


Abstract
The new web resource EviNet provides an easily run interface to network enrichment analysis for exploration of novel, experimentally defined gene sets. The major advantages of this analysis are (i) applicability to any genes found in the global network rather than only to those with pathway/ontology term annotations, (ii) ability to connect genes via different molecular mechanisms rather than within one high-throughput platform, and (iii) statistical power sufficient to detect enrichment of very small sets, down to individual genes. The users’ gene sets are either defined prior to upload or derived interactively from an uploaded file by differential expression criteria. The pathways and networks used in the analysis can be chosen from the collection menu. The calculation is typically done within seconds or minutes and the stable URL is provided immediately. The results are presented in both visual (network graphs) and tabular formats using jQuery libraries. Uploaded data and analysis results are kept in separated project directories not accessible by other users. EviNet is available at https://www.evinet.org/.
INTRODUCTION
Modern analyses of biological data is increasingly based on interactions between genes, proteins, and other biological molecules, combined into networks, otherwise called interactomes. Most often the task is to characterize a novel experimental or pathological condition through a set of genes with altered molecular features.
This approach of testing biological hypotheses in the network context requires running statistically adequate topology-based procedures. We proposed a method of network enrichment analysis (NEA) (1,2), where network topology is employed to evaluate functional impact of experimentally determined gene sets. NEA became a natural extension of the well-known pathway overrepresentation analysis (ORA) into the interactomics domain. The ORA methodology (3) has been elaborated during the last two decades (4,5). It utilizes the abundance of known functional gene sets (FGS), such as pathways, to characterize novel, experimentally defined altered gene sets (AGS). This is done by finding an overlap of the genes of FGS in the AGS and testing its statistical significance. Performance and applicability of ORA have been limited by incomplete pathway annotation of gene space and by only considering alterations observable within one platform, such as a transcriptomics microarray. NEA largely overcomes these limitations due to a key difference: while ORA counts the number of genes shared between an experimental list and a pathway, NEA considers network edges between any genes of both groups in the global network. This feature is surprisingly absent in most of the previously proposed algorithms for network enrichment analysis. Indeed, methods such as that of Ingenuity Pathway Analysis [Ingenuity® Systems, http://www.ingenuity.com], PheNetic (6), SteinerNet (7), ResponseNet (8,9) identify, in various ways, network modules (clusters, sub-networks etc.) that appear enriched in altered genes. Then ORA might be applied post-hoc to evaluate overlap between a module and each of the tested pathways.
A number of web tools are close to the idea of EviNet, such as e.g. EnrichNet (10). It also connects AGS and FGS in the network, with the difference that network paths of unlimited length are allowed and the node degrees are not explicitly accounted for. However, the web interface of EnrichNet is limited to submission of gene lists one by one. The same applies to a more recent web tool PathWAX (11), which is based on the same NEA approach as EviNet (path of length 1, i.e. accounts for only direct edges) but estimates confidence via network randomization – which is slower than the X2 statistic calculation (12) employed by EviNet. ToppGene (13) also enables analysis of a functionally relevant gene set (usually representing a disease) against a single gene by using unlimited path algorithms PageRank, HITS, and K-Step Markov. Another new tool, FunGeneNet (14) allows analyzing network enrichment using the NEA approach, but specifically within one user-submitted gene set. We note that in our resource this analysis is performed by default, in parallel with evaluating enrichment of AGS against FGSs. These alternative tools lack, to various extents, functionality for network visualization.
Another highly desirable feature is a dynamic re-definition of experimentally derived gene lists, e.g. via changing confidence thresholds. A web resource called NetVenn (15) has enabled flexibility of input for network analysis via Venn diagrams, although it identifies and presents network modules enriched in differentially expressed genes, thus performing the opposite task: gene set exploration rather than functional characterization via pathway enrichment.
The popular algorithms, such as PageRank, Random Walk with Restart and their modifications (16–19), were designed to operate along potentially unlimited network paths with edge weights decaying along a path, which was probabilistically modified by specific parameters.. While being commonly used today for biological enrichment analysis, these parameters were never, to the best of our knowledge, systematically optimized for biological networks. The network topology, such as node degree values, either does not affect ranking results and significance evaluation or is biologically counterintuitive. As an example PageRank, developed for Google search engine, considered hubs as most relevant nodes, which would not be the case for novel disease or drug target genes. By analyzing fixed paths of length 1 and by built-in accounting for node degree of genes and gene sets, the NEA approach reduces topological bias, makes the analysis generally faster, more transparent, biologically relevant, and easy in visualizing detailed gene–gene paths. We have also seen that NEA, given state-of-the-art global networks, is not inferior to the long path algorithms in terms of specificity and sensitivity (results to be published elsewhere). Such networks, possessing around one million edges and 10 to 20 thousands nodes as well as scale free topology, are available from the EviNet collection (STRING, FunCoup, PathwayCommons).
The new resource EviNet (https://www.evinet.org/) creates a user-friendly interface for gene network analysis in both hypothesis-driven and hypothesis-free research. It provides (i) a clearly defined, sequential analytic procedure, (ii) statistically rigorous hypothesis testing, (iii) biological interpretability and transparency for hypotheses and discoveries and (iv) high-quality visualization of findings, both as summarized and in depth details of gene–gene interactions and evidence behind them. At the same time, the output is maximally similar to ORA. While the web interface of EviNet is created using jQuery functionality (https://api.jquery.com/) and/or generated by perl scripts, the back-end employs PostgreSQL database engine and core functions of the R package NEArender (12). The latter is dedicated to NEA, but possesses a number of additional functions which, being irrelevant to the online analysis, are available for off-line users (12) (https://cran.r-project.org/web/packages/NEArender/). EviNet has been employed in a number of research projects (20), (21–25) as well as a platform for teaching systems biology. Here we demonstrate use of EviNet in an analysis of transcriptome dynamics upon embryonic stem cell differentiation by combining multiple differential expression criteria. It allowed corroborating known and revealing novel signaling genes and pathways potentially important for maintaining stem cell pluripotency, differentiation, and diversification toward either neuroectodermal or endodermal lineages.
MATERIALS AND METHODS
Cell differentiation
E14 mESCs were differentiated as described before (43).Total RNA was isolated from cells every 24 h between 0 and 3 days of differentiation condition (DDC) using the ZymoPure™ kit according to manufacturer's instructions. Three DDC cultures were enriched for neural progenitors using prominin1-based magnetic activated cell sorting (p-MACS) (Miltenyi Biotec).
RNA sequencing and quantification of gene expression
The differentiation stages 0d (ESC), 1d, 2d, 3d and 3d(sorted) were represented with 2, 2, 2, 2 and 3 biological replicates, respectively.
Reads were mapped with Tophat/2.0.4 (54) to the mouse genome assembly (build GRCm38). BAM files from samples run on different lanes were merged with samtools (55). Merged BAM files were sorted and duplicates removed using picard-tools/1.29 (http://broadinstitute.github.io/picard/). Mapping statistics were calculated from numbers obtained by running bam_stat.py (included in rseqc/2.3.6) on bam files with and without duplicates. Script GeneBody_coverage.py (included in rseqc/2.3.6) was run on bam files with duplicates (56). Gene count values were generated using htseq/0.6.1 on BAM files with duplicates included (57). FPKM values were not used in this analysis.
Analysis of differential expression
In order to compensate for heteroscedasticity (the correlation between mean and variance of gene expression profiles), the sample-specific count values were processed in R package limma (58) with function voom. After the empirical Bayesian variance interpretation, the DE and adjusted P-values were calculated with limma function topFC.
Example data
The file for Venn diagram generation with count, voom and differential expression values for this experimental series is available at EviNet.org in the public project ‘stemcell’ and in the menu Help.
Alternatively, example files with precompiled AGS collections can also be found in Help or in File directory of the demo project 'myveryfirstproject', ‘stemcell’.
Venn diagrams
The Venn diagrams overlay gene lists from different pairwise experimental contrasts. Such cross-comparisons allow the user to focus on gene sets characterizing specific processes. The server-side script re-reads the file with DE values and generates lists of genes that satisfy each contrast-specific set of filtering conditions. Each list corresponds to one ellipse on the Venn diagram, generated with R packages Vennerable (https://github.com/js229/Vennerable) and VennDiagram (59). Further, all possible overlaps in Venn diagrams (3, 7, and 15 in 2, 3 and 4-contrast analyses, respectively) are accompanied by corresponding gene lists, which pop up on the screen upon mouse clicks at the intersection areas. The lists also contain DE values and can be investigated by sorting, gene ID search etc. Users can change filtering criteria, followed by re-generation of the Venn diagram and the gene lists. Finally, the user chooses with checkboxes an arbitrary number of intersection gene lists, which will be treated as AGSs, and proceeds to the tabs Network, Functional Gene Sets, and Check and submit in order to execute NEA.
DATA ANALYSIS AND REQUIRED INPUT
An algorithm of the traditional, network-free gene set enrichment analysis requires two components: set(s) of experimentally derived genes/proteins that we term ‘altered gene sets’ (AGS) and a collection of gene/protein sets with previously characterized common functions (functional gene sets, FGS). A network enrichment analysis in addition requires a third component: a network where edges represent functional couplings, interactions, regulatory relationships etc. between genes and/or protein nodes (Figure 1). In order to be unbiased, such a network should be global, i.e. encompass all known nodes and edges rather than only those relevant to the analyzed AGS.
Figure 1.

Data flow of network enrichment analysis on EviNet.org. Three components shall be specified for an analysis on the server: (1) AGS, experimentally derived gene/protein lists in one of the three shown formats; (2) FGS, functional gene sets (typically pathways with well characterized biological function) selected from the FGS collection menu and (3) global network (also selected from the menu) where individual genes of AGS and FGS are supposed to be connected via edges (presence of all genes is not required, however AGS, FGS, and network must share the same name space). The program will identify available AGS-FGS edges, perform network enrichment analysis, produce graphical and tabular output, and store the results in Archive for future investigation. See details in ‘Data analysis and required input’.
Using the X2 based statistic, which was described in details elsewhere (1,12), enables quick and unbiased calculation of connectivity expected by chance between the given AGS and FGS from their cumulative node degrees NAGS and NFGS (the sums of connectivity values of the member nodes):
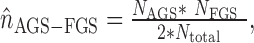 so that
so that 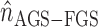 is then used for evaluating significance with:
is then used for evaluating significance with:
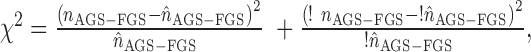 where nAGS-FGS is the actual connectivity and ‘!’ denote negation, i.e. the rest of network edges.
where nAGS-FGS is the actual connectivity and ‘!’ denote negation, i.e. the rest of network edges.
In this implementation, the time needed for a typical analysis of e.g. 10 AGSs versus 50 FGSs in a network of one million edges is around 20 s.
In the following text, we describe the three components and ways to define them for a particular analysis. Beyond that, new users of the web site can begin by running the animated demo analyses. The latter would sequentially fill all the required fields and thus facilitate overview and mastering of the options. Flowcharts in the Help menu explain the major analysis components and typical applications. The text sections below correspond to the numbered web page tabs, so that following this (although not fixed) order can facilitate the task for novel users.
Altered gene sets
A typical input to NEA should be one or multiple AGSs, in the form of gene or protein lists. Such lists can be submitted in one of three major ways, represented by three sections in the tab Altered gene sets: (i) a list pasted into the text box (minimally, a single ID); (ii) an uploaded file with predefined lists (list IDs must be present in a dedicated column) and (iii) an uploaded file with results of differential expression (DE) analysis that allows re-defining the lists by changing DE criteria. The latter option requires the file header to be in a standardized format, as explained in Help. Upon the file upload, the header is rendered into a set of web form controls, which allows to simultaneously consider up to four DE contrasts, apply desirable criteria and explore gene sets overlaps between specific DE lists.
For example, a user might possess transcriptomics data from experiments X, Y, Z and a control condition C. A DE analysis have compared these conditions using available replicates and suitable software tools, which resulted in fold change values and (adjusted) P-values for each contrast of interest: X versus C, Y versus C, and Z versus C. The DE lists can now be flexibly derived by choosing fold change and/or P-value cut-offs. After setting the criteria, the user can generate a Venn diagram of the three DE sets. This reveals gene groups that of particular interest in the given experimental design, such as e.g. differentially expressed in X versus C and Y versus C but not between Z versus C (dubbed ‘++-’). Given sensible DE criteria, each group (‘-+-’, ‘-++’, ‘+++’ etc.) will likely contain multiple genes. The respective Venn intersections (Figure 2) are clickable, resulting in pop-up tables of genes with respective DE values. These DE lists can be selected as AGS input for NEA. In our experience, this should address a frequent need to apply complex criteria to a combination of DE analyses and evaluate pathway enrichment in the overlap sets. In section ‘Transcriptomic changes during stem cell differentiation’ we analyze cell differentiation stages compared to the original embryonic stem cell state.
Figure 2.

Scheme of stem cell differentiation and analysis of differential gene expression. Mouse embryonic stem cells (mESCs) treated with retinoic acid (RA) and Shh agonist (SAG) were driven toward differentiation during at least three days. The cell transcriptomes of the days 1, 2, 3 were compared to that of the original mESCs. By day 3, neural progenitors emerged as a mixture population. From this mixture, neuroectodermal cells were derived by cell sorting and studied against the former. The four differential expression (DE) contrasts were selected and overlapped using the Venn diagram tool of evinet.org. As an example, genes DE in each of the four contrasts were chosen as the central intersection (table at the bottom) and forwarded to network enrichment analysis of evinet.org.
Network
The known part of the global gene network consists of physical interactions and protein-mediated gene regulation, which is learned experimentally (26). In addition, we and others developed Bayesian tools that reconstruct novel edges of functional coupling. This is done by integrating evidence from multiple high-throughput platforms and evaluating consistency of literature reports (27–29). In order to enable network analysis from different angles and consider particular molecular mechanisms, one can use functional links between genes and/or proteins from curated databases of protein complexes (30), signaling and metabolic pathways (31,32), protein phosphorylation (33,34), transcription factor binding (35) etc. Smaller networks have lower sensitivity due to having fewer gene nodes and edges (22). On the other hand, larger networks do gain from data integration: for instance, a pure protein interaction network is inferior to a FunCoup network, which also integrates gene co-expression, protein co-localization etc. At the same time, another data integration network STRING, being largely based on prokaryotic evidence (27), performs best with metabolic pathways, while being inferior to FunCoup in the cancer domain (22). In the tab ‘Network’, we provide a menu of such resources. It is also possible to choose a combination of networks for a particular analysis.
Functional gene sets
The usage of FGS is most similar to that in the ORA methodology. Each FGS (typically a pathway or a Gene Ontology term) can be viewed as a dimension in a functional space. Upon choosing one of the collections, such as BioCarta (36), KEGG (31), MetaCyc (37), Reactome (32), WikiPathways (38), Gene Ontology terms (39), or a custom collection used in previous research, the resulting network enrichment scores can be utilized either in an exploratory analysis (typically with few AGSs) or produce an FGS score matrix potentially informative on e.g. clinical phenotypes, when cohort patients are represented with AGSs as explained in details elsewhere (12). Alternatively, users can submit own, custom FGSs via the text box or by uploading files with predefined lists in the same way as it is done in the AGS section.
ANALYSIS OUTPUT AND AVAILABLE OPTIONS
When AGS, FGS, and network options have been defined in the tabs described above, the selected choices can be reviewed in the tab Check and submit. The results are generated by pressing the‘Submit and calculate’ button. If the analysis takes longer than a minute, the user can either bookmark the permanent URL in order to access it upon job completion or find the URL listed among previously performed analyses in the project archive.
The output is provided in graphical, detailed tabular, and matrix formats. The graph represents a map of significant AGS-FGS edges, which summarize respective single gene–gene connections. The tabular output provides an overview of node degree values as well as significance estimates. Normally distributed values needed for many downstream analyses are provided by recalculating the X2 statistic to z-scores. Matrices of z-scores, P-values (both raw and adjusted for multiple testing), and other potentially informative values can be obtained from the auxiliary menu in the top right corner of the tab. We should warn that adjustment for multiple testing by Benjamini and Hochberg (40) might be calculated properly only on larger numbers (e.g. hundreds) of tested hypotheses (AGSxFGS combinations). Otherwise the adjusted values may approach original P-values. For comparison with ORA, one can look at the second last column Shared genes displaying the number of genes that belong to both AGS and FGS, as well as the P-value estimated via ORA. This reveals the much higher sensitivity of NEA compared to ORA: the gene set overlap rarely exceeds 5–10 genes (usually 0 or 1), while tens and hundreds of AGS-FGS network edges are significant.
The graphical output is enabled with the jQuery plugin Cytoscape.js (41). The accompanying menu provides a comprehensive control over the graph features (edge/node content, layout, naming, coloring, filtering, size etc.) and saving presentation-quality Figure ures in PNG format. Since AGS-FGS edges summarize individual gene–gene connections, clicking on them retrieves respective sub-network views (both as graphs and tables) for further investigation of available evidence, such as edge confidence scores, links to literature, and other annotations. The Archive window displays a list of previous analyses in the current project, with records of actual parameters and URLs for restoring the results in a separate window.
Using the web site is open for everybody without registration or login. Each project with uploaded files is stored in a separate directory. However, registered users might obtain access to additional functionality via the project management system. Access to user files and analysis results can be privately shared between the project members. They may have different roles (administrator, read/write, read only).Administrators can make projects public, which might be convenient for publication purposes.
The web site works best with Google Chrome (v. 60 or later) and Mozilla Firefox (v. 57 or later) on Windows and Xubuntu. The site also works with Safari v. 11.1 (macOS Sierra v. 10.12.6) and Edge v. 38 (Windows 10). Some functions may not work with Internet Explorer 11.
Analysis of individual genes
A valuable feature of our NEA method is that it can be applied to single node AGS or FGS. An ORA based on estimating gene set overlap would not enable such tasks, whereas estimating significance of network connectivity of a single node against a multi-node set is fully possible in NEA. An arbitrary single network node is likely to have zero network connections to an arbitrary node set, and therefore statistical power of NEA would be lower compared to multi-node both AGS and FGS. However in practice almost any gene still reveals enrichment against a few specific FGSs. This feature can help in describing poorly characterized genes in the functional space, determining potential impact of cancer mutations etc. Increasing the AGS size - when it is feasible - can increase statistical power of NEA. For instance, analyzing AGSs of 300 most significant DE genes revealed a number of putative regulatory single nodes (23). An EviNet analysis can be turned gene-wise via check boxes at Check and submit tab or by submitting single nodes as separate AGSs or FGSs.
TRANSCRIPTOMIC CHANGES DURING STEM CELL DIFFERENTIATION
In order to test and demonstrate the web server functionality, we analyzed a dataset from RNA sequencing of our experimental series in mouse embryonic stem cells (mESCs). A non-differentiating stem cell condition was compared to mESCs cultured for 1, 2 or 3 days in a differentiating condition (1DDC, 2DDC, 3DDC). mESCs are pluripotent stem cells that can give rise to ectodermal, endodermal, and mesodermal cell lineages when cultured in appropriate differentiation conditions (42).We used a protocol that via treating cells with retinoic acid (RA) and Shh agonist (SAG) drives differentiation of mESCs towards a ventral hindbrain neural progenitor identity (43). In this differentiation condition mESCs recapitulate ventral hindbrain development and produce a population of neural progenitors with a minor presence of other cell types. This diversification occurs in a largely uniform mESCs culture upon treatment with the same morphogen cocktail. Both the cell identities in this population and the signaling cascades that govern this process are unknown (Figure 2).
First, we analyzed the transcriptome differences between mESCs in non-differentiating and differentiating conditions. We detected a dramatic and progressive decrease in expression of key regulators of pluripotency (Nanog, Pou5f1, Klf4) and upregulation of neuroectodermal (Sox1, Sox3, Nes, Prom1) and endodermal (Sox17, Sox7, Gata4) markers but not of mesoderm-specific genes (Brachyury(T), Eomes, Hand1) (44–46). In order to find genes involved in pluripotent state maintenance or differentiation progression, we generated a Venn diagram that compared three DE lists: mESCs versus 1DDC, mESCs versus 2DDC, and mESCs versus 3DDC. This yielded genes that were consistently either down- or up-regulated from 0 to 3DDC. In the Venn diagrams, the full intersection of the three DE contrasts delineated 65 genes downregulated throughout 1, 2, 3 DDC (log2(FC) < –2; FDR < 0.05) and therefore putatively involved in the maintenance of pluripotent state. On the other hand, 149 genes upregulated throughout 1, 2, 3 DDC; log2(FC) > 2; FDR < 0.05) might be involved in the differentiation progression. We note that typically up- and down-regulated genes are analyzed together, so that a pop up table can be automatically imported to the analysis as a single AGS. However in this case we the up- and down-regulated gene lists were of different biological nature, so that we copied them from the pop up tables and saved in a separate AGS file (these can be seen upon typing in ID ‘stemcell’ in the project box and pressing ENTER). The two AGSs were then used as input to NEA with the signaling KEGG pathways chosen as FGSs. For the network, we used a union of FunCoup (FClim), CORUM protein complexes, KEGG pathways, and the protein phosphorylation network PTMapper. This choice based on our previous benchmark where a similar union of human networks performed best (22).
This analysis (Figure 3) detected enrichment of FGSs previously associated with either the pluripotent state, such as JAK-STAT, TGF-beta, MAPK and VEGF signaling pathways (47–49) or with the differentiation state, such as WNT and Hedgehog signaling pathways (50,51). A detailed sub-network (inset at Figure 3) behind the AGS-FGS link ‘differentiation – Sonic Hedgehog signaling’ shows that the DE genes were connected to both the upstream (Gli genes) and deep downstream (Wnt and Bmp families) parts of the SHH cascade. For comparison, the trivial ORA detected with a formally significant, unadjusted P-value only two of the associations (TGF-beta and Hedgehog in the pluripotent and differentiation states, respectively). Importantly, the signaling pathways used in the NEA often overlapped with each other, so that the same genes could be behind enrichment of multiple FGS. As an example, Spry4 (sprouty homolog 4) (52) was downregulated 5–10-fold in the 1, 2, 3 DDC as compared to mESCs. This gene was connected in the global network to Cblb, Sos1, Sos2 (JAK-STAT signaling) and Ppp3r2, Siah1a (WNT signaling) and thus contributed to enrichment scores of respective FGSs. Spry4 belongs to Sprouty genes which encode negative regulators of the receptor tyrosine kinase signaling and therefore could play an important role in stemness (53). On the other hand, individual important FGS genes could also be connected to multiple AGS genes, likely affecting the up- and down-regulation. We identified such genes by using the option ‘Analyze the FGS genes/proteins individually’ at Check and submit tab. Although >85% of the signaling pathway genes did not have any network edges connecting them to the two AGSs, there was also a number of genes richly and significantly connected to the downregulation AGS, such as Fgfr1, Fgfr2, Fgfr4; Tgfb1, Tgfb2, Tgfb3, Tgfa, Igf1r and Pdgfrb. On the upregulation side, the most connected were Gria4, Grm3, Snap25, Camk2a, Shh, Ihh, Dhh and others, which could explain the pathway pattern at Figure 3. Again, we emphasize that many contributing FGS genes were not DE themselves. At the second stage, we searched for FGSs that might regulate the selection between neuroectoderm and endoderm differentiation lineages. In order to do that, we used transcriptomics data for the neuroectoderm population, derived by sorting three DDC culture. In this case, the DE values reflected a technical difference, i.e., a mixture of two (or even multiple) fractions versus one filtered fraction, rather than a transcriptional shift due to a biological transformation. Significant differential expression between the non-sorted and sorted populations was used as a criterion additional to the described above. We retrieved gene lists in the contrasts corresponding to 2DDC versus mESCs (log2(FC) > 1; FDR < 0.05) and 3DDC versus mESCs (log2(FC) > 1; FDR < 0.15), and either positive (log2(FC) > 1; FDR < 0.05) or negative (log2(FC) < –0.5; FDR < 0.10) fold changes the contrast between 3DDC and 3DDC(sorted) which provided us with AGSs specific for endodermal (N = 66) and neuroectodermal (N = 161) lineages, respectively. There were FGSs enriched against both AGSs commonly (Hedgehog, VEGF, JAK-STAT) and specifically against the neuroectodermal AGS (WNT, NOTCH, axon guidance) (Figure 4). Some of these pathways had been well known, while others appeared novel and potentially interesting. Clicking at the link between neuroectodermal AGS and NOTCH (inset at Figure 4) demonstrated that most influential DE genes were likely Dll1 and Dtx4.
Figure 3.

Network enrichment of KEGG signaling pathways against gene sets that showed ongoing up- and down-regulation during differentiation toward neuroectodermal progenitor identity. Differentiation and Pluripotency: AGSs of 149 and 65 genes that were respectively up- and down-regulated at least 4-fold at each of the 1, 2 and 3 DDC compared to mESCs. Yellow boxes: AGSs. Red boxes: FGSs. Circles in the inset: member genes of AGS (yellow) and FGS (magenta) or both (orange). Node size: log(node degree) in the global network (cumulative for FGS and individual for genes). Two-headed arrows: summaries of individual gene–gene links (undirected or of arbitrary direction) from the global network that connected AGS and FGS. Edge labels: the number of individual gene–gene links behind each enrichment, where edge transparency corresponds to confidence of network enrichment score (maximal allowed NEA FDR = 0.05 corresponds to the highest transparency) and edge thickness stands for the number of gene–gene links. Links in the inset: thickness denotes edge confidence. For simplicity, FunCoup edges with Final Bayesian Score < 5 (29) are not shown.
Figure 4.

Network enrichment of genes characterized by up-regulation during differentiation and by enrichment after sorting between neuroectoderm and endoderm. Neuroectoderm and Endoderm: AGS of genes upregulated at least 2-fold at both 2 DDC versus mESCs and three DDC versus mESCs, and then either up-regulated 1.41-fold (N = 161) or down-regulated 2-fold (N = 66) at three DDC versus three DDC(sorted), respectively. Map legend: same as in Figure 3.
The analysis can be recapitulated from the Venn diagram tab by setting the DE criteria as described above and using column mapping for the DE file as shown in Table 1 (the DE file is available for download from Help menu). There is also a simplified demo version of the Venn diagram analysis, which can be run from the Help.
Table 1. Mapping between experimental conditions and column identifiers in example file P.matrix.NoModNodiff_wESC.VENN.txt.
| Substring in file header | Meaning |
| WT_Nondiff_ESCs_Control | mESCs |
| WT_Nondiff_1_Control | 1 DDC, non-sorted culture |
| WT_Nondiff_2_Control | 2 DDC, non-sorted culture |
| WT_Nondiff_3_Control | 3 DDC, non-sorted culture |
| WT_Prog_3(sorted)_Control | sorted 3 DDC culture |
| -FC | log2(fold change) of DE |
| -P | DE P-value from eBayes |
| -FDR | False discovery rate (adjusted P-value, or q-value) |
CONCLUSION
We have introduced a novel web implementation of our method for network enrichment analysis. It is streamlined by (i) considering only single-step, direct links between AGS and FGS genes, (ii) ignoring intra-FGS and intra-AGS edges, (iii) disregarding edge confidence weights and directionality and (iv) replacing the network randomization step with the unbiased analytic estimation of expected network connectivity.
Compared to similar resources, the data flow and output of EviNet appears the most similar to the classical ORA. On the other hand, due to a much higher statistical power it can be used for purposes other than an exploratory analysis, such as evaluation of candidate disease genes, testing cancer mutations for being drivers, or building prognostic and predictive statistical models using patient cohorts. Among the alternative NEA matrix outputs, we recommend using the z-score matrix because its values would be normally distributed under true null and would thus fit downstream statistical analyses.
We incorporated ancillary functionality for flexible re-definition of DE gene list to be submitted to NEA. The Venn diagram tool facilitates understanding and analysis of the complex high-throughput experimental design. We demonstrated how significant functional connections identified by EviNet in the differentiating mESCs suggested potentially important roles in the integration of signaling for specification between neuroectoderm and endoderm cell fates.
DATA AVAILABILITY
The mESC dataset of 11 RNA sequencing samples is available from NCBI repository under GEO accession number GSE112698 using URL https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112698.
FUNDING
Swedish Foundation for Strategic research [SRL10-0030 to J.E.]; The Knut and Alice Wallenberg Foundation [KAW2011.0161, KAW2012.0101 to J.E.]; Swedish Research Council [D0415501 to J.E., 2016-04940 to A.A.]; Cancerfonden [110578 to J.E.]; Vera and Emil Kornells Stiftelse (to A.A.) as well as National Bioinformatics Infrastructure Sweden (NBIS), Science for Life Laboratory, the National Genomics Infrastructure (NGI), and computational facility Uppmax for providing assistance in massive parallel sequencing and computational infrastructure. Funding for open access charge: Vetenskapsrådet.
Conflict of interest statement. None declared.
REFERENCES
- 1. Alexeyenko A., Wassenberg D.M., Lobenhofer E.K., Yen J., Linney E., Sonnhammer E.L., Meyer J.N.. Dynamic zebrafish interactome reveals transcriptional mechanisms of dioxin toxicity. PLoS One. 2010; 5:e10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexeyenko A., Lee W., Pernemalm M., Guegan J., Dessen P., Lazar V., Lehtiö J., Pawitan Y.. Network enrichment analysis: extension of gene-set enrichment analysis to gene networks. BMC Bioinformatics. 2012; 13:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tavazoie S., Hughes J.D., Campbell M.J., Cho R.J., Church G.M.. Systematic determination of genetic network architecture. Nat. Genet. 1999; 22:281–285. [DOI] [PubMed] [Google Scholar]
- 5. Huang D.W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Maeyer D., Weytjens B., Renkens J., De Raedt L., Marchal K.. PheNetic: network-based interpretation of molecular profiling data. Nucleic Acids Res. 2015; 43:W244–W250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tuncbag N., McCallum S., Huang S.-S.C., Fraenkel E.. SteinerNet: a web server for integrating ‘omic’ data to discover hidden components of response pathways. Nucleic Acids Res. 2012; 40:W505–W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lan A., Smoly I.Y., Rapaport G., Lindquist S., Fraenkel E., Yeger-Lotem E.. ResponseNet: revealing signaling and regulatory networks linking genetic and transcriptomic screening data. Nucleic Acids Res. 2011; 39:W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Basha O., Tirman S., Eluk A., Yeger-Lotem E.. ResponseNet2.0: revealing signaling and regulatory pathways connecting your proteins and genes–now with human data. Nucleic Acids Res. 2013; 41:W198–W203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glaab E., Baudot A., Krasnogor N., Schneider R., Valencia A.. EnrichNet: network-based gene set enrichment analysis. Bioinforma Oxf. Engl. 2012; 28:i451–i457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ogris C., Helleday T., Sonnhammer E.L.L.. PathwAX: a web server for network crosstalk based pathway annotation. Nucleic Acids Res. 2016; 44:W105–W109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeggari A., Alexeyenko A.. NEArender: an R package for functional interpretation of ‘omics’ data via network enrichment analysis. BMC Bioinformatics. 2017; 18:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Bardes E.E., Aronow B.J., Jegga A.G.. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009; 37:W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tiys E.S., Ivanisenko T.V., Demenkov P.S., Ivanisenko V.A.. FunGeneNet: a web tool to estimate enrichment of functional interactions in experimental gene sets. BMC Genomics. 2018; 19:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y., Thilmony R., Gu Y.Q.. NetVenn: an integrated network analysis web platform for gene lists. Nucleic Acids Res. 2014; 42:W161–W166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brin S., Page L.. The anatomy of a large-scale hypertextual Web search engine. Comput. Netw. ISDN Syst. 1998; 30:107–117. [Google Scholar]
- 17. Guney E., Oliva B.. Exploiting protein-protein interaction networks for genome-wide disease-gene prioritization. PLoS One. 2012; 7:e43557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vanunu O., Magger O., Ruppin E., Shlomi T., Sharan R.. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010; 6:e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Köhler S., Bauer S., Horn D., Robinson P.N.. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008; 82:949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bersani C., Huss M., Giacomello S., Xu L.-D., Bianchi J., Eriksson S., Jerhammar F., Alexeyenko A., Vilborg A., Lundeberg J. et al. Genome-wide identification of Wig-1 mRNA targets by RIP-Seq analysis. Oncotarget. 2016; 7:1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akan P., Alexeyenko A., Costea P.I., Hedberg L., Solnestam B.W., Lundin S., Hällman J., Lundberg E., Uhlén M., Lundeberg J.. Comprehensive analysis of the genome transcriptome and proteome landscapes of three tumor cell lines. Genome Med. 2012; 4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merid S.K., Goranskaya D., Alexeyenko A.. Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinformatics. 2014; 15:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alexeyenko A., Alkasalias T., Pavlova T., Szekely L., Kashuba V., Rundqvist H., Wiklund P., Egevad L., Csermely P., Korcsmaros T. et al. Confrontation of fibroblasts with cancer cells in vitro: gene network analysis of transcriptome changes and differential capacity to inhibit tumor growth. J. Exp. Clin. Cancer Res. CR. 2015; 34:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alkasalias T., Alexeyenko A., Hennig K., Danielsson F., Lebbink R.J., Fielden M., Turunen S.P., Lehti K., Kashuba V., Madapura H. et al. RhoA knockout fibroblasts lose tumor-inhibitory capacity in vitro and promote tumor growth in vivo. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E1413–E1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Astakhova L., Ngara M., Babich O., Prosekov A., Asyakina L., Dyshlyuk L., Midtvedt T., Zhou X., Ernberg I., Matskova L.. Short Chain Fatty Acids (SCFA) reprogram gene expression in human malignant epithelial and lymphoid cells. PLoS One. 2016; 11:e0154102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur O., Anwar N., Schultz N., Bader G.D., Sander C.. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011; 39:D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. von Mering C., Jensen L.J., Kuhn M., Chaffron S., Doerks T., Krüger B., Snel B., Bork P.. STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007; 35:D358–D362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huttenhower C., Haley E.M., Hibbs M.A., Dumeaux V., Barrett D.R., Coller H.A., Troyanskaya O.G.. Exploring the human genome with functional maps. Genome Res. 2009; 19:1093–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexeyenko A., Sonnhammer E.L.L.. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009; 19:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doudieu O.N., Stümpflen V. et al. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2007; 36:D646–D650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M., Goto S., Kawashima S., Nakaya A.. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002; 30:42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014; 42:D472–D477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M.. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012; 40:D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Narushima Y., Kozuka-Hata H., Tsumoto K., Inoue J., Oyama M.. Quantitative phosphoproteomics-based molecular network description for high-resolution kinase-substrate interactome analysis. Bioinformatics. 2016; btw164. [DOI] [PubMed] [Google Scholar]
- 35. Bovolenta L.A., Acencio M.L., Lemke N.. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics. 2012; 13:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishimura D. BioCarta. Biotech Softw Internet Rep. 2001; 2:117–120. [Google Scholar]
- 37. Caspi R., Altman T., Billington R., Dreher K., Foerster H., Fulcher C.A., Holland T.A., Keseler I.M., Kothari A. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome databases. Nucleic Acids Res. 2014; 42:D459–D471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pico A.R., Kelder T., van Iersel M.P., Hanspers K., Conklin B.R., Evelo C.. WikiPathways: pathway editing for the people. PLoS Biol. 2008; 6:e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yosef Hochberg Y.B. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995; 1:289–300. [Google Scholar]
- 41. Franz M., Lopes C.T., Huck G., Dong Y., Sumer O., Bader G.D.. Cytoscape.js: a graph theory library for visualisation and analysis. Bioinforma Oxf. Engl. 2016; 32:309–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005; 19:1129–1255. [DOI] [PubMed] [Google Scholar]
- 43. Dias J.M., Alekseenko Z., Applequist J.M., Ericson J.. Tgfβ signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron. 2014; 84:927–939. [DOI] [PubMed] [Google Scholar]
- 44. Zhao S., Nichols J., Smith A.G., Li M.. SoxB transcription factors specify neuroectodermal lineage choice in ES cells. Mol. Cell. Neurosci. 2004; 27:332–342. [DOI] [PubMed] [Google Scholar]
- 45. Yasunaga M., Tada S., Torikai-Nishikawa S., Nakano Y., Okada M., Jakt L.M., Nishikawa S., Chiba T., Era T., Nishikawa S.. Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat. Biotechnol. 2005; 23:1542–1550. [DOI] [PubMed] [Google Scholar]
- 46. Loh Y.-H., Wu Q., Chew J.-L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006; 38:431–440. [DOI] [PubMed] [Google Scholar]
- 47. Pan G., Thomson J.A.. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007; 17:42–49. [DOI] [PubMed] [Google Scholar]
- 48. Niwa H., Ogawa K., Shimosato D., Adachi K.. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009; 460:118–122. [DOI] [PubMed] [Google Scholar]
- 49. Dalton S. Signaling networks in human pluripotent stem cells. Curr. Opin. Cell Biol. 2013; 25:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hitoshi S., Seaberg R.M., Koscik C., Alexson T., Kusunoki S., Kanazawa I., Tsuji S., van der Kooy D.. Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signaling. Genes Dev. 2004; 18:1806–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008; 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- 52. Ikeda M., Inoue F., Ohkoshi K., Yokoyama S., Tatemizo A., Tokunaga T., Furusawa T.. B-box and SPRY domain containing protein (BSPRY) is associated with the maintenance of mouse embryonic stem cell pluripotency and early embryonic development. J. Reprod. Dev. 2012; 58:691–699. [DOI] [PubMed] [Google Scholar]
- 53. Kim H.J., Bar-Sagi D.. Modulation of signalling by Sprouty: a developing story. Nat. Rev. Mol. Cell Biol. 2004; 5:441–450. [DOI] [PubMed] [Google Scholar]
- 54. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup . The sequence Alignment/Map format and SAMtools. Bioinforma Oxf. Engl. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L., Wang S., Li W.. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012; 28:2184–2185. [DOI] [PubMed] [Google Scholar]
- 57. Anders S., Pyl P.T., Huber W.. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen H., Boutros P.C.. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011; 12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The mESC dataset of 11 RNA sequencing samples is available from NCBI repository under GEO accession number GSE112698 using URL https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112698.