinated under physiological conditions (23). Moreover, none of these studies unambiguously identified the exact citrullination sites. Site localization can be greatly facilitated by the observation that collision-induced dissociation spectra of citrullinated peptides display a prominent and specific loss of isocyanic acid (43.0058 Da) from the modified amino acid. For instance, Jin et al. identified citrullination sites on glial fibrillary acid protein (GFAP), myelin basic protein, and neurogranin from human brain (24), demonstrating that the diagnostic neutral loss enables the identification of endogenous protein citrullination sites.
Despite the analytical progress over the past years, only relatively few and unambiguously assigned endogenous citrullination sites have been identified on human proteins, and no systematic analysis across human tissues has been conducted. In order to begin to map out the citrullinated human proteome, we have mined deep proteomes of 30 human tissues (Wang et al., manuscript in preparation) for endogenous protein citrullination. The analysis demonstrates that the main challenge is the distinction of citrullination from the frequently occurring deamidation of Asn and Gln residues. By using the neutral loss of isocyanic acid, the immonium ion of citrulline, manual spectrum interpretation as well as using reference spectra of synthetic citrullinated or deamidated peptides, we identified 375 bona fide citrullinated sites on 209 human proteins across 26 human tissues. Even though the present study is, by far, the most comprehensive report on human protein citrullination, the fact that most modified proteins are of high abundance suggests that protein citrullination is much more common than previously anticipated. The results also highlight the need for better methods for the enrichment of this low-abundance PTM in order to advance research on its presumably many biological functions.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
This study is based on the qualitative reanalysis of deep proteomic profiling of 30 human tissues with a focus on identifying and validating citrullinated proteins. The details of the data analysis workflow and the validation experiments are described in the following sections as well as the result section. Briefly, to identify bona fide citrullination sites in the background of ∼70 million tandem mass spectra, generated by 1,116 LC-MS/MS runs (36 strong anion exchange fractions for each tissue), we used standard database search statistics (1% FDR at peptide and protein level), followed by several filtering steps. All citrullination sites reported in this study were validated by either manual spectrum interpretation and/or synthetic peptide reference spectra.
Deep Human Tissue Proteome Data
As part of a separate study, we have created deep proteome data sets from 30 human tissues that are also used as part of the Human Protein Atlas Project (25). The details of this project will be published separately (Wang et al., manuscript in preparation). Here, we describe the results of mining this data for the specific purpose of identifying endogenous protein citrullination. Briefly, tissues from different donors and sexes were lysed in urea-containing buffer, and proteins were digested with trypsin. Digests of each tissue were separated into 36 fractions by strong anion exchange chromatography as described (26). Chromatographic fractions were analyzed by nanoLC-MS/MS on a Q-Exactive+ mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) using a 110-min gradient, resulting in the identification of 11,656 to 112,411 peptides and 8,759 to 12,418 protein groups per tissue (at 1% peptide and protein FDR). Each human tissue digest was analyzed once.
Data Analysis
The data processing steps are illustrated in Fig. 1 and Fig. S1. Briefly, Mascot Distiller (version 2.5.1, Matrix Science, London) was used to process 1,116 Orbitrap .raw files into peak list files (.mgf). These were searched using Mascot (version 2.5.1, Matrix Science, London, UK) against the UniProtKB-Swiss-Prot complete human proteome, including the canonical and isoform sequences database (download from www.uniprot.org, version August 2016, 42,158 entries) and supplemented with sequences of common contaminants. The search parameters were as follows: fixed modification of Cys (carbamidomethylation); variable modification of methionine oxidation, N-terminal acetylation, N-terminal Gln to pyro-Glu conversion, and deamidation of Arg (citrullination), Asn, and Gln. The neutral loss of isocyanic acid (HNCO, 43.0058 Da) was added to the definition of citrullination in the search algorithm. The enzyme specificity was set to trypsin, allowing for up to two missed cleavage sites. Peptide mass tolerance was set to 10 ppm and fragment ion mass tolerance to 0.05 Da. The target decoy option of Mascot was enabled. Mascot search results (DAT files) were imported into Scaffold (version 4.7.3, Proteome Software, Inc., Portland, OR). Peptide identifications were initially accepted if they had greater than 90.0% probability to achieve an FDR less than 1.0% by the Scaffold Local FDR algorithm. Protein identifications were initially accepted if they had greater than 99.0% probability to achieve an FDR less than 1.0% and contained at least one identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (27). Proteins sharing significant peptide evidence were grouped into clusters. Filters including removing PSMs with C-terminal citrullination (3,981 PSMs), and poor quality PSMs (Mascot ion score below identity score; 3,078 PSMs) were applied to decrease the number of spectra required to be manually inspected. Next, all spectra assigned to citrullinated PSMs were extracted from the Mascot peak list files, these were searched again against Swissprot (see above), and the results file was imported into Scaffold for visualization and further evaluation of spectra. The list of identified citrullinated peptides and proteins is shown in Table S1. Candidate citrullinated PSMs were inspected and validated manually, placing emphasis on the overall spectrum quality, presence of modified fragment ions, and the detection of neutral loss ions. Based on these criteria, the citrullinated PSMs were categorized into poor quality, ambiguous, and valid (MI, manual inspection). See Table S2 for a breakdown of PSMs assigned to the different categories.
Fig. 1.
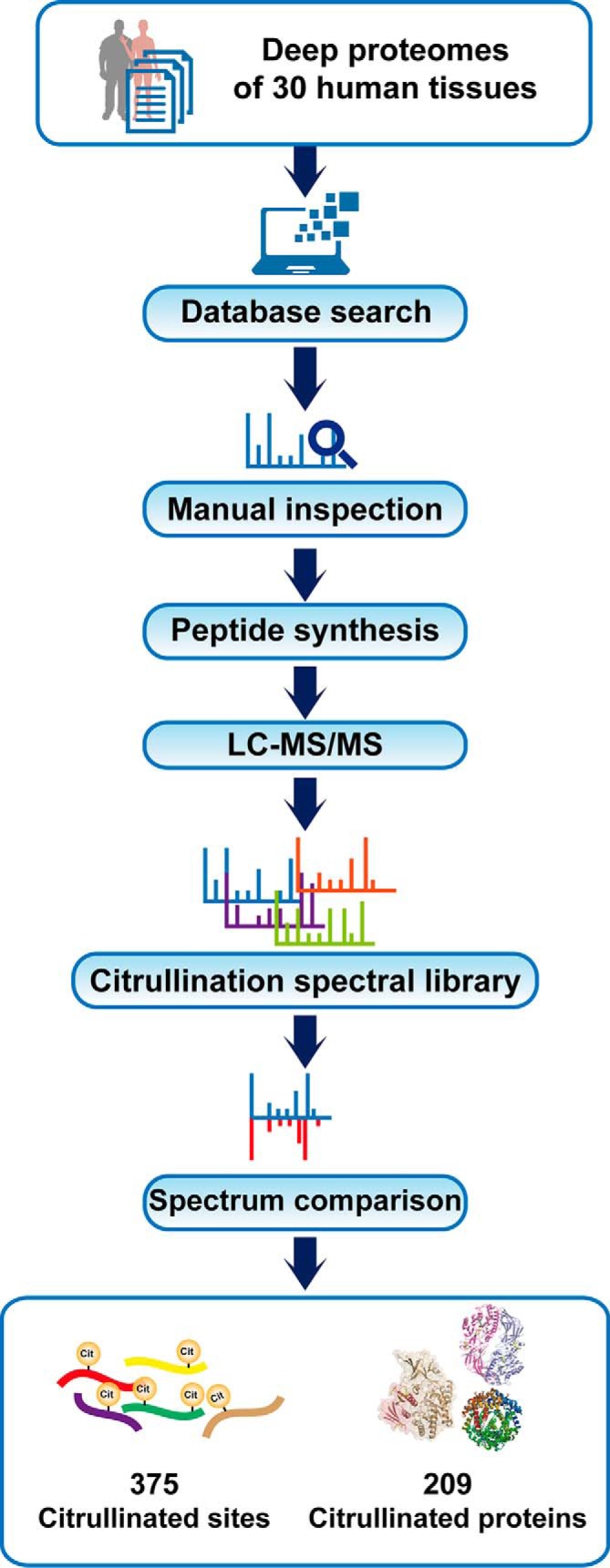
Workflow for the identification and validation of citrullinated peptides in human tissue proteomes.
Synthetic Peptides
To validate potential citrullinated peptides and to distinguish these from otherwise deamidated peptides, a total of 2,248 unique citrullinated (form short list and long list, Fig. S1), and 1,312 deamidated peptides were synthesized in a total of 25 separate pools, minimizing the number of isobaric peptides in each pool (Table S3). This list of synthetic peptides was built from the following criteria. We included 276 peptides that were valid by manual inspection (length of 7–25 amino acids; designated as “short list”). A second “long list” contained 3,506 peptides of all database search results (poor quality/ambiguous/valid), including 2,194 Cit peptides and 1,312 deamidated peptides. Please, note that 222 Cit peptides in the short list were also covered by the long list.
LC-MS/MS Measurement and Data Analysis of Synthetic Peptides
All synthetic peptide pools were analyzed by nanoLC-MS/MS on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) as previously described (28). Briefly, each peptide pool was measured using a survey method consisting of an Orbitrap full MS scan (60,000 resolution, 5 × 105 automatic gain control (AGC) target, 50 ms maximum injection time, 360–1,300 m/z, profile mode), followed by MS2 events with a duty cycle of 2 s for the most intense precursors and a dynamic exclusion set to 5 s as follows: (i) HCD scan with 28% normalized collision energy and Orbitrap readout (15,000 resolution, 1 × 105 AGC target, 22 ms maximum injection time, inject ions for all available parallelizable time enabled, 1.3 m/z isolation width, centroid mode); (ii) CID scan with 35% normalized collision energy and ion trap readout (rapid mode, 3 × 104 AGC target, 0.25 activation Q, 22-ms maximum injection time, inject ions for all available parallelizable time enabled, 1.3 m/z isolation width, centroid mode). For the purpose of this study, only the MS2 spectra from HCD (NCE 28) fragmentation event with Orbitrap readout were used as reference spectra. The MS data were processed by Mascot distiller, and all .raw files were searched by Mascot using the same parameters, database, and acceptance criteria indicated above for the human tissue data. In addition, we required that PSMs of synthetic peptides matched to the correct pool in which they were synthesized. The results file was imported into Scaffold for visualization and further evaluation of spectra. The list of synthetic peptides identification is shown in Table S1.
Spectrum Comparisons
As part of the validation process, we systematically compared HCD spectra of endogenous and synthetic peptides using spectral-contrast-angle analysis (29) between spectra extracted and aligned from .mgf files that were identified as the same modified sequence with matching precursor charge state using a custom R script. We applied a cutoff of 0.5 for the spectral-contrast-angle and chose the best matching spectrum (i.e. highest correlation) of each endogenous peptide for subsequent manual inspection. This cutoff was chosen based on the observation that most PSMs passed manual inspection in case they had a spectral-contrast-angle analysis of 0.5 or higher. Spectra passed examination if most of the intense fragment ions in the spectra matched to the reference spectra. However, since the spectral-contrast-angle analysis does not take the total number of matched peaks into account, spectra with few intense peaks were disregarded even if they showed high spectral-contrast-angle correlation with reference spectra.
PADs Expression Profiling
MS-based protein quantification of PAD isoform expression acrossin the 30 human tissues followed the intensity-based absolute quantification (iBAQ) approach (30) implemented in MaxQuant (31, 32) (version 1.5.3.30) (Wang et al., manuscript in preparation). Briefly, iBAQ values (Fig. 6B, left y axis) for a given protein are derived from the summed intensities of the precursor peptides that map to each protein and divided by the number of theoretically observable peptides. Therefore, iBAQ values are proportional to the molar quantities of proteins in a sample, which can be used to roughly estimate the relative abundance of the proteins within each sample.
Fig. 6.

Analysis of protein citrullination across 30 human tissues. (A) Distribution of Cit peptides and proteins as validated by manual inspection (MI), comparison to synthetic reference peptide spectra (spectrum comparison, SC), or both. (B) Comparison of PAD isoform expression and distribution of Cit PSMs and proteins across 30 human tissues. PSMs, peptide spectrum matches; iBAQ, intensity-based absolute quantification.
Motif Analysis
pLOGO (33) was used to investigate and visualize the substrate motif preference of PADs. Based on the list of validated endogenous citrullinated peptides, the amino acid frequencies ranging from -6 to +6 residues around the citrullination site were analyzed and compared with the respective frequencies of all sequences in Uniprot as background. Motifs were filtered for statistical significance (p = 0.05; Bonferroni corrected).
Gene Ontology (GO) Enrichment Analysis
GO analysis was performed using GOrilla (34). For the combined analysis of all Cit proteins identified in tissues, all genes from the human genome (21,042 entries) were used as background set. For the analysis of citrullinated proteins identified in individual tissues, only genes were included as background that were identified in their respective proteomes. The p value threshold was set to 10e-05.
RESULTS AND DISCUSSION
Mining Deep Human Tissue Proteomes for Protein Citrullination
The workflow for the identification and validation of citrullination sites in human protein is shown in Fig. 1 (see Fig. S1 for more details). After combined database searching of 1,116 peak list files, ∼9 million PSMs, including 13,031 putative citrullinated PSMs (Cit-PSMs) were identified across all tissues (Figs. S1 and S2, Table S2). We note that these only represent 0.14% of all PSMs (ranging from 0.03–0.4% depending on the tissue). This not only indicates that citrullination is an overall low abundance modification, it also suggests that most of the 13,031 putative Cit-PSMs may be false matches given the FDR criteria applied to the data (1% at peptide and protein level). Therefore, additional evidence is required in order to identify bona fide citrullination events. We next removed all PSMs in which the C-terminal arginine residue was marked as the citrullination site (C-term Cit) because previous biochemical work has shown that the catalytic activity of trypsin for benzyl citrulline is 105-fold lower than that for benzyl arginine (36) therefore resulting in a missed cleavage at each citrullination site. Indeed Bennike et al. showed that more than 50% of false-positive citrullination annotations in data from the analysis of the synovial fluid of a RA patient could be removed by requiring a missed cleavage (37). Consistent with this observation, the rate of (erroneous) C-terminal citrullination annotation ranged from 10–48% between human tissues (Table S2, Fig. S2B). We also removed all PSMs for which the Mascot ion score was below the identity threshold thus going beyond a simple FDR cutoff as well as PSMs of low signal-to-noise ratio or that contained few fragment ions. Depending on the tissue analyzed, these filtering steps removed 22–85% of all putative Cit-PSMs illustrating the limitations of current database search algorithms with respect to unambiguously identifying low frequency PTMs.
Modification Verification by Manual Spectrum Interpretation
All remaining spectra were manually inspected for the presence of diagnostic ions (neutral loss of 43.0058 Da, immonium ion at 130.0975 m/z), fragment ion coverage of the putative citrullination site or the presence of Asn or Gln residues that may be deamidated and thus providing an alternative interpretation for these PSMs. Fig. 2 illustrates the manual spectrum validation process by four examples. The HCD spectrum of the peptide GDFSSANN(R+1)DNTYNR of fibrinogen alpha chain (Fig. 2A; R+1 denoting the citrullinated residue) displays a near complete y-ion series, including the citrullination site, and each y-ion that contains the site shows a loss of isocyanic acid. This PSM therefore represents a bona fide citrullinated peptide and also localizes the modification site. A strong Cit-immonium ion is also detected, lending further support for the presence of the modification. Figs. 2B–D show examples of ambiguous citrullination assignments. The HCD spectrum of E(R+1)YFD(R+1)INENDPEYIR (Fig. 2B) shows the diagnostic loss of 43 Da from the precursor ion but no y- or b-ions identifying the modification site. The data shown in Fig. 2C for the peptide ALE(R+1)GLQDEDGYR show no neutral loss, but the peptide does not contain Asn or Gln residues leaving a possibility that the peptide might be citrullinated. Finally, the HCD spectrum shown for N(R+1)SSAVDPEPQVK does contain an N-terminal Asn residue that may or may not be deamidated instead of the assigned citrullinated residue, but no information is present in the spectrum that would resolve this ambiguity. Based on the above, manual spectrum interpretation resulted in 2,091 valid and 3,881 ambiguous Cit-PSMs. Across all 30 tissues, the validation rate of PSMs by MI ranged from 0–57% (average of 9%) and 32–73% of all Cit-PSMs remained ambiguous at this stage (average of 56%; Table S2, Fig. S2).
Fig. 2.

Example HCD tandem mass spectra illustrating criteria for the validation of peptide citrullination. (A) A valid Cit peptide is characterized by the detection of fragment ions covering the presumed Cit modification site as well as the detection of more than one neutral loss ion from Cit fragment ions. NL, neutral loss of isocyanic acid. (B) Ambiguous assignments may be represented by a single diagnostic neutral loss ion and the absence of site-determining fragment ions. Here, the y11 ion determining the Cit site is missing, but a neutral loss from the precursor ion as well as a b2/b3 pair can be detected that localizes the Cit site. (C) An ambiguous assignment is also made when no neutral loss ion is detected but the peptide sequence does not contain any residues that may be deamidated (N or Q). (D) Finally, an ambiguous assignment is also made when the sequence not covered by fragment ions contains N/Q residues, raising the probability that the peptide may be deamidated rather than citrullinated.
Modification Verification by Comparison to Synthetic Peptides
One way of independently validating the verification process is to compare the experimental spectrum of a PTM peptide to that of a synthetic standard. As part of the ProteomeTools project (28), we are generating hundreds of thousands of modified peptides for this purpose. Based on the data at hand, we attempted synthesis of 276 citrullinated peptides (validated by MI) as well as 2,194 further candidate citrullinated peptides from the result of database searching. In addition, we included 1,312 peptides in which we replaced Asn and Gln residues with Asp or Glu, respectively, in putative citrullinated peptides in order to assess how often citrullination may be confused with deamidation. Example HCD spectra for an instructive case are shown in Fig. 3. The spectrum of the endogenous peptide E(R+1)YFD(R+1)INENDPEYIR (Fig. 3A) did not allow the unambiguous assignment of two citrullination sites on this peptide because the characteristic loss of isocyanic acid was only observed from the precursor ion, and there are two further Asn residues that, if deamidated, would offer an alternative explanation. However, the HCD spectrum of the corresponding synthetic reference peptide (Fig. 3B) is essentially identical to that of the endogenous peptide (spectral-contrast-angle correlation of 0.76; Table S4), indicating that the database search made the correct assignment. This is supported by the spectra of the synthetic peptides shown in Figs. 3C–F. The presence of Asn deamidation in these sequences drastically changes the appearance of the respective HCD spectra (and often the precursor ion charge state) thus ruling out that the endogenous peptide is deamidated instead of citrullinated. We next performed systematic comparisons of all spectra of the 1,567 successfully synthetized citrullinated and deamidated peptides with the respective endogenous peptides using the spectral-contrast-angle approach (Table S4). Manual inspection showed that HCD spectra with spectral contrast angles of >0.5 mostly represent genuine Cit-PSMs, resulting in the validation of 216 citrullinated peptides. When combining the results of manual inspection and spectral comparison to synthetic peptides, we obtained 375 validated citrullination sites on 209 proteins (Table S5).
Fig. 3.
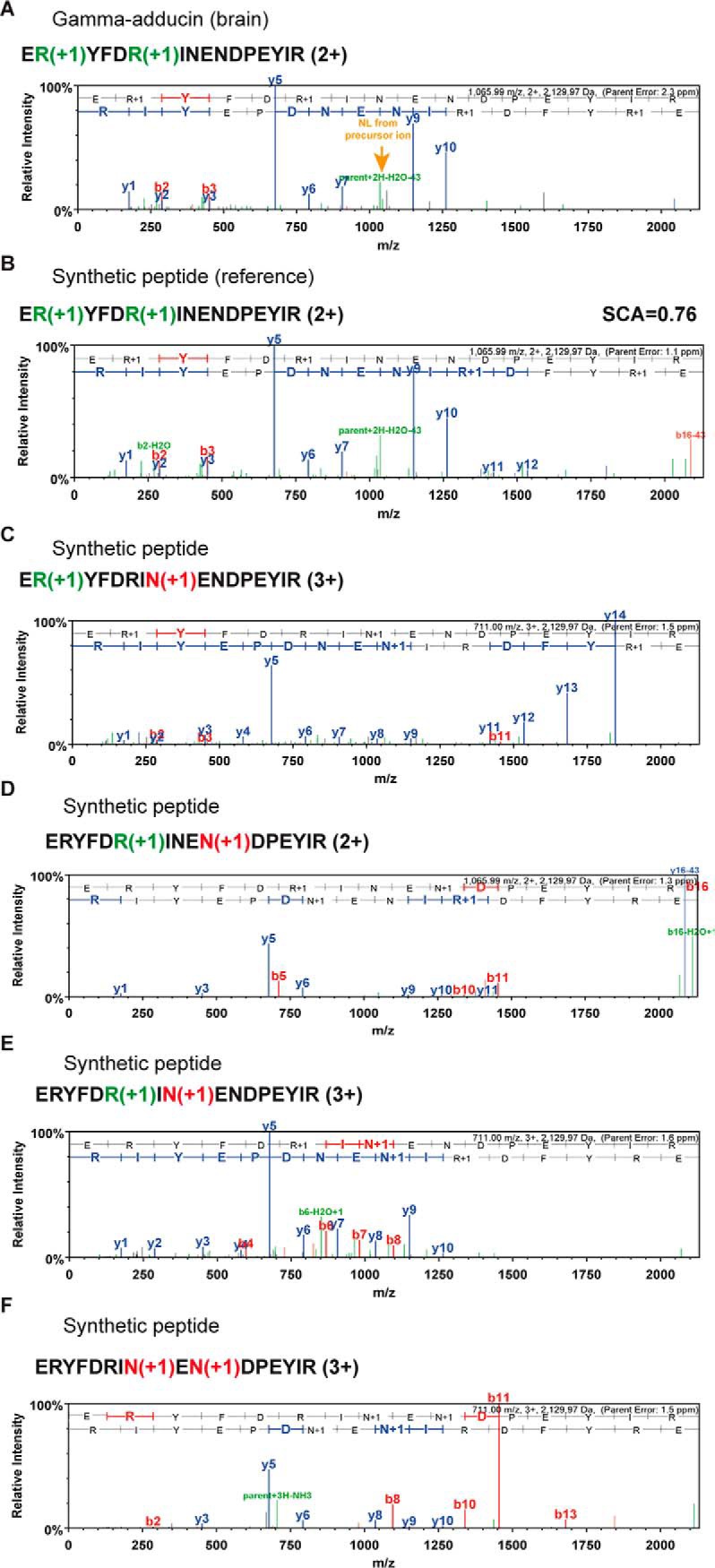
Validation of peptide citrullination by synthetic peptides. (A) HCD spectrum of an endogenously doubly citrullinated peptide from gamma-adducin from human brain. (B) Reference spectrum of the corresponding synthetic peptide. The two spectra are virtually identical, thus validating the Cit modification. (C and D) Spectra of synthetic peptides in which citrullinated Arg residues are replaced by deamidated Asn residues. These spectra are very different from the endogenous peptide shown in Panel A, thus illustrating how such permutations can aid in the validation of Cit peptides. R(+1), citrullination; N(+1), deamination.
Value of Diagnostic Fragment Ions
Considering the very large number of MS2 spectra and search results from our deep proteome data, manual validation is not feasible in many cases and may also not be free of error. The presence of ions diagnostic for citrullination may thus be useful to automate the process of identifying and validating the modification akin to the value of such ions for other modifications (e.g. loss of 64 Da from oxidized methionine, immonium ions of phosphotyrosine, or acetylated lysine residues (38)). To test this hypothesis, we extracted 295,368 MS2 spectra from 1,116 raw MS files that contained a putative citrulline immonium ion (130.0975 m/z; 0.01 m/z tolerance). Database searching of these spectra identified merely 84 Cit-PSMs of which 39 passed manual inspection representing 19 citrullinated peptides (Table 1, Fig. 4A). This is far lower than the number of valid citrullinated peptides obtained using all MS2 spectra, indicating that the detection of a putative citrullination immonium ion is not very diagnostic even though PSMs that do contain such an immonium ion tend to have a higher validation rate than PSMs that do not (∼50%; Fig. 4B). This was confirmed by the separate analysis of the corresponding synthetic peptides for which less than 20% of all spectra contained the immonium ion (Fig. 4C). In contrast, the neutral loss of isocyanic acid from the precursor ion was observed in a third of all spectra, and the loss from fragment ions was detectable in nearly 94% of all citrullinated spectra, and 98% of all validated citrullinated spectra contained at least one of these three fragment ions. Still, care has to be taken as the loss of isocyanic acid is not entirely specific for citrulline. Tandem mass spectra of carbamylated lysine (i.e. homocitrulline) residues that can be formed when using urea-containing lysis buffers show the same loss of isocyanic acid owing to the structurally very similar side chains of citrulline and homocitrulline ((39); see Fig. S3 for an example). However, this would only confound the analysis in very rare cases. Citrullination and homocitrullination (mass difference of a 14 Da for the extra CH2 group in homocitrulline) can only be confused in cases in which a peptide contains at least two missed cleavage sites, of which one represents a potential citrulline and the other a homocitrulline or vice versa.
Table I. Data summary for the identification of protein citrullination in human tissues.
| Search approach | Total PSMs | Cit PSMs | aC-term Cit | bBelow identity score | cAmbiguous | dValid (MI) | Valid peptides (MI) | Valid proteins |
|---|---|---|---|---|---|---|---|---|
| Database search | 8,988,146 | 13,031 | 3,981 | 3,078 | 3,881 | 2,091 | 301 | 154 |
| Immonium ion | 295,838 | 84 | 21 | 14 | 10 | 39 | 19 | 17 |
a PSMs with Cit annotation at the C-terminus of a tryptic peptide were categorically rejected because trypsin would not generate such a peptide.
b PSMs with Mascot ion scores below the identity threshold were discarded to reduce spurious PSMs.
c PSMs with otherwise ambiguous spectra, such as incomplete Cit-containing fragment ion series and sequence ambiguities regarding deamindation/citrullination.
d PSMs with at least two neutral loss ions from Cit fragment ions in MS2 spectra. MI, manual inspection.
Fig. 4.

Diagnostic fragment ions in HCD spectra of citrullinated peptides. (A) Comparison of the number of unique Cit peptides identified by database searching (all tissues) to those whose HCD spectra contain citrulline immonium ion (130.0975 m/z). It is apparent that only a minority of all Cit peptide spectra contain the immonium ion. (B) In case a Cit immonium ion can be detected, there is a higher chance that the peptide is a genuine Cit peptide. MI, manual inspection. (C) Comparison of the prevalence of diagnostic ions in HCD spectra of synthetic peptides. It is apparent, that the neutral loss (NL) ion is of more diagnostic value than the detection of the immonium ion.
Decision Tree for Citrullination Identification
As mentioned above, current database search algorithms strongly overestimate the number of potential citrullinated peptides in large data sets. The results from the systematic and careful analysis performed here confirmed earlier individual observations but also enabled us to formulate a decision tree that could be incorporated into search engines in the future in order to automate the correct assignment of protein citrullination in large data sets (Fig. 5). First, it is mandatory to specify deamidation as a variable modification because we found this to be a major source of error. Second, the neutral loss of 43 Da should be specified in the modification table of the search engine akin to the loss of 64 Da from oxidized methionine, and future versions of search engines should use this neutral loss for scoring. Third, any modification to the C-terminal arginine should be categorically rejected because trypsin will not generate such a cleavage. Fourth, particularly in very large data sets, individual PSM scores should be considered rather than global or local FDR cutoffs in order to reduce the number or random matches. Fifth, we found it important to manually inspect spectra for the presence of diagnostic ions with the neutral loss of isocyanic acid being more valuable (albeit not entirely specific) than the detection of the immonium ion. This step should be straightforward to automate in a search engine. Sixth, any remaining candidates should be checked for the presence of N/Q residues not covered by fragment ions to eliminate the modification assignment to remaining deamidated PSMs, and the (rare) case of the conversion of lysine into homocitrulline should also be considered.
Fig. 5.

Decision tree summarizing the experience obtained in this study and aiding the identification and validation of citrullinated peptides from HCD spectra. NL, neutral loss of isocyanic acid.
The Landscape of Protein Citrullination of Human Tissues
Having established a rigorous method for the identification of citrullinated peptides, we examined the distribution of the modification across the 30 tissues investigated (Fig. 6 and Fig. S2A, Table S5). The modification was identified in 26 tissues, and although we cannot make strong quantitative statements, we note that the number of identified modified proteins and peptides varied drastically (Fig. S2). The largest number of citrullinated proteins was found in the brain consistent with previous studies reporting high levels of citrullination in this organ and its role in the development of the central nervous system (40, 41). Our data cover many previously identified citrullinated proteins, but >80% of the identified modifications sites were new, and, for 56% of the proteins, citrullination was detected for the first time. For example, myelin basic protein and GFAP are the most well-studied citrullinated proteins in the brain. Hypercitrullination of these proteins shows an association with multiple sclerosis and Alzheimer's disease (42, 43). We identified 11 and 16 in vivo citrullination sites of myelin basic protein and GFAP from brain, respectively. Ten of the 11 sites of myelin basic protein were reported in a previous publication or in Uniprot (24), showing the consistency of our analysis with the current state-of-the-art. GFAP is a major component of intermediate filaments in astrocytes and a known substrate of PAD2. Interestingly, we identified 14 novel citrullination sites on GFAP (Table S5), suggesting a stronger role of the modification for GFAP biology than hitherto anticipated. Surprisingly, even among the most highly citrullinated proteins in the brain, we found citrullination for the first time (CNP, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase; six sites, Fig. 7A).
Fig. 7.

Discovery of citrullinated proteins across human tissues. (A) Ranked list of the ten most highly citrullinated proteins in this study. Numbers on top of columns indicate the number of tissues in which the protein was found to be citrullinated. (B) Ranked list of how many Cit proteins were detected in how many tissues. (C) Abundance distributions of all proteins identified in each of 26 human tissues in which citrullination was detected. Blue circles indicate citrullinated proteins, and blue horizontal lines indicate the median abundance of the (likely partially) modified proteins.
The second highest number of citrullinated peptides was identified in lung tissue. Previous studies have shown that (chronic) inflammation induced by smoking and environmental stimuli such as infection can increase the levels of inflammatory cytokines such as TNF-α in the lung and trigger protein citrullination (44, 45). The notion that citrullination is an inflammation-dependent process (46) may be substantiated by our observation that the third highest levels of citrullination was found in placenta, a tissue with strong systemic inflammatory responses during normal pregnancy and acute inflammation during labor (47). Interestingly, the majority of proteins was only found in a single tissue in a citrullinated form (Fig. 7B). While this may have technical reasons (i.e. low abundance of the modification, varying sampling depth in each proteome, stochastic nature of data-dependent acquisition), it is also possible that the modification is regulated in an organ-specific fashion. To this end, we examined the expression levels of PAD isoforms across the organs as these enzymes are the only known proteins that can catalyze the conversion from arginine to citrulline. As shown in Fig. 6B, the expression of PAD enzymes (estimated by the iBAQ approach) varied greatly between tissues, consistent with earlier reports (8, 48) and immunohistochemistry data available for the PAD enzymes in the Protein Atlas Project (25). For example, in the brain, the expression of PAD2 is 1,000-fold higher than that of PAD4 and at least fivefold higher than in other PAD2-expressing tissues. PAD4 was also found in a number of tissues with the highest relative expression in fat tissue and the spleen. The latter may be rationalized by the presence of a large number of white blood cells in which PAD4 is highly expressed (49). The relatively high PAD4 levels in fat and the detection of citrullinated proteins in this tissue are novel observations, and one can only speculate what this may mean in terms of biology. However, a previous study has reported that PAD-mediated histone hypercitrullination can induce the formation of macrophage extracellular traps in inflamed adipose tissue of obese patients (50), making yet another link of citrullination to inflammatory processes. Somewhat surprisingly, we found that the expression of PAD enzymes was not strongly correlated with the number of identified Cit-PSMs or peptides (Fig. 6B). As no activity of PAD6 could so far be established in vitro or in vivo (1), the citrullination sites identified in this study most likely stems from enzymatic activity of PAD1–4. Consistent with information in the literature, the data suggest that PAD2 may be responsible for most of the PAD activity in several tissues (51). A possible explanation for the lack of a clear relationship between citrullination levels and PAD protein expression levels is that the enzymatic activity of PADs requires calcium as a co-factor, which is regulated by intracellular calcium ion levels. One may speculate that the physiological intracellular concentration of calcium (10–100 nm) may not always be high enough to activate these enzymes (7, 18, 19) but that some tissues maintain a pool of these proteins to be able to activate these rapidly if required. Given the reasonably large number of identified citrullination sites (375), we performed a motif analysis to investigate if PAD enzymes show preferences for certain arginines within a sequence (Figs. S4A–C). Although no particularly strong motif was observed in this analysis, we found that Asp and Ser were represented more frequently at the -1 position, and Asp and Gly residues were overrepresented at the +1 position consistent with previous studies arriving at similar conclusions (52, 53).
Given that we did not perform any enrichment for citrullinated proteins or peptides, the question arose as to how comprehensive our analysis of the modification may be. When plotting the abundance of all proteins in all tissues (Fig. 7C), it became apparent that most proteins for which we identified citrullination sites were also of very high abundance. This strongly suggests that there could be many more citrullination sites on endogenous human proteins than represented by our or any other study performed to date and highlighting the strong need for the development of specific enrichment methods that may enable to describe the landscape of protein citrullination more comprehensively. The citrullination motif mentioned above might enable the generation of citrullination-specific antibodies that might address this need. We identified a total of 209 citrullinated proteins, and more than half of these have not been reported before to be modified in this way. As most of them were only identified in one tissue (∼72%) (Fig. 7B), one might speculate that protein citrullination may regulate or be regulated by distinct physiological factors in different tissues. We note here that, for most citrullination sites, we also identified the unmodified peptide, indicating that the modification is substoichiometric, which in turn could be the reason for why most modification sites were only identified in one or few tissues. That said, we also found that a few citrullinated proteins were identified in many tissues, possibly indicating a broader biological role.
Combined or tissue-centric GO term analysis of citrullinated proteins did not return strong associations. The majority of citrullinated proteins was associated with the very broad categories of cytoskeleton organization or RNA binding. Still, some interesting observations on individual proteins could be made. As mentioned in the introduction, citrullination participates in epigenetic and gene expression control. Interestingly, in our data, we found that several RNA-binding proteins were identified in more than one tissue and with more than one citrullination site. Citrullinated RNA-binding motif protein X chromosome has been reported (23) and was identified across 14 tissues with 10 citrullination sites in our study (Fig. 7A). The protein has been reported to regulate tissue-specific gene transcription and alternative splicing of several pre-mRNAs (54, 55). As none of the citrullination sites are located in the N- and C-terminal RNA-binding regions, citrullination of RNA-binding motif protein X chromosome might not disturb its RNA-binding ability but potentially affect the structural stability or cellular localization of the protein because two of the citrullination sites, R223 and R232, are localized in a region essential for the association with nascent RNA polymerase II (RNAPII) and nuclear localization (55). Another example is TATA-binding protein-associated factor 2N (TAF15), which was identified in seven tissues with six citrullination sites. TAF15, together with RNAPII, is part of the transcription preinitiation complex at distinct promoters (56). It is interesting that all of the identified citrullination sites are located between residues 428–550. The possible reason is that TAF15 contains 21 tandem repeats of the sequence DR[S,G]GGYGG between residues 407–575, and the arginine in this sequence is flanked by Asp, Gly, and Ser residues which are highly favored substrates of PAD2 and PAD4 according to our motif analysis and the literature (52, 53). Anti-citrullinated protein antibodies are present in the majority of patients with early RA. Hence, the identification of novel citrullinated proteins and sites (i.e. potential epitopes) could aid in the development of novel or more refined diagnostic targets for RA. In particular, citrullinated fibrinogen alpha chain, filaggrin, Type II collagen, α-enolase, and vimentin are well-known autoantigens in RA patients (10). In our data, citrullinated fibrinogen alpha chain, filaggrin, collagen, and vimentin were also found to be citrullinated in tissues under healthy conditions. Particularly, citrullinated fibrinogen alpha chain was found in 16 tissues (nine sites), suggesting that site stoichiometry needs considering when using or developing diagnostic markers of RA based on citrullination autoantigens.
In conclusion, this study demonstrated that protein citrullination is much more frequent than hitherto anticipated and that it is possible to unambiguously identify this modification by mass-spectrometry-based proteomics provided that care is exercised during data analysis. The data also enabled us to propose changes to database search engines so that the analysis of protein citrullination may be facilitated in the future. Although this study reports the largest number of citrullination site to date, the potentially many biological roles of the modification need to be further examined in the future. This would be greatly aided by the development of chromatographic or antibody-based methods for the enrichment of citrullinated peptides or proteins akin to what has been achieved for other PTMs.
DATA AVAILABILITY
The MS proteomics data, Mascot search results, and Scaffold files with annotated spectra have been deposited with the ProteomeXchange Consortium via the PRIDE (35) partner repository with the data set identifier PXD008970.
Supplementary Material
Footnotes
* This work was in part funded by the German Federal Ministry of Education and Research (BMBF; grant no. 031L0008A; Proteome-Tools project) and also was supported by the German Academic Exchange Service (DAAD; grant no 57261473) and The Ministry of Science Technology of Taiwan for providing scholarships to C.-Y.L.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PAD
- peptidylarginine deiminases
- Cit
- citrullinations/citrullinated
- RA
- rheumatoid arthritis
- HCD
- high energy collision dissociation
- PSM
- peptide-spectrum match
- FDR
- false discovery rate
- iBAQ
- intensity-based absolute quantification
- MI
- manual inspection.
REFERENCES
- 1. Hirofumi T., Gomi T., Knuckley B., Thompson P. R., Vugrek O., Hirata K., Miyahara T., Shinoda K., Hounoki H., Sugiyama E., Usui I., Urakaze M., Tobe K., Ishimoto T., Inoue R., Tanaka A., Mano H., Ogawa H., and Mori H. (2011) Purification of enzymatically inactive peptidylarginine deiminase type 6 from mouse ovary that reveals hexameric structure different from other dimeric isoforms. Adv. Biosci. Biotechnol. 2, 7 [Google Scholar]
- 2. Nakashima K., Hagiwara T., Ishigami A., Nagata S., Asaga H., Kuramoto M., Senshu T., and Yamada M. (1999) Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3). J. Biol. Chem. 274, 27786–27792 [DOI] [PubMed] [Google Scholar]
- 3. Kanno T., Kawada A., Yamanouchi J., Yosida-Noro C., Yoshiki A., Shiraiwa M., Kusakabe M., Manabe M., Tezuka T., and Takahara H. (2000) Human peptidylarginine deiminase type III: Molecular cloning and nucleotide sequence of the cDNA, properties of the recombinant enzyme, and immunohistochemical localization in human skin. J. Invest. Dermatol. 115, 813–823 [DOI] [PubMed] [Google Scholar]
- 4. Ishigami A., Ohsawa T., Asaga H., Akiyama K., Kuramoto M., and Maruyama N. (2002) Human peptidylarginine deiminase type II: Molecular cloning, gene organization, and expression in human skin. Arch. Biochem. Biophys. 407, 25–31 [DOI] [PubMed] [Google Scholar]
- 5. Guerrin M., Ishigami A., Méchin M. C., Nachat R., Valmary S., Sebbag M., Simon M., Senshu T., and Serre G. (2003) cDNA cloning, gene organization and expression analysis of human peptidylarginine deiminase type I. Biochem. J. 370, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J., Dai J., Zhao E., Lin Y., Zeng L., Chen J., Zheng H., Wang Y., Li X., Ying K., Xie Y., and Mao Y. (2004) cDNA cloning, gene organization and expression analysis of human peptidylarginine deiminase type VI. Acta Biochim. Polonica 51, 1051–1058 [PubMed] [Google Scholar]
- 7. György B., Tóth E., Tarcsa E., Falus A., and Buzás E. I. (2006) Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 38, 1662–1677 [DOI] [PubMed] [Google Scholar]
- 8. Witalison E. E., Thompson P. R., and Hofseth L. J. (2015) Protein arginine deiminases and associated citrullination: Physiological functions and diseases associated with dysregulation. Curr. Drug Targets 16, 700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schellekens G. A., de Jong B. A., van den Hoogen F. H., van de Putte L. B., and van Venrooij W. J. (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 101, 273–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wegner N., Lundberg K., Kinloch A., Fisher B., Malmström V., Feldmann M., and Venables P. J. (2010) Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 233, 34–54 [DOI] [PubMed] [Google Scholar]
- 11. Pruijn G. J., Wiik A., and van Venrooij W. J. (2010) The use of citrullinated peptides and proteins for the diagnosis of rheumatoid arthritis. Arthritis Res. Therapy 12, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puszczewicz M., and Iwaszkiewicz C. (2011) Role of anti-citrullinated protein antibodies in diagnosis and prognosis of rheumatoid arthritis. Arch. Med. Sci. 7, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fuhrmann J., and Thompson P. R. (2016) Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem. Biol. 11, 654–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee Y. H., Coonrod S. A., Kraus W. L., Jelinek M. A., and Stallcup M. R. (2005) Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc. Natl. Acad. Sci. U.S.A. 102, 3611–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snijders A. P., Hautbergue G. M., Bloom A., Williamson J. C., Minshull T. C., Phillips H. L., Mihaylov S. R., Gjerde D. T., Hornby D. P., Wilson S. A., Hurd P. J., and Dickman M. J. (2015) Arginine methylation and citrullination of splicing factor proline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. RNA 21, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Ceuleneer M., Van Steendam K., Dhaenens M., and Deforce D. (2012) In vivo relevance of citrullinated proteins and the challenges in their detection. Proteomics 12, 752–760 [DOI] [PubMed] [Google Scholar]
- 17. Hensen S. M., and Pruijn G. J. (2014) Methods for the detection of peptidylarginine deiminase (PAD) activity and protein citrullination. Mol. Cell Proteomics 13, 388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y. L., Lee C. Y., Huang Y. N., Chen H. Y., Liu G. Y., and Hung H. C. (2017) Probing the roles of calcium-binding sites during the folding of human peptidylarginine deiminase 4. Scientific Rep. 7, 2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knuckley B., Causey C. P., Jones J. E., Bhatia M., Dreyton C. J., Osborne T. C., Takahara H., and Thompson P. R. (2010) Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 49, 4852–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tutturen A. E., Holm A., Jørgensen M., Stadtmüller P., Rise F., and Fleckenstein B. (2010) A technique for the specific enrichment of citrulline-containing peptides. Anal. Biochem. 403, 43–51 [DOI] [PubMed] [Google Scholar]
- 21. Tutturen A. E., Holm A., and Fleckenstein B. (2013) Specific biotinylation and sensitive enrichment of citrullinated peptides. Anal. Bioanal. Chem. 405, 9321–9331 [DOI] [PubMed] [Google Scholar]
- 22. Tutturen A. E., Fleckenstein B., and de Souza G. A. (2014) Assessing the citrullinome in rheumatoid arthritis synovial fluid with and without enrichment of citrullinated peptides. J. Proteome Res. 13, 2867–2873 [DOI] [PubMed] [Google Scholar]
- 23. Lewallen D. M., Bicker K. L., Subramanian V., Clancy K. W., Slade D. J., Martell J., Dreyton C. J., Sokolove J., Weerapana E., and Thompson P. R. (2015) Chemical proteomic platform to identify citrullinated proteins. ACS Chem. Biol. 10, 2520–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin Z., Fu Z., Yang J., Troncosco J., Everett A. D., and Van Eyk J. E. (2013) Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics 13, 2682–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Pontén F. (2015) Proteomics. tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 26. Ruprecht B., Wang D., Chiozzi R. Z., Li L. H., Hahne H., and Kuster B. (2017) Hydrophilic strong anion exchange (hSAX) chromatography enables deep fractionation of tissue proteomes. Methods Mol. Biol. 1550, 69–82 [DOI] [PubMed] [Google Scholar]
- 27. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 28. Zolg D. P., Wilhelm M., Schnatbaum K., Zerweck J., Knaute T., Delanghe B., Bailey D. J., Gessulat S., Ehrlich H. C., Weininger M., Yu P., Schlegl J., Kramer K., Schmidt T., Kusebauch U., Deutsch E. W., Aebersold R., Moritz R. L., Wenschuh H., Moehring T., Aiche S., Huhmer A., Reimer U., and Kuster B. (2017) Building proteome tools based on a complete synthetic human proteome. Nat. Methods 14, 259–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wan K. X., Vidavsky I., and Gross M. L. (2002) Comparing similar spectra: From similarity index to spectral contrast angle. J. Am. Soc. Mass Spectr. 13, 85–88 [DOI] [PubMed] [Google Scholar]
- 30. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 32. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protocols 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 33. O'Shea J. P., Chou M. F., Quader S. A., Ryan J. K., Church G. M., and Schwartz D. (2013) pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 10, 1211–1212 [DOI] [PubMed] [Google Scholar]
- 34. Eden E., Navon R., Steinfeld I., Lipson D., and Yakhini Z. (2009) GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanborn B. M., and Hein G. E. (1968) The interaction of trypsin with neutral substrates and modifiers. Biochemistry 7, 3616–3624 [DOI] [PubMed] [Google Scholar]
- 37. Bennike T., Lauridsen K. B., Olesen M. K., Andersen V., Birkelund S., and Stensballe A. (2013) Optimizing the identification of citrullinated peptides by mass spectrometry: Utilizing the inability of trypsin to cleave after citrullinated amino acids. J. Proteomics Bioinform. 6, 288–295 [Google Scholar]
- 38. Falick A. M., Hines W. M., Medzihradszky K. F., Baldwin M. A., and Gibson B. W. (1993) Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry. J. Am. Soc. Mass Spectr. 4, 882–893 [DOI] [PubMed] [Google Scholar]
- 39. Zee-Yong Park R. S., Judy Clark M., John Clark I., Yates J. R. III (2007) Assigning in vivo carbamylation and acetylation in human lens proteins using tandem mass spectrometry and database searching. Int. J. Mass Spectr. 259, 13 [Google Scholar]
- 40. Pritzker L. B., Nguyen T. A., and Moscarello M. A. (1999) The developmental expression and activity of peptidylarginine deiminase in the mouse. Neurosci. Lett. 266, 161–164 [DOI] [PubMed] [Google Scholar]
- 41. Nicholas A. P., King J. L., Sambandam T., Echols J. D., Gupta K. B., McInnis C., and Whitaker J. N. (2003) Immunohistochemical localization of citrullinated proteins in adult rat brain. J. Comparat. Neurol. 459, 251–266 [DOI] [PubMed] [Google Scholar]
- 42. Acharya N. K., Nagele E. P., Han M., Coretti N. J., DeMarshall C., Kosciuk M. C., Boulos P. A., and Nagele R. G. (2012) Neuronal PAD4 expression and protein citrullination: Possible role in production of autoantibodies associated with neurodegenerative disease. J. Autoimmun. 38, 369–380 [DOI] [PubMed] [Google Scholar]
- 43. Moscarello M. A., Mastronardi F. G., and Wood D. D. (2007) The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem. Res. 32, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bawadekar M., Gendron-Fitzpatrick A., Rebernick R., Shim D., Warner T. F., Nicholas A. P., Lundblad L. K., Thompson P. R., and Shelef M. A. (2016) Tumor necrosis factor alpha, citrullination, and peptidylarginine deiminase 4 in lung and joint inflammation. Arthritis Res. Ther. 18, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moldoveanu B., Otmishi P., Jani P., Walker J., Sarmiento X., Guardiola J., Saad M., and Yu J. (2009) Inflammatory mechanisms in the lung. J. Inflamm. Res. 2, 1–11 [PMC free article] [PubMed] [Google Scholar]
- 46. Makrygiannakis D., af Klint E., Lundberg I. E., Löfberg R., Ulfgren A. K., Klareskog L., and Catrina A. I. (2006) Citrullination is an inflammation-dependent process. Ann. Rheumat. Diseases 65, 1219–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Redman C. W., and Sargent I. L. (2004) Preeclampsia and the systemic inflammatory response. Sem. Nephrol. 24, 565–570 [DOI] [PubMed] [Google Scholar]
- 48. Nachat R., Méchin M. C., Takahara H., Chavanas S., Charveron M., Serre G., and Simon M. (2005) Peptidylarginine deiminase isoforms 1–3 are expressed in the epidermis and involved in the deimination of K1 and filaggrin. J. Invest. Dermatol. 124, 384–393 [DOI] [PubMed] [Google Scholar]
- 49. Asaga H., Nakashima K., Senshu T., Ishigami A., and Yamada M. (2001) Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J. Leukocyte Biol. 70, 46–51 [PubMed] [Google Scholar]
- 50. Mohanan S., Horibata S., McElwee J. L., Dannenberg A. J., and Coonrod S. A. (2013) Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: A preliminary study. Frontiers Immunol. 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Beers J. J., Zendman A. J., Raijmakers R., Stammen-Vogelzangs J., and Pruijn G. J. (2013) Peptidylarginine deiminase expression and activity in PAD2 knock-out and PAD4-low mice. Biochimie (Paris) 95, 299–308 [DOI] [PubMed] [Google Scholar]
- 52. Assohou-Luty C., Raijmakers R., Benckhuijsen W. E., Stammen-Vogelzangs J., de Ru A., van Veelen P. A., Franken K. L., Drijfhout J. W., and Pruijn G. J. (2014) The human peptidylarginine deiminases type 2 and type 4 have distinct substrate specificities. Biochim. Biophys. Acta 1844, 829–836 [DOI] [PubMed] [Google Scholar]
- 53. Olson J. S., Lubner J. M., Meyer D. J., and Grant J. E. (2017) An in silico analysis of primary and secondary structure specificity determinants for human peptidylarginine deiminase types 2 and 4. Computat. Biol. Chem. 70, 107–115 [DOI] [PubMed] [Google Scholar]
- 54. Heinrich B., Zhang Z., Raitskin O., Hiller M., Benderska N., Hartmann A. M., Bracco L., Elliott D., Ben-Ari S., Soreq H., Sperling J., Sperling R., and Stamm S. (2009) Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J. Biol. Chem. 284, 14303–14315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanhoush R., Beenders B., Perrin C., Moreau J., Bellini M., and Penrad-Mobayed M. (2010) Novel domains in the hnRNP G/RBMX protein with distinct roles in RNA binding and targeting nascent transcripts. Nucleus 1, 109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jobert L., Argentini M., and Tora L. (2009) PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Experimental Cell Res. 315, 1273–1286 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data, Mascot search results, and Scaffold files with annotated spectra have been deposited with the ProteomeXchange Consortium via the PRIDE (35) partner repository with the data set identifier PXD008970.