

Abstract
The signaling lipid phosphatidylinositol 3,5-bisphosphate, PI(3,5)P2, functions in vesicular trafficking through the endo-lysosomal compartment. Cellular levels of PI(3,5)P2 are regulated by an enzyme complex comprised of the kinase PIKFYVE, the phosphatase FIG4, and the scaffold protein VAC14. Mutations of human FIG4 cause inherited disorders including Charcot-Marie-Tooth disease type 4J, polymicrogyria with epilepsy, and Yunis-Varón syndrome. Constitutive Fig4−/− mice exhibit intention tremor, spongiform degeneration of neural tissue, hypomyelination, and juvenile lethality. To determine whether PI(3,5)P2 is required in the adult, we generated Fig4flox/−; CAG-creER mice and carried out tamoxifen-induced gene ablation. Global ablation in adulthood leads to wasting, tremor, and motor impairment. Death follows within 2 months of tamoxifen treatment, demonstrating a life-long requirement for Fig4. Histological examinations of the sciatic nerve revealed profound Wallerian degeneration of myelinated fibers, but not C-fiber axons in Remak bundles. In optic nerve sections, myelinated fibers appear morphologically intact and carry compound action potentials at normal velocity and amplitude. However, when iKO mice are challenged with a chemical white matter lesion, repair of damaged CNS myelin is significantly delayed, demonstrating a novel role for Fig4 in remyelination. Thus, in the adult PNS Fig4 is required to protect myelinated axons from Wallerian degeneration. In the adult CNS, Fig4 is dispensable for fiber stability and nerve conduction, but is required for the timely repair of damaged white matter. The greater vulnerability of the PNS to Fig4 deficiency in the mouse is consistent with clinical observations in patients with Charcot-Marie-Tooth disease.
Introduction
The biosynthetic complex that generates phosphatidylinositol-3,5-bisphosphate [PI(3,5)P2] is evolutionarily conserved in eukaryotes and includes the 5-phosphate kinase PIKFYVE, the 5-phosphatase FIG4, and the scaffold protein VAC14. The complex is maintained by protein-protein interaction and is destabilized by loss of FIG4 or VAC14 (1,2). The biosynthetic complex is localized to early endosomes, late endosomes and lysosomes, where PI(3,5)P2 is thought to specify organelle identity and contribute to ion homeostasis, vesicle fission, nutrient sensing and autophagy (3,4). Mutations of FIG4 and VAC14 result in recessive inherited disorders in human and mouse. Complete loss-of-function of FIG4 or VAC14 results in the multisystem, lethal Yunis-Varón Syndrome (5,6). Partial loss-of-function of FIG4 manifests as anatomical CNS defects (polymycrogyria), epilepsy, psychotic behaviors, calossal dysplasia and neurodegeneration (7). Bi-allelic missense mutations of VAC14 are responsible for recessive pediatric neurological disorders (8). Human patients with the FIG4 missense mutation p.Ile41Thr in combination with a null FIG4 allele present with Charcot-Marie-Tooth type 4J (CMT4J), a severe peripheral neuropathy (9–11). The pathology of CMT4J in human patients suggests both demyelinating and neurodegenerative contributions. The presence of ‘onion bulb’ myelin structures in nerve biopsies and reduced sciatic nerve conduction velocity indicate that CMT4J is a demyelinating neuropathy. Impairment of upper and lower motor neuron function, proximal muscle weakness and sensory deficits are indicative of neuronal damage (10,11). While most CMT4J patients do not exhibit CNS deficits, recently two families have been identified where FIG4 variants manifest in CNS pathology (12,13). These observations suggest differential requirements for PI(3,5)P2 in the CNS versus PNS.
Studies in the mouse have demonstrated the particular vulnerability of the nervous system to PI(3,5)P2 deficiency. Morphologically, in Fig4 null mice there is spongiform degeneration of the brain with extensive vacuolization that is not seen in other tissues with the exception of spleen (9). Strikingly, the lethal wasting disorder of global null mice can be rescued by neuron-specific expression of a Fig4 cDNA transgene (14). Overexpression of the FIG4 patient mutation p.Ile41Thr in neurons of transgenic mice also rescues lethality (2). Conversely, neuron-specific knockout of a floxed allele of Fig4 reproduces the lethal wasting disorder (14). Interestingly, a neuron-specific transgene lacking phosphatase activity can partially rescue the Fig4 null mouse (15). Mice with missense mutations of Vac14 exhibit deficits similar to Fig4 mutants (1,16).
In the CNS, myelin is produced by oligodendrocytes (OLs). OL precursor cells (OPCs) arise early in development, migrate extensively, and are broadly distributed in the brain and spinal cord throughout life. After OPC differentiation and maturation into myelin producing cells, OLs form internodes along axons. One mature OL can form internodes on up to 50 different axons. Mature OLs do not contribute to white matter repair (17). Instead, myelin repair is carried out by OPCs, tissue resident progenitors that differentiate into myelin producing OLs (18). In the developing murine PNS, Schwann cell precursors arise from neural crest cells around E13 and undergo differentiation into mature Schwann cells that form myelin or non-myelinating Schwann cells that ensheath multiple axons, called Remak bundles. In contrast to OLs, myelin producing Schwann cells commit to myelinate a single axon, forming only one internode (19). After nerve damage, Schwann cells and Remak cells are capable of de-differentiation and adopt a repair phenotype supporting regeneration (20,21,22).
We previously carried out detailed analysis of the role of PI(3,5)P2 biosynthesis in myelin development, both in the PNS and CNS. Cell-specific inactivation of the biosynthetic complex in OPC/OLs or in Schwann cells results in hypomyelination of the CNS or PNS, respectively (9,14,16,23). While PI(3,5)P2 is clearly required for myelin synthesis during development, its role in myelin stability, turnover, and repair in the adult has not yet been examined. Here, we carried out inducible knock-out of Fig4 (iKO) in adult mice and found that global loss of Fig4 leads to animal wasting and death within 2 months, recapitulating at a later time point the early postnatal effects of constitutive loss of Fig4. Myelinated fibers were rapidly and severely affected in the mature PNS but not in the CNS, suggesting differential vulnerability to reduced levels of PI(3,5)P2. Additional studies demonstrated that repair of a chemically induced white matter lesion in the mature CNS is dependent on Fig4 function. Therapeutic strategies for treating the disorders of PI(3,5)P2 biosynthesis will have to take into account the continuing requirements of the adult CNS and PNS.
Results
Neonatal knockout of Fig4 recapitulates the neurological defects of constitutive Fig4−/− null mice
To assess the effects of Fig4 knockout in vivo, we combined the floxed allele of Fig4 (14) with the tamoxifen-inducible CreER expressed under a ubiquitous promoter (CAG-CreER) (24). In the absence of tamoxifen, there was minimal deletion of the floxed allele in tail and brain biopsies of Fig4flox/−, CAG-CreER mice, as previously reported (25). To evaluate the effectiveness of tamoxifen induction in these mice, we administered 2 doses of tamoxifen to Fig4flox/−, CAG-CreER mice on postnatal (P) days P3 and P4 and analyzed the effect at P22. Mice are designated iKO(3→22) to specify the first day of tamoxifen treatment and the day of analysis. As expected, iKO(3→22) brains exhibited massive spongiform degeneration in neocortical layers IV and V (Fig. 1A and B) and thinning of myelinated fibers in the corpus callosum and white matter in cerebellar lobules (Fig. 1A–D). Western blotting of cerebellar lysates revealed a substantial decrease in FIG4 protein, myelin-associated glycoprotein (MAG), and myelin basic protein (MBP) (Fig. 1E). A hallmark of Fig4−/− cells in the OL lineage is the accumulation of large peri-nuclear LAMP1+ vacuoles (16). OPCs/OLs were isolated from iKO(3→9) pups by immunopanning with anti-PDGFRα to capture OPCs (Fig. 1F and F’), O4 antibody to capture immature OLs (Fig. 1G and G’) and anti-MOG to capture mature OLs (Fig. 1H and H’) followed by four days in culture. Many OLs in the iKO cultures contained enlarged LAMP1+ perinuclear vacuoles, identified by double-fluorescence immunolabeling with anti-MAG and anti-LAMP1 (Fig. 1F’, G’ and H’). OPCs/OLs from Fig4 control pups did not contain enlarged LAMP1+ vacuoles (Fig. 1F, G and H). This recapitulation of the phenotypes of constitutive Fig4 null mice, including regional spongiform degeneration, CNS hypomyelination and vacuoles in the OL lineage (9,16), demonstrates effective deletion of the floxed Fig4 allele by tamoxifen activation of CAG-CreER.
Figure 1.

Global neonatal inducible ablation of Fig4 leads to CNS degeneration, hypomyelination, and perinuclear accumulation of LAMP1+ vacuoles. (A, B) Sagittal sections of the P22 neocortex of Fig4 control (Fig4+/+ or Fig4+/flox) and Fig4 iKO(3→22) mice stained with Fluoromyelin Green. In the Fig4 iKO(3→22) neocortex, arrowheads point to extensive spongiform degeneration in deep cortical layers. Myelin staining in the corpus callosum (CC) is reduced in iKO mice. (C, D) Sagittal sections of Fig4 control and Fig4 iKO(3→22) cerebellum. Arrows in D point to cerebellar lobules and deep cerebellar nuclei with reduced Fluoromyelin staining. Scale bar, A–D = 200 μm; n = 2 mice per genotype. (E) Western blots of cerebellar lysates prepared from Fig4 control (n = 2) and Fig4 iKO(3→22) mice (n = 2), probed with antibodies specific for FIG4, myelin-associated glycoprotein (MAG) and myelin basic protein (MBP). Class III β-tubulin, detected with the TuJ1 antibody, is shown as a loading control. (F–H’) Representative images of primary OLs isolated from Fig4 control and Fig4 iKO(3→9) pups by immuno-panning with (F, F’) anti-PDGFRα to capture OPCs, (G, G’) the O4 antibody to capture immature OLs and (H, H’) anti-MOG to capture myelinating OL. After four days in vitro, cultures were fixed and stained with anti-MAG (green), anti-LAMP1 (red) and the nuclear dye DAPI (blue). LAMP1+ structures in OLs are marked by arrows and shown at higher magnification in inserts. Scale bar, F–H’ = 20 μm.
Deletion of Fig4 in adult mice causes rapid deterioration and death
To determine whether loss of Fig4 function in adult animals would lead to pathology similar to neonatal inactivation, Fig4flox/−; CAG-CreER mice between the ages of 60 and 90 days were treated with intraperitoneal tamoxifen (75 mg/kg) for five consecutive days. Mice were examined every third day with the tail suspension test and gait analysis was monitored. As early as 10 days after the first tamoxifen dose, abnormal forelimb and hindlimb clasping was observed (not shown). After 20 days, iKO mice exhibited reduced body weight (Fig. 2A) and after 30 days there was truncal twisting and abnormal hindlimb clasping (Fig. 2B and C). While control littermates splayed their hindlimbs 90% of the test time, this was reduced to 5% in iKO mice (Fig. 2F). At 60 days after tamoxifen treatment, the average weight of Fig4 iKO (60→120) mice was 16 ± 0.8 g, less than half the weight of littermate controls (33 ± 1 g) (Fig. 2A). Fig4 iKO mice also displayed variable dilution of pigmentation, ranging from occasional white patches on the rump and abdomen to an almost completely white fur (Fig. 2D and E), reminiscent of the hypo-pigmentation of Fig4 constitutive null mice (9). The severe gait impairment and ‘swimming’ movement of limbs observed at in iKO (60→120) mice also closely resembles the defects of juvenile mice with the constitutive null Fig4 mutation (Supplementary Material, Videos S1 and S2). Two months after tamoxifen treatment, Fig4 iKO(60→120) and iKO (90→150) mice became progressively lethargic and non-responsive, with severely compromised gait, intention tremor, and hunched posture (Supplementary Material, Videos). All of the tamoxifen treated mice died or were terminated as moribund between 60 and 65 days after initiation of treatment. Inactivation of Fig4 in adult mice thus recapitulates the lethal disorder seen in constitutive null mice, with a similar time course.
Figure 2.
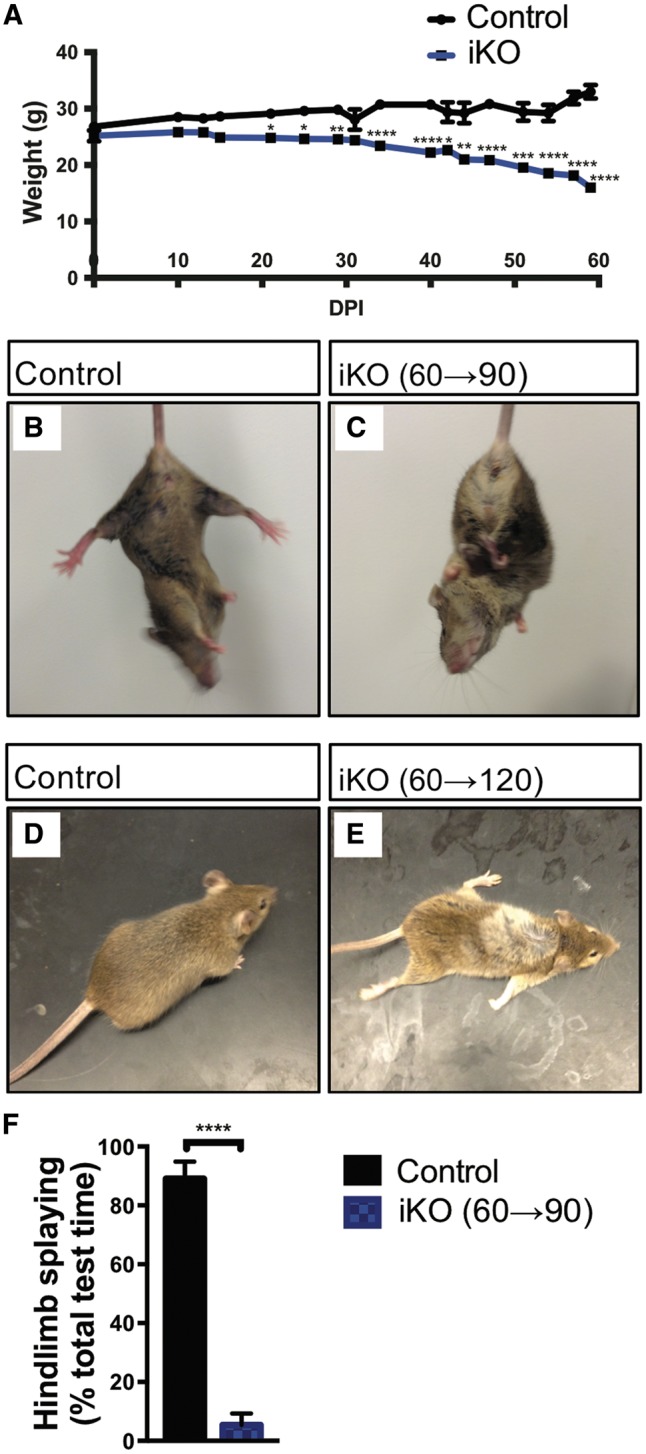
Global adult inducible Fig4 deletion results in severe motor defects, weight loss, and death. (A) The graph shows animal weight (g) as function of days post tamoxifen injection for Fig4 control and adult Fig4 iKO mice. Day 0 denotes the first tamoxifen injection when mice were between P60 and P90. Fig4 control mice, n = 10 (day 0) and n = 8 (day 60). For iKO mice, n = 5 (day 0) and n = 3 (day 60). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. 2-way ANOVA with Sidak correction. (B, C) Tail suspension test with Fig4 control and adult Fig4 iKO(60→90) mice. Control mice display typical hindlimb spreading, whereas iKO mice show abnormal clasping and truncal twisting. (D, E) Representative images of Fig4 control and adult Fig4 iKO(60→120) mice. iKO mice show spotty loss of fur pigmentation. (F) Quantification of tail suspension test. Animals were held by their tails for 15s and time displaying hindlimb splaying was recorded. Data presented as percentage of total time hindlimb splaying, mean ± SEM, ****P < 0.0001, unpaired Student t-test. n = 3 (control) and n = 5 (iKO).
Spongiform degeneration of the CNS after knockout of Fig4 in adult mice
A hallmark of constitutive Fig4 deficiency is a region-specific spongiform degeneration of the CNS. Deep neocortical layers, deep cerebellar nuclei, brainstem, spinal motoneurons and dorsal root ganglia (DRGs) are severely affected (9,16). To determine whether CNS spongiform degeneration occurs after Fig4 knockout starting at P21 or at P60, brain structures were examined in iKO(21→81) and iKO(60→120) mice (Fig. 3). In constitutive null mice, extensive degeneration is apparent in neocortical layers IV and V, deep cerebellar nuclei, brainstem, spinal motoneurons and DRGs (Fig. 3). When Fig4 knockout was induced at P21 or at P60, there was only minimal vacuolization in the neocortex, deep cerebellar nuclei, and brainstem (Fig. 3). Vacuolization of the spinal motoneurons and DRGs in iKO(21→81) and iKO(60→120) mice, however, was comparable in severity to constitutive Fig4 null mutants (Fig. 3), demonstrating greater vulnerability of these neurons which have long peripheral projections.
Figure 3.
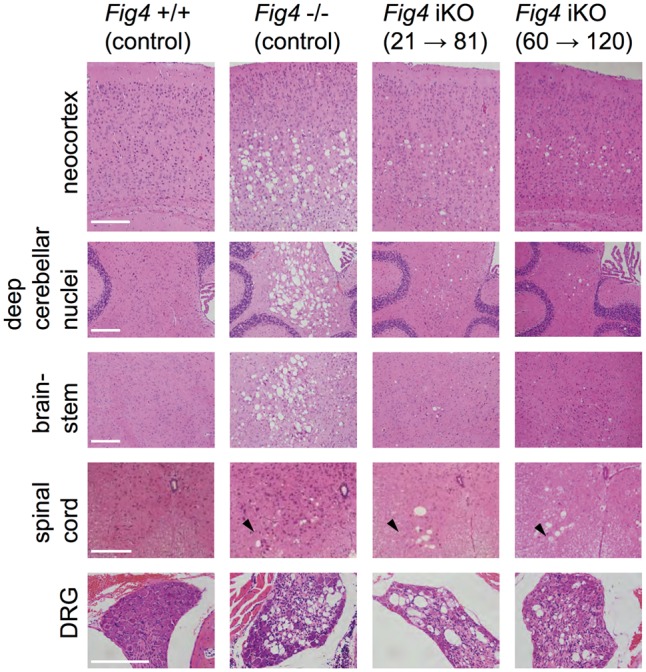
Spongiform degeneration in global adult inducible Fig4 knockout mice. Hematoxylin/eosin stained tissue sections of mouse neocortex, deep cerebellar nuclei, brainstem, spinal cord and dorsal root ganglion (DRG). Tissue sections of mice with the following genotypes are shown: wildtype mice (Fig4+/+ control), Fig4 germline null mice (Fig4−/− control), Fig4 iKO(21→81) and Fig4 iKO(60→120). Most notable are the large vacuolar (sponge-like) structures in different regions of the Fig4−/− nervous system, including deep layers of the neocortex, deep cerebellar nuclei, brainstem, ventral horn of the spinal cord and DRGs. A milder but similar phenotype is observed in the neocortex, cerebellum, and brainstem of Fig4 iKO(21→80) and Fig4 iKO(60→120) mice. DRGs and the ventral horn of the spinal cord (arrowhead) in Fig4−/− and iKO mice show numerous large vacuoles. Scale bar = 200 μm.
Abundance of CNS myelin is not reduced in adult Fig4 iKO mice
To determine whether FIG4 is necessary for myelin stability in the fully developed adult brain, whole brain membranes were isolated from Fig4 iKO(60→120) and age-matched control mice and subjected to immunoblotting with antibodies specific for the myelin markers MAG, MBP, proteolipid protein (PLP), and 2′3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (Fig. 4A). When compared to brain membranes from Fig4 control mice, there was a 73 ± 5% reduction in FIG4 protein in the iKO mice (Fig. 4A and B). However, there were no significant differences in the abundance of the myelin proteins MAG, MBP, CNPase, and PLP (Fig. 4A–F). Thus, in adult mice, integrity of CNS myelin is maintained for a 2-month period without continued expression of Fig4. In a similar vein, Western blot analysis of Fig4 iKO (21→81) forebrain and cerebellar lysates revealed no significant reduction in MAG. This suggests that Fig4 iKO after P21 no longer leads to a reduction in CNS myelin (Supplementary Material, Fig. S1).
Figure 4.

Myelin abundance is not affected by adult inducible Fig4 deletion after 60 days. (A) Representative Western blots of brain membranes isolated from adult Fig4 control and Fig4 iKO(60→120) mice, probed with antibodies specific for FIG4 and the myelin proteins MAG, CNPase, MBP and PLP. Neuronal classIII β-tubulin is shown as loading control. (B–F) Quantification of FIG4 and myelin protein signals normalized to classIII β-tubulin. Relative protein intensities compared to Fig4 control brains are shown as mean value ± SEM. N = 4 per genotype. Unpaired Student t-test, ****P < 0.0001; n.s. = not significant.
Normal structure and function of optic nerve fibers in Fig4 iKO mice
To independently confirm the results of the Western blots in Figure 4, we examined the structure of optic nerves by TEM. No defects were observed in myelin thickness or structure, or in the number of myelinated axons (Fig. 5A and B). To evaluate optic nerve function, we recorded compound action potentials (CAPs) from Fig4 iKO and littermate controls (Fig. 5C and D). A mature optic nerve fires a multi-peak CAP with the peaks corresponding to myelinated axonal populations of different diameter (26). The conduction velocity and amplitude of each peak can be resolved by fitting the CAP as a sum of three of four Gaussians (27). In constitutive Fig4−/− null mice, there is a significant increase in the population of slowly conducting fibers with a concomitant decrease in fast conducting populations (26). These changes were not observed in the optic nerve of adult Fig4 iKO mutants. Neither conduction velocity nor amplitude differed from controls (Fig. 5E and F). Taken together, the normal abundance of myelin proteins, the structural integrity of myelin sheaths in optic nerve cross sections, and the normal electrophysiological properties demonstrate persistence of CNS myelin during the 2 months survival of adult iKO mice.
Figure 5.

Optic nerve ultrastructure and conduction are not affected in adult Fig4 iKO60d mice. (A, B) Representative TEM optic nerve cross section micrographs of adult Fig4 control and iKO(60→120) mice. Scale bar = 1 μm, n = 4 mice per genotype. (C, D) Representative optic nerve CAP traces recorded from adult Fig4 control and iKO(60→120) mice show three characteristic peaks. (E) Quantification of conduction velocities and (F) amplitudes for each peak within CAP traces identified by Gaussian fit. Data are shown as mean ± SEM. Multiple unpaired Student t-tests, n = 5 nerves (Fig4 control), n = 6 nerves (iKO).
Defective repair of CNS myelin after adult knockout of Fig4
Mbp mRNA is strongly upregulated in myelin producing OLs and transported into internodes (28) making it a useful marker for white matter repair (29). Wildtype and Fig4 iKO mice were injected with tamoxifen at P60 followed by stereotaxic injection of lysolecithin into the corpus callosum at P70. Three weeks later, at P91, white matter repair was assessed by in situ hybridization of Mbp and electron microscopy (Fig. 6A). Serial sections through the lesion were stained with a digoxigenin labeled cRNA probe specific for Mbp (Fig. 6B and C). The center of the lesion was defined as the section with the largest circumference of intensely Mbp labeled area (Fig. 6B and C, white dotted line). Quantification revealed a comparable lesion size in Fig4 control and iKO mice (Fig. 6D). The area that failed to undergo repair was defined as the inner rim of elevated Mbp labeling (Fig. 6B and C, yellow dotted line). The remyelinated area was significantly reduced in the iKO mice (Fig. 6E). At the ultrastructural level, myelinated fibers in the corpus callosum of Fig4 control and iKO mice, prior to LPC injection, are indistinguishable (Fig. 6F and H). At 21 days after LPC administration, wildtype mice exhibit extensive remyelination of the lesion, as indicated by thinly myelinated axons (Fig. 6G). However, there was little evidence of remyelination in the iKO mice (Fig. 6I). The non-myelinated axons in the iKO mice are surrounded by glial cells with the characteristic vacuolization of Fig4 null cells (Fig. 6I, arrowheads). Together these data suggest that in adult mice Fig4 is necessary for axon remyelination and the timely repair of damaged white matter.
Figure 6.

In the adult CNS Fig4 is required for white matter repair. (A) Timeline showing tamoxifen administration, five consecutive injections at P60-65, stereotaxic injection of LPC in the corpus callosum (P70) and animal analysis (P91). (B, C) Coronal sections through the lesion center of adult Fig4 control and iKO(60→91) mice stained for Mbp transcript. The white dashed line demarcates the lesion area in the corpus callosum. The dashed yellow line indicates the non-myelinated area in the lesion core. Scale bar = 200μm. Quantification of the percent remyelination (white area − yellow area)/(white area), reveled less complete myelination in iKO mic; n = 6 (control) and n = 8 (iKO). (F, H) Electron photomicrographs through the corpus callosum of Fig4 control and iKO(60→91) mice. (G, I) Electron photomicrographs through the lesion core of Fig4 control and iKO(60→91) mice. Arrows in G point to re-myelinated axons. Fig4 iKO show extensive vacuolization in the lesion area. Scale bar, 2 μm. N = 4 mice per genotype.
Severe peripheral nerve degeneration after knockout of Fig4 in adult mice
To determine whether ablation of Fig4 in adult mice causes PNS damage, sciatic nerves were examined 2 months after tamoxifen treatment in iKO(60→120) mice. Toluidine blue staining of semi-thin transverse sections from wildtype sciatic nerve demonstrated structurally intact nerve architecture with numerous myelinated fibers, large and small in caliber (Fig. 7A). In longitudinal sections, internodes, separated by nodes of Ranvier, are readily detected in wildtype nerves (Fig. 7C). In contrast, sciatic nerve sections of iKO(60→120) mice presented with gross histological abnormalities including reduced fiber density, myelin disintegration and appearance of myelin ovoids, characteristic of Wallerian degeneration (Fig. 7B and B’). Longitudinal sections of mutant nerves revealed fiber fragmentation, disintegration of internodes and the presence of myelin ovoids (Fig. 7D). In iKO, but not control nerves, macrophages are present that phagocytose myelin debris (Fig. 7D’). At the ultrastructural level, cross sections of P120 wildtype sciatic nerves show regularly formed and compact myelin sheath (Fig. 7E). Analysis of iKO(60→120) sciatic nerves revealed typical signs of Wallerian degeneration, including axonal drop-out and enlarged myelin profiles. The presence of tomacula, or myelin swelling, was prominent on the adaxonal side of myelinated fibers, while abaxonal myelin lamellae were much less affected (Fig. 7F). The presence of tomacula is an indication that axoplasm is lost and suggests that nearby axon fragmentation has occurred. In support of axonal pathology, electron-dense inclusions, characteristic of dystrophic axons, are present in iKO mice (Fig. 7F). For quantification of Wallerian degeneration, sciatic nerve fibers were binned into four categories with increasing morphological defects, ranging from (1) normal appearing fibers, (2) mild disintegration of myelin sheaths, (3) formation of large myelin out folds with clear signs of axonal degeneration and (4) fully disintegrated fibers. As shown in Figure 7G, iKO mice exhibit a significant increase in degenerated fibers.
Figure 7.

Adult Fig4 iKO mice exhibit severe Wallerian degeneration of the sciatic nerve. (A–D’) Semi-thin transverse (A, B, B’) and longitudinal (C, D, D’) sections of sciatic nerve prepared from adult Fig4 control and iKO(60→120) mice stained with toluidine. In iKO nerves, myelin ovoids (arrow in B’) and macrophages that phagocytose myelin debris (arrowhead in D’) are shown. Scale bar = 2 μm (A, B, D, E) and 0.5 μm (C, F). (E–I) Representative TEM photomicrographs of cross sciatic nerve main trunk sections isolated from adult Fig4 control and Fig4 iKO(60→120) mice. Note, B’ and D’ are higher magnification views on B and D, respectively. (E) Fig4 control nerves demonstrate robust myelination of large caliber fibers. (F) Sciatic nerves from iKO mice show adaxonal myelin swelling and disintegration of myelin sheaths (asterisk). Fibers of iKO sciatic nerves frequently are filled with electron-dense inclusions, characteristic of dystrophic axons (arrow in F). (G) Quantification of fiber degeneration in the sciatic nerve of Fig4 control (n = 4) and iKO (n = 5) mice. Degeneration index 1 = intact fibers, 2 = mild disintegration of myelin sheaths, 3 = formation of large myelin outfolds with clear signs of axonal degeneration, and 4 = degenerated fibers. ***P < 0.001, ** P < 0.01, *P < 0.5. Multiple t-tests corrected for multiple comparisons using Holm-Sidak. (H, I) High magnification of Remak bundles revealed no obvious differences between Fig4 control and iKO(60→120) nerves. H, I, Scale bar = 1 μm. (J) Quantification of axon number per Remak bundle (n = 2 mice per genotype, 141 bundles for control and 60 bundles for iKO mice) and (K) quantification of C-fiber axon diameter (n = 2 mice per genotype, 200 axons for control and 200 axons for iKO mice). Unpaired Student’s t-test comparing WT to iKO, P = 0.2933, axon number per Remak bundle; P = 0.787, axon diameter.
Small-caliber C-fiber axons in the sciatic nerve are not myelinated but contacted by non-myelinating Schwann cells (23,30). One non-myelinating Schwann cell can ensheath multiple C-fibers to form a Remak bundle. In Fig4 control and iKO sciatic nerves Remak bundles are present and appear largely intact (Fig. 7H and 7I). Quantification of axons per Remak bundle (Fig. 7J), and C-fiber axon diameter (Fig. 7K), revealed no significant differences between WT and iKO mice. Taken together, these data indicate that in the adult PNS Fig4 is necessary for the stability of myelinated fibers, but not Remak bundles.
Discussion
PI(3,5)P2 is a low abundance membrane phosphoinositide with important function in vesicular trafficking through the endo-lysosomal compartment. Perturbation of PI(3,5)P2 homeostasis causes severe developmental defects in humans and mice (2,5,8,9,15,16). Here we demonstrate that PI(3,5)P2 homeostasis is required for survival of adult mice. As early as 10 days after Fig4 deletion, there were defects in gait and limb coordination. These were followed by gradual decline that included weight loss, dilution of pigmentation, development of ‘swimming’ gait, intention tremor, and death within 2 months. This ‘wasting’ phenotype is likely due to impaired function of the nervous system, as many of these defects are also observed in Fig4flox/−; SynCre mice (14). As in juvenile spontaneous Fig4−/− null mice, we observed region-specific spongiform degeneration in the adult iKO nervous system, including the neocortex, deep cerebellar nuclei, DRGs, and spinal motoneurons. At the ultrastructural level, sciatic nerves of adult Fig4 iKO(60→120) mice showed Wallerian degeneration of myelinated fibers with prominent axonal pathology and disintegration of myelin profiles. Typical for damaged PNS tissue, macrophages accumulate in the sciatic nerve of iKO mice to phagocytose myelin debris (31). Interestingly, in iKO sciatic nerves, C-fiber axons in Remak bundles appear morphologically intact, suggesting they are less vulnerable to Fig4 deficiency. Remarkably, while myelinated fibers in the PNS are severely affected, in the same animals, CNS myelin is spared, with no signs of fiber degeneration or delay in nerve conduction. However, repair of CNS white matter is impaired in the adult iKO mice. Taken together, our data demonstrate that Fig4 function is required for survival, and the dependence on Fig4 is greater for myelinated fibers in the PNS than in the CNS.
Adult knockout of Fig4 leads to severely shortened lifespan
Germline inactivation of any component of the PI(3,5)P2 biosynthetic complex in mice results in decreased survival. Constitutive null Fig4 mice survive for up to 8 weeks, Vac14 null mutants die neonatally, and Pikfyve null mice do not survive past E3.5 in utero (9,32,33). The most obvious pathology in Fig4 null mutants is in the nervous system (9), and muscle atrophy is secondary to neurodegeneration (34). We now demonstrate that induced deletion of Fig4 in adult mice leads to premature death. The cause of death is not fully understood, but the most obvious defect is the spongiform degeneration of neural tissues. Both germline and adult Fig4 inducible knockouts may die due to impaired breathing as a result of compromised nerve conduction in the phrenic nerve. Moribund animals were frequently observed to be gasping for air shortly before being euthanized. Consistent with the view that neuronal Fig4 is required for survival, Fig4flox/−, SynCre have a shorter lifespan than Fig4flox/−, Olig2Cre and Fig4flox/−, PDGFRaCreER mice (16). Conversely, lifespan is extended in Fig4−/− mice expressing transgenic Fig4 in neurons (Fig4−/−, NSE-Fig4) (14).
Fig4 is required in the adult PNS
Reminiscent of human CMT4J patients, PNS pathology in adult Fig4 iKO mice exhibits signs of both neuronal and glial contributions. Our findings clearly demonstrate the essential role of Fig4, and by extension of PI(3,5)P2, in the adult PNS. The severe degeneration of the sciatic nerve in adult iKO mice is very similar to the pathology reported for juvenile Fig4−/− mice (9). Our experiments using CAG-CreER do not distinguish between dependence of fiber integrity on Fig4 expression in sensory neurons, motoneurons or Schwann cells. However, developmental studies have demonstrated that knockout of Fig4flox in motoneurons with Hb9-Cre causes extensive neuronal vacuolization and a mild myelination phenotype (16). In Fig4flox/flox; Olig2-Cre mice, spinal motoneurons are Fig4 deficient and show large vacuoles in axons that project toward the ventral root (16). Fig4 conditional knockout in Schwann cells with P0-Cre causes severe defects sciatic nerve myelination (22), indicating that global loss of Fig4 results in combined axonal and demyelinating pathology. Loss of Fig4 in adult mice causes vacuolization in the ventral horn of the spinal cord, suggesting life-long vulnerability of spinal motoneurons. Accumulating evidence suggests that FIG4 mutations may also be associated with amyotrophic lateral sclerosis (4,35,36).
Based on the severe DRG vacuolization in adult Fig4 iKO mice, Fig4 function appears to play an essential role in sensory neurons throughout life. The appearance of abaxonal myelin out-foldings and formation of tomacula in the sciatic nerve are suggestive of axonal drop out and neurodegeneration. Mice and human patients deficient in FIG4 have both motor and sensory abnormalities. In human CMT4J patients, the sensory defects are relatively mild and motor defects are severe and progress to loss of ambulation. Our data further suggest differential vulnerability between myelinated fibers and axons ensheathed by Remak Schwann cells. Future studies will have to address whether Fig4 deficiency affects C-fiber function.
Fig4 function in the adult CNS
The spongiform degeneration and impaired CNS myelination in adult Fig4 iKO mice is less severe than in the constitutive global null mouse, and the function of CNS myelin is largely retained. The retention of CNS myelination is unlikely to result from incomplete deletion of Fig4 in OLs, since inducible Fig4 ablation in neonatal and juvenile iKO mice recapitulates the major phenotypes of Fig4−/− mice and primary OL prepared from iKO mice displayed enlarged vacuoles similar to germline Fig4−/− OL cultures. The CNS may be protected by the greater stability (reduced turnover) of its myelin proteins (37). However, recent advances in in vivo monitoring of OLs in the CNS indicate that remodeling of CNS myelin persists in the adult (38). The 2 months survival period after ablation of Fig4 in our mice may be too short a time for significant turnover of CNS myelin. Additional studies assessing long-term Fig4 deletion specifically in the OL lineage could determine whether Fig4 is dispensable for myelin maintenance in the CNS. It is important to differentiate between myelin maintenance by existing OLs, and myelin replacement which involves adult OPC migration and differentiation into myelinating OLs and is likely a slower process (38).
FIG4 is required for adult CNS myelin repair
Our data suggest that FIG4 is required for myelin repair but not maintenance in the CNS. These observations are consistent with a recapitulation of FIG4-depenent development-like events during repair of adult white matter and axon remyelination. Notably, OPCs undergo proliferation and differentiation during repair of damaged myelin. Given the critical role of PI(3,5)P2 biosynthesis in OPC differentiation (16), the repair defect after ablation of Fig4 expression may reflect arrest of OPC differentiation at the lesion site.
We have demonstrated a continuing requirement for Fig4 expression in the adult mouse, as indicated by the dramatic decline in weight and neural function after FIG4 deletion. The 2 month progression to death is similar to the postnatal decline of global Fig4 null mice, which only survive for 6–8 weeks. Both neurons and glia are dependent on Fig4 expression, with an intriguing interdependence in CNS myelination (16). The wide range of dysfunction and clinical severity in patients with deficiency of PI(3,5)P2 biosynthesis is consistent with differential dependence of various cell types on this universal signaling molecule. Further studies of patient mutations and animal models will be required to completely elucidate the pathogenic mechanisms underlying PI(3,5)P2 deficiency.
Materials and Methods
Mouse genetics
All mice were housed and cared for in accordance with NIH guidelines, and all research was conducted with the approval of the University of Michigan Committee on Use and Care of Animals. Both male and female mice were used in experiments. To obtain global inducible deletion of Fig4, a congenic line of Fig4+/− mice on the C57BL/6J strain background were crossed with the CAG-CreER tamoxifen inducible line on the C57BL/6J background (#004682, Jackson Laboratories). Fig4+/−, CAG-CreER progeny were crossed with Fig4flox/floxmice on the C3HeB/J strain background. Fig4 controls contained at least one WT Fig4 allele and were treated with tamoxifen in most cases to control for potential toxicity.
Cre induction
Tamoxifen (Sigma-Aldrich) was dissolved at a concentration of 10 mg/ml in a 910: 90 mixture of Sunflower seed oil and 100% Ethanol. Mice were treated at P21 or between P60-P90 with 75 mg/kg of tamoxifen by daily IP injections for 5 days. For early postnatal studies, 4-hydroxytamoxifen was dissolved at 10 mg/ml in pure ethanol and administered to P3 pups on 2 consecutive days as described (16).
Tail suspension test
The tail suspension test was performed as described by (39).
Hematoxylin/eosin staining
Tissue slices were stained with H&E as previously described (14).
Toluidine blue staining and transmission electron microscopy
Transmission electron microscopy (TEM) was performed as described (26). The main trunk of the sciatic nerve was used for ultrastructural analysis. Toluidine blue images were taken using 40× oil objective on Zeiss Axio Examiner inverted microscope (Zeiss) and tiled images were stitched using ZEN software.
Optic nerve recordings
CAP recordings of acutely isolated optic nerves were performed as reported previously (26,40).
Brain membrane preparation
Membrane isolation from whole brain was performed as described previously (26).
Western blotting
Western blotting was performed as described previously (16).
OPC/OL cultures
Primary OPCs/OLs were isolated by immunopanning from P10 (7 days after the first 4-hydroxytamoxifen injection) Fig CAG-CreER and Fig4flox/−, CAG-CreER pups as reported previously (16). PDGFR+, O4+ and MOG+ cells were allowed to differentiate in T3 supplemented culture medium for 4 days. Cells were fixed and stained for lysosomal marker LAMP1 (Abcam, 1: 1000) and MAG (Millipore, 1: 200) as described previously (16). Cells were imaged using Zeiss Axio Examiner inverted microscope (Zeiss, Germany) equipped with Axiocam 503 and Apotome. Z-stacks and apotome optical sections were collected and maximum intensity projections were generated with ZEN and FIJI.
Immunohistochemistry
For Fluoromyelin Green staining, animals were perfused with cold PBS and brains were rapidly dissected and post-fixed in 4% PFA for 2 h at 4°C. Following the cryoprotection in 30% sucrose, 40 μm sagittal free-floating sections in PBS were prepared using a cryostat (Leica). Fluoromyelin Green staining was performed according to manufacturer’s instructions (Thermo Scientific). Brain sections were imaged using Zeiss Axio Examiner and tiled images were stitched together with ZEN.
Lysophosphatidylcholine injection
Ten days after the first tamoxifen dose, male and female P70-P90 mice were prepared for stereotaxic injection of LPC as described (29). Briefly, mice were anesthetized with 4% isoflurane, secured in stereotaxic stage (Stoelting 51730D) and kept under 2% isoflurane anesthesia during surgery. A syringe (5µl-hamilton) was loaded with 1% LPC (Sigma, L4129) in PBS (Gibco, 10010023), mounted on a motorized stereotaxic pump (Quintessential Stereotaxic injector, 53311), and used for intracranial injection into the corpus callosum using the following coordinates, AP: 1.25 mm, LR: ±1 mm, D: 2.25 mm. Over a duration of 1 min 0.5 µl of 1% LPC solution was injected on the ipsilateral site and 0.5 µl PBS on the contralateral side then the needle was kept in place for 2 min before retraction. Mice were treated with 3 doses of 70 µl of buprenorphine (0.3 mg/ml) every 12 h post-surgery, brains were harvested at 21 post injection.
Quantification of lesion size and myelin repair
Serial sections of the corpus callosum, containing the LPC and PBS injection sites were mounted onto glass coverslips and stained by ISH with digoxigenin-labeled cRNA probes specific for Mbp, Mag, Plp and Pdgfra (27). For quantification of the white matter lesion area, the same intensity cutoff was set by Image J threshold for all brain sections and used to measure the size of the lesion. The outer rim of the strongly Mbp+ region (lesionout) was traced with the ImageJ freehand drawing tool. The inner rim facing the Mbp- region (lesionin) was traced as well. For each animal examined, the size of the initial lesion area (lesionout) in µm2 and remyelinated area (lesionout-lesionin) in µm2 was calculated by averaging the measurement from two sections at the lesion core. The lesion core was defined as the section with the largest lesion area (lesionout). To determine remyelination, the ratio of (lesionout-lesionin)/(lesionout) in % was calculated (29).
Statistical analysis
Statistical analysis was performed using Graphad Prism and Microsoft Excel. For Western blotting, intensity of the control sample was set as 1. For electrophysiological studies, the analysis was performed using Clampfit (Molecular Devices) and Origin Pro. For TEM quantification, 10 images were taken at random throughout the main train of sciatic nerves and percentage of myelinated fibers was quantified.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank Jonah Chan and Bradley Zuchero for providing O4 and MOG hybridoma cell lines, respectively, The University of Michigan Microscopy and Imaging Core for assistance with electron microscopy, and Samuel Pappas for discussion of the hindlimb clasping phenotype.
Conflict of Interest statement. None declared.
Funding
This work was supported by NIH T32 NS07222, Training in Clinical and Basic Neuroscience (AK), NIH T32-GM113900, Training Program in Translational Research (LH), NIH R01 NS081281 (M.M. and R.G.) and the Dr. Miriam and Sheldon G. Adelson Foundation in Neurorepair and Rehabilitation (RG). This work was supported by the NIH T32-GM007315 Cellular and Molecular Biology Training Grant (YM).
References
- 1. Jin N., Chow C.Y., Liu L., Zolov S.N., Bronson R., Davisson M., Petersen J.L., Zhang Y., Park S., Duex J.E.. et al. (2008) VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J., 27, 3221–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lenk G.M., Ferguson C.J., Chow C.Y., Jin N., Jones J.M., Grant A.E., Zolov S.N., Winters J.J., Giger R.J., Dowling J.J., Weisman L.S., Meisler M.H. (2011) Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet., 7, e1002104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCartney A.J., Zhang Y., Weisman L.S. (2014) Phosphatidylinositol 3, 5-bisphosphate: low abundance, high significance. BioEssays, 36, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin N., Lang M.J., Weisman L.S. (2016) Phosphatidylinositol 3,5-bisphosphate: regulation of cellular events in space and time. Biochem. Soc. Trans., 44, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campeau P.M., Lenk G.M., Lu J.T., Bae Y., Burrage L., Turnpenny P., Roman Corona-Rivera J., Morandi L., Mora M., Reutter H.. et al. (2013) Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet., 92, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lines M.A., Ito Y., Kernohan K.D., Mears W., Hurteau-Miller J., Venkateswaran S., Ward L., Khatchadourian K., McClintock J., Bhola P.. et al. (2017) Yunis-Varon syndrome caused by biallelic VAC14 mutations. Eur. J. Hum. Genet., 25, 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baulac S., Lenk G.M., Dufresnois B., Ouled Amar Bencheikh B., Couarch P., Renard J., Larson P.A., Ferguson C.J., Noe E., Poirier K.. et al. (2014) Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology, 82, 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenk G.M., Szymanska K., Debska-Vielhaber G., Rydzanicz M., Walczak A., Bekiesinska-Figatowska M., Vielhaber S., Hallmann K., Stawinski P., Buehring S.. et al. (2016) Biallelic mutations of VAC14 in pediatric-onset neurological disease. Am. J. Hum. Genet., 99, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chow C.Y., Zhang Y., Dowling J.J., Jin N., Adamska M., Shiga K., Szigeti K., Shy M.E., Li J., Zhang X.. et al. (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature, 448, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang X., Chow C.Y., Sahenk Z., Shy M.E., Meisler M.H., Li J. (2008) Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain, 131, 1990–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicholson G., Lenk G.M., Reddel S.W., Grant A.E., Towne C.F., Ferguson C.J., Simpson E., Scheuerle A., Yasick M., Hoffman S.. et al. (2011) Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain, 134, 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertolin C., Querin G., Bozzoni V., Martinelli I., De Bortoli M., Rampazzo A., Gellera C., Pegoraro E., Soraru G. (2018) New FIG4 gene mutations causing aggressive ALS. Eur. J. Neurol., 25, e41–e42. [DOI] [PubMed] [Google Scholar]
- 13. Orengo J.P., Khemani P., Day J.W., Li J., Siskind C.E. (2018) Charcot Marie Tooth disease type 4J with complex central nervous system features. Ann. Clin. Transl. Neurol., 5, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferguson C.J., Lenk G.M., Jones J.M., Grant A.E., Winters J.J., Dowling J.J., Giger R.J., Meisler M.H. (2012) Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum. Mol. Genet., 21, 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lenk G.M., Frei C.M., Miller A.C., Wallen R.C., Mironova Y.A., Giger R.J., Meisler M.H. (2016) Rescue of neurodegeneration in the Fig4 null mouse by a catalytically inactive FIG4 transgene. Hum. Mol. Genet., 25, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mironova Y.A., Lenk G.M., Lin J.P., Lee S.J., Twiss J.L., Vaccari I., Bolino A., Havton L.A., Min S.H., Abrams C.S.. et al. (2016) PI(3,5)P2 biosynthesis regulates oligodendrocyte differentiation by intrinsic and extrinsic mechanisms. Elife, 5, doi: 10.7554/eLife.13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crawford A.H., Tripathi R.B., Foerster S., McKenzie I., Kougioumtzidou E., Grist M., Richardson W.D., Franklin R.J. (2016) Pre-existing mature oligodendrocytes do not contribute to remyelination following toxin-induced spinal cord demyelination. Am. J. Pathol., 186, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franklin R.J., Ffrench-Constant C. (2008) Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci., 9, 839–855. [DOI] [PubMed] [Google Scholar]
- 19. Woodhoo A., Sommer L. (2008) Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia, 56, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 20. Hall S. (2005) The response to injury in the peripheral nervous system. J. Bone Joint Surg. Br., 87, 1309–1319. [DOI] [PubMed] [Google Scholar]
- 21. Gomez-Sanchez J.A., Pilch K.S., van der Lans M., Fazal S.V., Benito C., Wagstaff L.J., Mirsky R., Jessen K.R. (2017) After nerve injury, lineage tracing shows that myelin and remak schwann cells elongate extensively and branch to form repair Schwann cells, which shorten radically on remyelination. J. Neurosci., 37, 9086–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kidd G.J., Ohno N., Trapp B.D. (2013) Biology of Schwann cells. Handb. Clin. Neurol., 115, 55–79. [DOI] [PubMed] [Google Scholar]
- 23. Vaccari I., Carbone A., Previtali S.C., Mironova Y.A., Alberizzi V., Noseda R., Rivellini C., Bianchi F., Del Carro U., D'Antonio M.. et al. (2015) Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum. Mol. Genet., 24, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayashi S., McMahon A.P. (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol., 244, 305–318. [DOI] [PubMed] [Google Scholar]
- 25. Mirantes C., Eritja N., Dosil M.A., Santacana M., Pallares J., Gatius S., Bergada L., Maiques O., Matias-Guiu X., Dolcet X. (2013) An inducible knockout mouse to model the cell-autonomous role of PTEN in initiating endometrial, prostate and thyroid neoplasias. Dis. Model. Mech., 6, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winters J.J., Ferguson C.J., Lenk G.M., Giger-Mateeva V.I., Shrager P., Meisler M.H., Giger R.J. (2011) Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. J. Neurosci., 31, 17736–17751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carbajal K.S., Mironova Y., Ulrich-Lewis J.T., Kulkarni D., Grifka-Walk H.M., Huber A.K., Shrager P., Giger R.J., Segal B.M. (2015) The cell diversity in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol., 195, 2552–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ainger K., Avossa D., Morgan F., Hill S.J., Barry C., Barbarese E., Carson J.H. (1993) Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell. Biol., 123, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin J.P., Mironova Y.A., Shrager P., Giger R.J. (2017) LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair. Elife, 6, doi: 10.7554/eLife.30498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murinson B.B., Archer D.R., Li Y., Griffin J.W. (2005) Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C-fiber afferents. J. Neurosci., 25, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hirata K., Kawabuchi M. (2002) Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc. Res. Tech., 57, 541–547. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y., Zolov S.N., Chow C.Y., Slutsky S.G., Richardson S.C., Piper R.C., Yang B., Nau J.J., Westrick R.J., Morrison S.J., Meisler M.H., Weisman L.S. (2007) Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc. Natl. Acad. Sci. U S A., 104, 17518–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ikonomov O.C., Sbrissa D., Delvecchio K., Xie Y., Jin J.P., Rappolee D., Shisheva A. (2011) The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve−/− embryos but normality of PIKfyve+/− mice. J. Biol. Chem., 286, 13404–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reifler A., Lenk G.M., Li X., Groom L., Brooks S.V., Wilson D., Bowerson M., Dirksen R.T., Meisler M.H., Dowling J.J. (2013) Murine Fig4 is dispensable for muscle development but required for muscle function. Skelet. Muscle, 3, 21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chow C.Y., Landers J.E., Bergren S.K., Sapp P.C., Grant A.E., Jones J.M., Everett L., Lenk G.M., McKenna-Yasek D.M., Weisman L.S.. et al. (2009) Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet., 84, 85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Osmanovic A., Rangnau I., Kosfeld A., Abdulla S., Janssen C., Auber B., Raab P., Preller M., Petri S., Weber R.G. (2017) FIG4 variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur. J. Hum. Genet., 25, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savas J.N., Toyama B.H., Xu T., Yates J.R. 3rd, Hetzer M.W. (2012) Extremely long-lived nuclear pore proteins in the rat brain. Science, 335, 942.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snaidero N., Mobius W., Czopka T., Hekking L.H., Mathisen C., Verkleij D., Goebbels S., Edgar J., Merkler D., Lyons D.A., Nave K.A., Simons M. (2014) Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell, 156, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weisheit C.E., Dauer W.T. (2015) A novel conditional knock-in approach defines molecular and circuit effects of the DYT1 dystonia mutation. Hum. Mol. Genet., 24, 6459–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen C., Westenbroek R.E., Xu X., Edwards C.A., Sorenson D.R., Chen Y., McEwen D.P., O'Malley H.A., Bharucha V., Meadows L.S.. et al. (2004) Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci., 24, 4030–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.