


Abstract
MOLEonline is an interactive, web-based application for the detection and characterization of channels (pores and tunnels) within biomacromolecular structures. The updated version of MOLEonline overcomes limitations of the previous version by incorporating the recently developed LiteMol Viewer visualization engine and providing a simple, fully interactive user experience. The application enables two modes of calculation: one is dedicated to the analysis of channels while the other was specifically designed for transmembrane pores. As the application can use both PDB and mmCIF formats, it can be leveraged to analyze a wide spectrum of biomacromolecular structures, e.g. stemming from NMR, X-ray and cryo-EM techniques. The tool is interconnected with other bioinformatics tools (e.g., PDBe, CSA, ChannelsDB, OPM, UniProt) to help both setup and the analysis of acquired results. MOLEonline provides unprecedented analytics for the detection and structural characterization of channels, as well as information about their numerous physicochemical features. Here we present the application of MOLEonline for structural analyses of α-hemolysin and transient receptor potential mucolipin 1 (TRMP1) pores. The MOLEonline application is freely available via the Internet at https://mole.upol.cz.
INTRODUCTION
Channels, which comprise tunnels and pores, are important features of biomolecular structures. The difference between these two types of channels is that tunnels connect an active site to the surface whereas pores have openings on opposing sides of a biomacromolecule. More than 64% of enzymes include buried active sites that are connected to the outside environment through tunnels, a feature that enables the transport of substrates and products (1). Internal enzyme channels are crucial to the catalysis of complex reactions (2,3), and mutations in substrate access tunnels can affect both enzyme activity and substrate specificity (4,5). Biomolecular channels are also involved in the trafficking of large molecules; for example, the recent characterization of the complete elongation complex of RNA polymerase II demonstrates channels that enable the entry of DNA and the exit of DNA and RNA (6). Another example is the 10 nm long polypeptide tunnel that directs a nascent protein out of the ribosome (7,8).
Pores also assist many biologically important processes, i.e. they facilitate and control the transportation of water, ions and molecules across biomembranes (9,10). Progress in crystallization and cryo-electron microscopy techniques has greatly increased the amount of structural information concerning transmembrane proteins. The available structures are crucial to our comprehension of many biologically relevant processes such as channel opening and gating of various receptors (11–13) and the structural basis of lipopolysaccharide transport (14).
The importance of channels in biomacromolecular structures calls for the development of tools that can both detect and analyze these structural features. Many software tools, which provide varying degrees of functionality and accessibility, have been developed (15). Among these solutions, web-based tools are highly appreciated by structural biologists because of their accessibility on various devices, platforms and operating systems, and the ability to integrate them with other bioinformatics tools and pipelines. MOLEonline is a popular tool providing highly interactive functionality; however, its previous version (16) suffered from certain drawbacks that ultimately motivated a complete rebuild. In its current form, MOLEonline enables full and interactive functionality with two modes: one for the analysis of tunnels and the other dedicated to the analysis of transmembrane pores. In addition to calculating the geometry of identified channels, MOLEonline also provides information about channel-lining residues and their physicochemical properties. Finally, MOLEonline is interconnected with the ChannelsDB database (17) to facilitate the comparison of identified structural features with those deposited in the database.
DESCRIPTION OF THE TOOL
The MOLE channel search algorithm is based on our previous approach, which has been published in detail (18). However, the updated version includes new features, together with an original user experience (UX) design, that are accessible via web-based interface. The calculation setup proceeds through an interactive interface that comprises only a few components (Figure 1). The biomolecular structure, including the computed channels or pores, is then visualized by the LiteMol Viewer JavaScript application (19).
Figure 1.

The new user experience (UX) design of MOLEonline, demonstrated using a screenshot of the result for human aquaporin (PDB ID: 4CSK).
Input structure
The structure can be uploaded directly from the PDBe database (20) or as a PDB or mmCIF file. MOLEonline can select a biologically active unit according to PDBe annotation.
Calculation modes
The next step requires the user to select the mode of calculation. MOLEonline offers both Channels mode (default), which is used for the identification of tunnels and merged pores, and Pore mode, which is designed for the characterization of transmembrane pores. The Pore mode computation starts with a definition of the membrane region according to information from the Orientations of Proteins in Membranes (OPM) database (http://opm.phar.umich.edu/) (21) or calculations by the MEMEMBED program (for structures without OPM membrane annotation) (22). The planes of membrane region boundaries are further used for the definition of starting and end points along the normal near the center of the macromolecule. The Beta structure parameter is recommended for transmembrane regions including a β-barrel structure because it facilitates prediction of the membrane region. The Pore mode calculations are more time consuming because they include the identification of transmembrane regions.
Start and end points
Channels mode calculations require the selection of a starting point. The starting point can be estimated based on the selection of certain residues or specific XYZ coordinates. The residues can be selected by typed command or clicking on a specific amino acid sequence or 3D structure. Catalytic site amino acids, uploaded automatically from the CSA database, can also be used as the starting point (23). The automatic detection of starting points uses the deepest positions in the calculated cavities, which are visualized as blue spheres. The updated version of MOLEonline allows users to select starting points based on the positions of specified cofactors. The starting point can also be selected based on geometrical features, described via PatternQuery language (24). MOLEonline also allows users to select end points, and it is possible to select two points on different sides and find the best path connecting them. The end points can be specified in the same way as the starting points, and it is possible to select them by directly clicking on a site on the surface of the biomolecule. This enables users to tune MOLE automatic detection to their specific needs (Figure 2).
Figure 2.

Interactive selection of start and end points via residues or XYZ coordinates. The end point can be selected as a facet on the surface using Ctrl + left mouse click.
Calculation of channels
The channel search algorithm of MOLE was described in detail elsewhere (18). Shortly, the MOLE algorithm involves seven steps: (i) computation of the Delaunay triangulation/Voronoi diagram for the atomic centers; (ii) construction of the molecular surface; (iii) identification of cavities; (iv) identification of possible channel start points; (v) identification of possible channel end points; (vi) localization of channels and (vii) filtering of the localized channels.
The user can utilize the default MOLE parameters or modify them upon request. The cavity parameters Probe Radius and Interior Threshold define the alpha-shape surfaces of the biomacromolecule and minimal beta-shape cavities within them, respectively. The channel parameters Origin Radius and Surface Cover Radius can be used to optimize the starting point position to the closest tetrahedron or end point on the surface, respectively. PatternQuery language can be used not only for the definition of start and end points, but also to exclude specific groups or residues (e.g. individual heteroatoms, known bottleneck amino acids, etc.) that may block and/or hide channels. The channel localization feature uses Weight Function for channel pruning. The original Length and Radius weight function prefers channels with minimal length and maximal radius. The added Length weight function prefers short channels and thus discourages channels on the surface of the biomacromolecule, which was a recognized artifact of the MOLE 2.0 calculation. However, the Length weight function might identify passages that are too narrow; therefore, we have further added the Voronoi scale weight function, which evaluates differences between the geometrical centers of tetrahedrons and the resulting channel spheres. The weight is calculated as
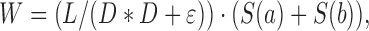 |
(1) |
where L is length of edge, D a distance to the closest vdW sphere, ϵ small number to avoid division by zero and S(a) and S(b) are distances between center of geometrical centers of two closest tetrahedrons and resulting channel spheres. As such, the Voronoi scale weight function prefers tetrahedrons that face towards the interior of the biomolecule. Further options allow merging channels into the pores, adjusting tolerance to bottlenecks and discarding channels that are too similar according to characteristics of their tetrahedrons. More details on method and tips on channel calculation parameters are shown on the MOLEonline webpage (https://mole.upol.cz/method and https://mole.upol.cz/FAQ) and can be found in (15).
Visualization and analysis of results
Calculated channels are sorted according to their type into Tunnels, Pores and Paths (links two user-selected points) and then categorized further according to their length. Channels originating from the same cavity are assigned into one set. The channels can be visualized in LiteMol Viewer and, upon selection, their properties are shown in the lower left corner of the webpage (Figure 1). Two independent plots show channel profiles, i.e. radius vs. distance/length. In addition to the Radius of the maximally inscribed ball, the user can also choose Free Radius and BRadius. Free Radius illustrates the radius of the maximally inscribed ball based on the protein backbone (i.e. ignoring amino acid side chains), the position of which can be modified by amino acid side-chain movement or virtual glycine mutation. BRadius adds a value of root-mean square fluctuations (RMSF) of the residues along the channel (calculated from B-factors stored within the structure) to the initially calculated Radius to reflect local flexibility. It should be noted that this approach reflects the flexibility in postprocessing, an analysis on set of molecular structures, e.g. from NMR or molecular dynamics should be carried out for inherent inclusion of structural dynamics. Each profile can be colored according to the physicochemical properties (16,18) of the lining amino acids. The user can select Charge, Hydrophobicity, Hydropathy, Polarity, Lipophilicity (log P, log D), Solubility (log S) or Mutability. Lipophilicity and Solubility are calculated as either octanol/water partition coefficient (log P), octanol/water distribution coefficient at pH 7.4 (log D) or water solubility (log S) prediction of individual sidechain or mainchain fragments truncated at Cβ. The Chemicalize website (https://chemicalize.com/; ChemAxon, Budapest, Hungary) provides these values. The plots are composed of interactive layers that allow users to select and visualize surrounding residues in the LiteMol Viewer individually or multiple layers together. The overall results for all considered channels are also summarized.
To ensure privacy, the results of each calculation are stored as an individual submission, with all of the parameters within a hashed job ID. Individual submission history can be browsed in the bottom right corner. The user can also compare their own results with the annotated channels deposited in ChannelsDB (if available). Where available, the residue annotation obtained from the UniProt API (25–27) is also shown. The user can also send their results, as an input of channel annotation, to ChannelsDB using a simple form available on the top of the page.
All results can be downloaded as a report in various formats—either macromolecule itself in mmCIF format or channel coordinates in PDB format or combined in PyMol, VMD and Chimera visualization scripts, or as a JSON or ZIP files containing all information for further textual analysis. The ZIP file contains all the results in a variety of formats for further analysis and visualization in popular molecular browsers such as Pymol, Chimera or VMD. A report of the calculation can also be downloaded in PDF format.
RESULTS AND DISCUSSION
Examples of usage
MOLEonline 2.0 (16) was designed for analyzing channels in biomolecules. We kept this functionality in the updated version and introduced options that made the application more informative and easier to use. We significantly extended the ability of MOLEonline to analyze transmembrane pores. Due to limited space, we have listed all updated features on the MOLEonline web page (https://mole.upol.cz/updates). Here, we have provided examples of the pore analysis functionality and would like to note that examples of channel analysis are available on the MOLEonline web page (https://mole.upol.cz/examples).
α-Hemolysin pore
Staphylococcus aureus secretes α-hemolysin as water-soluble monomers, which consecutively form a heptameric ring-like structure with a central pore (27) that can cause uncontrolled permeation across a membrane. MOLEonline calculated the transmembrane pore of α-hemolysin (PDB ID: 7AHL (28)) to be 88.1 Å long with a bottleneck radius of 5.3 Å (Figure 3). As seen in Figure 3, the pore constricts from the stem (within the membrane region) towards the vestibule at the cap of the structure. The widest part of the pore has a diameter of 15.8 Å. The α-hemolysin pore enables the uncontrolled transportation of water and ions across a membrane, causing membrane depolarization and subsequent cell death. It is permeable to both cations and anions, but has a preference towards anions (29). The cation flux depends on cation size, with potassium cations demonstrating the highest conductance (30). The uncharged and mainly nonpolar interior of the pore (Figure 3) may accelerate the movement of charged species. It is important to note that the positively charged bottleneck in the rim of the pore may contribute to the preferred transport of anions.
Figure 3.

The transmembrane pore of α-hemolysin (PDB ID: 7AHL (28)). The blue and red dashed lines delimit the membrane region (also in the profile). The profile depicts hydropathy and charge along the pore. The pore is divided into three parts: (i) Stem consisting of β-barrel membrane domain (with trans-opening); (ii) Rim—lipid membrane anchoring domain and (iii) Cap—external domain (with cis-opening). The figure was generated in Pymol software (31) from a MOLEonline report.
Transient receptor potential mucolipin 1 (TRMPL1)
TRPML1 pores are voltage-gated channels composed of six transmembrane parts (32) that belong to the group of thoroidal pores. The TRPML1 pore plays a significant role in transferring Ca2+ ions from the lysosomal lumen to the cytoplasm (33). Moreover, it is a homotetrameric cation channel (Figure 4) involved in Ca2+ signaling and maintaining homeostasis within lysosomes (12). The channel, in closed conformation, shows apo structure (PDB ID: 5WJ5) whereas the open conformation is a structure in which mucolipin synthetic agonist 1 is bound (PDB ID: 5WJ9). MOLEonline identified a significant difference in pore radius between the closed and open TRMPL1 conformations (Figure 4). The central pore is considerably wider (4.5 Å) when the agonist is bound (open form) than when the pore is in closed form (3.8 Å). It should be noted that the negatively charged Ca2+ selectivity filter containing D471 and D472 can be detected at the lumenal side of the lysosome based on the reported physicochemical properties.
Figure 4.

(A) The channel (closed conformation) of TRPML1 (PDB ID: 5WJ5 (12)). The blue and red dashed lines delimit the membrane region. The profiles depict polarity and charge along the pore. (B) The channel in open conformation (PDB ID: 5WJ9 (12)). The pore is divided into three main parts: Activation gate; Central cavity and Selectivity filter. The figure was generated using Pymol software (31).
Limitations
Despite our best efforts, MOLEonline suffers from a few inherent limitations. MOLEonline was designed to analyze static structures identified through X-ray, NMR or cryo-EM experiments, so it cannot directly provide any information about the breathing motions of detected channels. In addition, ligand passage along the channel/pore may cause significant digestive motions (34), and this should be taken into account during the interpretation of results. Moreover, the physicochemical properties are calculated only from amino acids lining the channel/pore and the reported values do not cover nonstandard amino acids, cofactors and nucleic acids. MOLEonline cannot detect pores in which the diameter is smaller or the same as the length due to the used MOLE algorithm for cavities identification. Nevertheless, MOLEonline is able to calculate channels in structures containing up to ∼60k residues in reasonable time.
CONCLUSIONS
MOLEonline enables interactive, platform-independent analyses of channels, tunnels and pores. These structures are visualized by LiteMol Viewer with unprecedented performance. MOLEonline can detect, visualize and analyze features and physicochemical properties of biomolecular channels and now also enables the detection and characterization of transmembrane pores. Furthermore, the application is interconnected with the ChannelsDB database to allow users to identify the biological function of detected channels and pores by comparing their results with annotated structures.
DATA AVAILABILITY
MOLEonline is available free of charge at https://mole.upol.cz.
FUNDING
ELIXIR CZ research infrastructure project (MEYS) [LM2015047 to L.P., D.S., R.S.V., D.T., V.B. and K.B.]; Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 [LQ1601 to L.P., D.S., R.S.V., D.T. and J.K.]; ELIXIR-EXCELERATE project, which received funding from the European Union's Horizon 2020 research and innovation programme [676559]; Czech Science Foundation [P208/12/G016 to M.O. and V.N.]; Ministry of Education, Youth and Sports of the Czech Republic [project NPUII LO1305 to M.O. and K.B.]; Palacký University Olomouc [IGA_PrF_2018_032 to V.N. and V.B.]. Funding for the open access charge: Czech Science Foundation [P208/12/G016].
Conflict of interest statement. None declared.
REFERENCES
- 1. Pravda L., Berka K., Svobodová Vařeková R., Sehnal D., Banáš P., Laskowski R.A., Koča J., Otyepka M.. Anatomy of enzyme channels. BMC Bioinformatics. 2014; 15:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang X., Holden H.M., Raushel F.M.. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 2001; 70:149–180. [DOI] [PubMed] [Google Scholar]
- 3. Raushel F.M., Thoden J.B., Holden H.M.. Enzymes with molecular tunnels. Acc. Chem. Res. 2003; 36:539–548. [DOI] [PubMed] [Google Scholar]
- 4. Sykora J., Brezovsky J., Koudelakova T., Lahoda M., Fortova A., Chernovets T., Chaloupkova R., Stepankova V., Prokop Z., Smatanova I.K. et al. Dynamics and hydration explain failed functional transformation in dehalogenase design. Nat. Chem. Biol. 2014; 10:428–430. [DOI] [PubMed] [Google Scholar]
- 5. Kingsley L.J., Lill M.A.. Substrate tunnels in enzymes: Structure-function relationships and computational methodology. Proteins Struct. Funct. Bioinform. 2015; 83:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehara H., Yokoyama T., Shigematsu H., Yokoyama S., Shirouzu M., Sekine S.. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science. 2017; 357:921–924. [DOI] [PubMed] [Google Scholar]
- 7. Voss N.R., Gerstein M., Steitz T.A., Moore P.B.. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 2006; 360:893–906. [DOI] [PubMed] [Google Scholar]
- 8. Petřek M., Košinová P., Koča J., Otyepka M.. MOLE: a Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure. 2007; 15:1357–1363. [DOI] [PubMed] [Google Scholar]
- 9. Smart O.S., Neduvelil J.G., Wang X., Wallace B.A., Sansom M.S.P.. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996; 14:354–360. [DOI] [PubMed] [Google Scholar]
- 10. Oliva R., Thornton J.M., Pellegrini-Calace M.. PoreLogo: A new tool to analyse, visualize and compare channels in transmembrane proteins. Bioinformatics. 2009; 25:3183–3184. [DOI] [PubMed] [Google Scholar]
- 11. Twomey E.C., Yelshanskaya M.V., Grassucci R.A., Frank J., Sobolevsky A.I.. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature. 2017; 549:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmiege P., Fine M., Blobel G., Li X.. Human TRPML1 channel structures in open and closed conformations. Nature. 2017; 550:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tarvin R.D., Borghese C.M., Sachs W., Santos J.C., Lu Y., O’Connell L.A., Cannatella D.C., Harris R.A., Zakon H.H.. Interacting amino acid replacements allow poison frogs to evolve epibatidine resistance. Science. 2017; 357:1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mi W., Li Y., Yoon S.H., Ernst R.K., Walz T., Liao M.. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature. 2017; 549:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koča J., Svobodová Vařeková R., Pravda L., Berka K., Geidl S., Sehnal D., Otyepka M.. Structural Bioinformatics Tools for Drug Design. 2016; 1st ednCham: Springer International Publishing. [Google Scholar]
- 16. Berka K., Hanák O., Sehnal D., Banáš P., Navrátilová V., Jaiswal D., Ionescu C.-M., Svobodová Vařeková R., Koča J., Otyepka M.. MOLEonline 2.0: interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 2012; 40:W222–W227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pravda L., Sehnal D., Svobodová Vařeková R., Navrátilová V., Toušek D., Berka K., Otyepka M., Koča J.. ChannelsDB: database of biomacromolecular tunnels and pores. Nucleic Acids Res. 2018; 46:D399–D405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sehnal D., Svobodová Vařeková R., Berka K., Pravda L., Navrátilová V., Banáš P., Ionescu C.-M., Otyepka M., Koča J.. MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013; 5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sehnal D., Deshpande M., Vařeková R.S., Mir S., Berka K., Midlik A., Pravda L., Velankar S., Koča J.. LiteMol suite: interactive web-based visualization of large-scale macromolecular structure data. Nat. Methods. 2017; 14:1121–1122. [DOI] [PubMed] [Google Scholar]
- 20. Mir S., Alhroub Y., Anyango S., Armstrong D.R., Berrisford J.M., Clark A.R., Conroy M.J., Dana J.M., Deshpande M., Gupta D. et al. PDBe: towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res. 2018; 46:D486–D492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L.. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012; 40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nugent T., Jones D.T.. Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinformatics. 2013; 14:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porter C.T., Bartlett G.J., Thornton J.M.. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004; 32:D129–D133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sehnal D., Pravda L., Svobodová Vařeková R., Ionescu C.-M., Koča J.. PatternQuery: web application for fast detection of biomacromolecular structural patterns in the entire Protein Data Bank. Nucleic Acids Res. 2015; 43:W383–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nightingale A., Antunes R., Alpi E., Bursteinas B., Gonzales L., Liu W., Luo J., Qi G., Turner E., Martin M.. The proteins API: accessing key integrated protein and genome information. Nucleic Acids Res. 2017; 45:W539–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Velankar S., Dana J.M., Jacobsen J., Van Ginkel G., Gane P.J., Luo J., Oldfield T.J., O’Donovan C., Martin M.J., Kleywegt G.J.. SIFTS: structure integration with function, taxonomy and sequences resource. Nucleic Acids Res. 2013; 41:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valeva A., Pongs J., Bhakdi S., Palmer M.. Staphylococcal α-toxin: the role of the N-terminus in formation of the heptameric pore - a fluorescence study. Biochim. Biophys. Acta - Biomembr. 1997; 1325:281–286. [DOI] [PubMed] [Google Scholar]
- 28. Galdiero S., Gouaux E.. High resolution crystallographic studies of α-hemolysin-phospholipid complexes define heptamer-lipid head group interactions: implication for understanding protein-lipid interactions. Protein Sci. 2004; 13:1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Menestrina G., Dalla Serra M., Comai M., Coraiola M., Viero G., Werner S., Colin D.A., Monteil H., Prévost G.. Ion channels and bacterial Infection: the case of β-barrel pore-forming protein toxins of staphylococcus aureus. FEBS Lett. 2003; 552:54–60. [DOI] [PubMed] [Google Scholar]
- 30. Bhattacharya S., Muzard J., Payet L., Mathé J., Bockelmann U., Aksimentiev A., Viasnoff V.. Rectification of the current in α-hemolysin pore depends on the cation type: the alkali series probed by molecular dynamics simulations and experiments. J. Phys. Chem. C. 2011; 115:4255–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schrodinger LLC The PyMOL Molecular Graphics System. 2010. [Google Scholar]
- 32. Gees M., Colsoul B., Nilius B.. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010; 2:a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li M., Zhang W.K., Benvin N.M., Zhou X., Su D., Li H., Wang S., Michailidis I.E., Tong L., Li X. et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat. Struct. Mol. Biol. 2017; 24:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paloncýová M., Navrátilová V., Berka K., Laio A., Otyepka M.. Role of enzyme flexibility in ligand access and egress to active site: bias-exchange metadynamics study of 1,3,7-Trimethyluric acid in cytochrome P450 3A4. J. Chem. Theory Comput. 2016; 12:2101–2109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
MOLEonline is available free of charge at https://mole.upol.cz.