


Abstract
Somatic genome mutations occur due to combinations of various intrinsic/extrinsic mutational processes and DNA repair mechanisms. Different molecular processes frequently generate different signatures of somatic mutations in their own favored contexts. As a result, the regional somatic mutation rate is dependent on the local DNA sequence, the DNA replication/RNA transcription dynamics and epigenomic chromatin organization landscape in the genome. Here, we propose an online computational framework, termed Mutalisk, which correlates somatic mutations with various genomic, transcriptional and epigenomic features in order to understand mutational processes that contribute to the generation of the mutations. This user-friendly tool explores the presence of localized hypermutations (kataegis), dissects the spectrum of mutations into the maximum likelihood combination of known mutational signatures and associates the mutation density with numerous regulatory elements in the genome. As a result, global patterns of somatic mutations in any query sample can be efficiently screened, thus enabling a deeper understanding of various mutagenic factors. This tool will facilitate more effective downstream analyses of cancer genome sequences to elucidate the diversity of mutational processes underlying the development and clonal evolution of cancer cells. Mutalisk is freely available at http://mutalisk.org.
INTRODUCTION
Somatic genome mutations cause cancers (1). Early studies in the 1980s isolated a number of cancer-causing DNA sequence alterations. For example the single G:C>T:A nucleotide substitution causes a glycine to valine change in codon 12 of the HRAS gene in bladder cancer cell-lines (2,3). This fundamental discovery provoked great enthusiasm for the study of genes or mutations underlying cancer development. In the 2000s, completion of the Human Genome Project (4) enabled more efficient means of studying mutations in cancer; targeted gene sequencing studies have revealed many additional cancer genes, including BRAF, PIK3CA and IDH1 (5–7). At last, the revolution of high-throughput genome sequencing technologies over the past decade has tremendously accelerated genome-wide analysis of somatic mutations in population-scale cancer cohorts. As a result, we now have a comprehensive and unbiased mutation catalog harboring over 43 million base substitutions through whole-genomes obtained from thousands of cancer samples (https://www.biorxiv.org/content/early/2017/08/24/179705). Today, whole-genome sequencing analyses of cancer genomes are widely applicable to the study of cancer biology. These studies also provide important clinical implications (8).
Human tissues accumulate somatic mutations throughout the lifetime, even from the very first cell divisions of human life (9). The extensively studied heterogeneity of the mutational landscape between different tumor types dictates the wide variability of the mutational history of somatic cells in different tissues (10,11). Many intrinsic processes (e.g. spontaneous 5-methylcytosine deamination, DNA polymerase error, impairment in DNA repair pathways or misregulation of APOBEC enzymes) and mutagenesis from extrinsic causes such as physical and chemical carcinogens (e.g. ultraviolet (UV) light, tobacco smoking, aristolochic acid or temozolomide) are well known mechanisms of somatic mutations (10,12,13). These different mutational processes often generate distinct mutational patterns in terms of their base alteration spectra and their associated nucleotide contexts, known as the mutational signature. For example, APOBEC-mediated mutagenesis preferentially generates C:G>T:A and C:G>G:C base substitutions preferentially at the TpCpA and TpCpT sequence contexts in early-replicating regions (14). Occasionally, these mutations show a pattern of physically localized hypermutation termed kataegis (15). In addition, UV-mediated mutations are preferentially single C:G>T:A and double CC:GG>TT:AA base substitutions prevalent in dipyrimidine sequence contexts with a strong transcriptional strand bias (16). Furthermore, regional mutational rates are strongly associated with epigenomic features such as chromatin organization and DNA replication strand/timing (17). However, many biological mechanisms underlying the differential distribution of mutations across the genome remain to be discovered.
To this end, analyzing the correlations between somatic mutations and various genomic, transcriptional and epigenomic variables as well as determining the accurate decomposition of mutational signatures are essential steps. However, these processes require collection of reference datasets obtained from many heterogeneous studies. While mutational signature decomposition has been developed in various software packages (18,19) and as a web-based analysis tool (20), user-friendly toolkit that associates somatic mutations with various regulatory elements in the genome as well as with mutational signatures remains unavailable, to the best of our knowledge. Here, we developed an online tool (Mutalisk: MUTation AnaLysIS toolKit), which outputs genome/epigenome associations and mutational signatures from lists of somatic mutations. All of the results are provided with elegant vector graphics and statistical significance levels. The user can download the results in pdf and text formats, which can be inserted directly into the user’s own manuscript or can be used for additional downstream analyses.
MATERIALS AND METHODS
Mutalisk correlates somatic mutations in query sample with the physical location of the genome, regional DNA sequence contexts, and the functional elements in the genome, such as DNA replication, RNA transcription and epigenome landmarks. To do this, Mutalisk consists of four major functional modules: (i) localized hypermutation analysis, (ii) mutational signature decomposition, (iii) transcriptional strand bias analysis and (iv) epigenome association analysis. Mutalisk is developed using php and R scripts. In this section, we describe the methods and datasets used for each analysis. Figure 1 shows a comprehensive overview of the Mutalisk analysis pipeline.
Figure 1.

Overview of the Mutalisk analysis pipeline. The Mutalisk pipeline consists of (A) the input module, (B) the processing module and (C) the output module. Mutalisk takes vcf files as input. The mutation data are analyzed for (1) localized hypermutation (kataegis), (2) decomposition of mutational signatures, (3) transcriptional strand bias, and (4)-(6) genomic and epigenomic modifications. Mutalisk outputs the results of these analyses in a single view with graphs and statistical significance levels.
Inputs and options
Mutalisk takes variant call format (vcf) files as inputs and requires the user to specify the build of the human reference genome sequences (either in GRCh37 or GRCh38) (Figure 1A and Supplementary Figure S1). Multiple input files can be uploaded and analyzed in parallel independently. For the decomposition of mutational signatures, users can select either the linear regression or the multinomial method. The signature decomposition can be thoroughly conducted using (i) 30 currently known standard signatures, (ii) 65 provisional signatures or (iii) user-defined signatures (see ‘Mutational signature decomposition’ section for more details). Alternatively, Mutalisk provides a list of 40 cancer types with the mutational signatures known to be present in the corresponding cancer type so that the decoupling process can be more efficiently accomplished with proper biological context. To analyze the epigenomic association, Mutalisk uses diverse heterogeneous chromatin landscape from the ENCODE project (21) and users can choose one reference cell type where the landscape was explored. Current version of Mutalisk offers a total of 31 cell lines, 18 of which are cancer cell lines that cover 11 unique tissue types: e.g. A549 (lung adenocarcinoma), Dnd41 (T-cell leukemia), GM12878 (immortalized B lymphocyte), HeLa-S3 (cervix adenocarcinoma), HepG2 (hepatocellular carcinoma), HUVEC (human umbilical vein endothelial cells), K562 (chronic myeloid leukemia), and NHEK (normal human epidermal keratinocytes) (Supplementary Table S7).
Localized hypermutation analysis
Localized hypermutation (kataegis) can be visually inspected by rainfall plots as shown previously (15). For each vcf file, Mutalisk analyzes the genomic distances between each mutation (intermutation distance) and generates a rainfall plot using karyoploteR (22) (Figure 1B.1).
Mutational signature decomposition
Decomposition
With somatic mutations in the uploaded vcf file, Mutalisk constructs its mutational spectrum by categorization of the mutations into 96 substitution classes while considering sequence context as previously suggested (six types of substitutions (C>A, C>G, C>T, T>A, T>C, T>G; referred to as the pyrimidine of the mutated Watson–Crick base pair) * four possible immediate upstream bases * four possible immediate downstream bases) (18). To decompose the mutational spectrum, 30 known standard mutational signatures are primarily used (available at http://cancer.sanger.ac.uk/cancergenome/assets/signatures_probabilities.txt). We also provide the option to use the 65 provisional mutational signatures established from the Pancancer Pancancer Analysis of Whole Genomes (PCAWG) project (https://www.biorxiv.org/content/early/2017/07/12/162784). Alternatively, users are able to upload their own signatures that can be used in the signature decomposition (Supplementary Table S8). Mutalisk employs a greedy algorithm to identify relevant mutational signatures underlying the observed mutational profile. Mutalisk identifies a maximum of seven mutational signatures as we conventionally expect seven signatures at most from a specific somatic tissue (9,18,23–25). For each set of signatures, a decomposition model is generated by the maximum likelihood estimation method using the optim function in R software by minimizing a constrained function, either by a linear function as previously suggested (9,23) or by −2× natural logarithm of likelihood ratio (multinomial test) depending on the user’s specification. Practically, these two independent methods show very similar performance and are complementary to each other. To discourage overfitting, Mutalisk uses Bayesian information criterion (BIC) and an optimal number of mutational signatures is selected. At last, the observed mutational spectrum across the 96 mutation classes and the expected spectrum from the final combination of known signatures are compared by cosine similarity score (Figure 1B.2). The confidence interval of the relative contribution of each mutational signature is then calculated by bootstrapping methods. For more details on the decomposition analysis method, refer to Supplementary Method 1.
Compilation of data on regulatory elements
In Mutalisk, regional mutation rates are correlated with various functional elements in the genome as reported previously (17). Reference dataset for the functional elements were collected from (i) RefSeq gene annotation (transcriptional strand bias), (ii) human genome sequence itself (GC-content), and (iii) the ENCODE Project (21) for DNA replication timing (Repli-seq), DNase I hypersensitivity regions and a series of histone modifications. These data were downloaded from the University of California Santa Cruz (UCSC) Genome Browser golden path (ftp://hgdownload.soe.ucsc.edu/goldenPath) (26). Of note, the ENCODE datasets are cell-line specific and we collected data from all available cancer cell lines. Each feature was pre-processed to be mapped against the user uploaded list of mutations. Specific details are described below.
Transcriptional strand bias analysis
Using the RefSeq Gene dataset, we annotated transcribed (non-coding) and untranscribed (coding) strand of the expressed regions in the human genome (Figure 1B.3).
GC-content
The ratio of the nucleotides guanine (G) and cytosine (C) was calculated in each 1-kilobase (Kb) window. Subsequently, each bin was labeled as a low (≤25th percentile), intermediate (from 25th to 75th percentile), or high (≥75th percentile) level (Figure 1B.4).
DNA replication timing (Repli-seq)
For each cell line, the percentage-normalized signal data corresponding to the six cell cycle phases of G1/G1b, S1, S2, S3, S4 and G2 at 1-kb intervals were categorized into one of four enrichment levels: early, intermediate, late, and unknown. The following formulae were used to label the i-th 1kb interval:
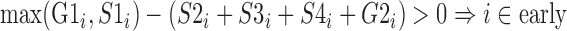 |
 |
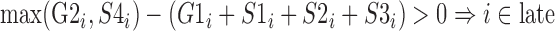 |
Genomic positions unlabeled from the formulae above were defined as unknown. Genomic regions with any missing signal from Repli-seq were also classified as unknown (Figure 1B.5).
Histone modification and DNase I hypersensitivity
For each feature, the sum of read depth was obtained for each 160-base-pair (bp) window across the genome. Windows overlapping with any simple repeats or microsatellites were excluded. The 160-bp windows were then summed at 1-megabase (Mb) intervals (17). The summed read density for each 1-Mb interval was normalized based on the maximum read density for the given feature. The signal at each 1-Mb interval was then categorized into a low (≤25th percentile), intermediate (from the 25th to 75th percentiles), or high (≥75th percentile) level based on the normalized intensity of the peaks (Figure 1B.6).
Transcriptional strand bias analysis
Using the RefSeq Gene dataset, Mutalisk annotates the transcriptional strand information (i.e. transcribed or untranscribed) of the somatically mutated pyrimidine base (the reference allele in C or T). We calculate the enrichment of each mutation class (i.e. in the six classes of C>A, C>G, C>T, T>A, T>C and T>G base substitutions and in more detail with the 96 subclasses). The confidence intervals of the enrichment are then calculated using exact Poisson tests.
Genomic and epigenomic modification analysis
Each somatic mutation is mapped to genomic regions of varying intensity for each feature. These are the low, intermediate or high level for the histone modification features, DNase I hypersensitivity and GC-content. DNA replication timing, on the other hand, is classified as early, intermediate or late phase.
For the analysis of transcriptional strand bias, DNA replication timing, GC-content and histone modification, goodness of fit tests are performed to assess whether the distribution of the observed mutations is significantly different from the expected proportions. Chi-square tests determine the statistical significance of the observed distribution of mutations against the expected proportions (Supplementary Tables S3 and 4).
Associations between somatic mutation and regulatory elements
Correlation coefficient
Mutalisk calculates the Pearson correlation coefficient r between the rate of somatic mutations and the intensities of the tested functional elements of the genome using the cor function in R.
Percentage of explained variance
To quantify the extent to which the genomic and epigenomic properties can explain the somatic mutation-rate variation, Mutalisk calculates the percentage of explained variance for each vcf file using a previously reported method (17). This is achieved by forward feature selection where we iteratively select the genomic and epigenomic features with the lowest Akaike information criterion after fitting each remaining feature and the mutation data by a generalized least-squares estimation method. The percentage of explained variance is obtained from the linear regression model of these features.
Outputs
For each analysis, Mutalisk results are provided with elegant vector graphics and output text files for the analyses of localized hypermutations, decomposition of mutational signatures, and associations between somatic mutation density levels and a comprehensive set of genomic and epigenomic features (Figure 1C). More specifically, the Mutalisk outputs are (i) a rainfall plot of the mutations from the localized hypermutation analysis, (ii) a summary view of each set of the decomposed signatures from the mutational signature decomposition, (iii) statistical significance of the association between each regulatory element and the varying rate of mutations across the genome (see Supplementary Figures S2–11 and Table S6). All of the graphics and annotated vcf files are freely downloadable from Mutalisk.
RESULTS
Evaluation of Mutalisk
To evaluate the accuracy of the analyses performed by Mutalisk, we compared the Mutalisk outputs against the results published in previous studies (16,17,23).
Localized hypermutation
In the analysis of somatic mutations using an example dataset (somatic mutations from a lung adenocarcinoma) (8), three kataegis regions were clearly seen in 5p, 6p and Xq (Figure 2A, left panel, shown with three arrows) as reported previously. Furthermore, in the rainfall plot of the COLO-829 melanoma sample, we observed a universally higher rate of mutations across all chromosomes as well as domination of the CC>TT dinucleotide mutation type (mutations with intermutation distance = 1), reflective of DNA damage due to ultraviolet light (Figure 2A, right panel; shown with an arrow) (16).
Figure 2.

Mutalisk analysis results: (A) Rainfall plot of lung cancer mutations exhibiting kataegis (left) and melanoma mutations (right). (B) Detailed results of the mutational signature decomposition of the COLO-829 melanoma sample. (C) Cosine similarities between Mutalisk and Nik-Zainal et al. (24) mutational signatures for each of the 560 breast cancer samples. (D) Transcriptional strand bias analysis results of the COLO-829 melanoma mutations. (E) Detailed results of the epigenomic modification analysis of H3K9me3 and the COLO-829 mutations (**P-value < 0.05 and ***P-value < 0.01) and (F) Pearson correlation coefficients and percentages of explained variance for the regulatory elements and the COLO-829 mutations.
Mutational signature
Figure 2B shows the mutational signature decomposition results generated by Mutalisk using a COLO-829 melanoma sample (16). Based on the weight assigned to each signature included in the decomposition model, users can obtain the number of mutations attributed to each signature as well as the cosine similarity score between the observed distribution of mutations and the decomposed distribution of mutations. To evaluate the accuracy of the mutational decomposition results in Mutalisk, we used whole-genome sequences from 560 breast cancers and their reported signatures as references (23). The signatures from Mutalisk were mostly very similar to those reported, with an average cosine similarity score of 0.927 (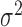 = 0.104) (Figure 2C). Additional details are summarized in Supplementary Tables S1 and 2. Furthermore, using the 560 breast cancer samples (23) we benchmarked the performance of Mutalisk's mutational signature decomposition against two other tools: deconstructSigs (19) and MutaGene (20). Out of the three tools, Mutalisk decomposition results were the closest to the results obtained by Nik-Zainal et al. based on median cosine similarity scores (Supplementary Figure S12).
= 0.104) (Figure 2C). Additional details are summarized in Supplementary Tables S1 and 2. Furthermore, using the 560 breast cancer samples (23) we benchmarked the performance of Mutalisk's mutational signature decomposition against two other tools: deconstructSigs (19) and MutaGene (20). Out of the three tools, Mutalisk decomposition results were the closest to the results obtained by Nik-Zainal et al. based on median cosine similarity scores (Supplementary Figure S12).
Transcriptional strand bias
Figure 2D shows the transcriptional strand bias of somatic mutations in the COLO-829 melanoma cell line (16). C>T mutations were highly enriched in the untranscribed regions, indicative of transcription-coupled nucleotide excision repair (Figure 2B and D).
Genomic and epigenomic modification
Figure 2E shows the positive correlation between regional rate of the COLO-829 somatic mutations and the histone modification (H3K9me3) as calculated in Mutalisk. Consistent with a previous paper (17), H3K9me3 was one of the most positively correlated chromatin features among various histone marks. The Pearson correlation coefficient between each of the 17 histone modification features and the melanoma somatic mutations as well as the percentages of explained variance of these features ordered by forward feature selection are shown in Figure 2F. The correlations coefficients were commensurate with those reported in the previous report (17).
DISCUSSION
Mutalisk is a convenient and publicly available mutation analytics tool that facilitates investigations of the mutational profiles of cancer genomes while simultaneously assessing the influence of diverse genetic and epigenetic properties on the degree of somatic mutation-rate variation. This online tool is very easy to use. Only the upload of a mutation list and a few simple clicks are necessary for all of the downstream analyses. Computation of the analysis depends on both the input data size and input conditions, with the linear regression decomposition method generating results the fastest. From the analysis, users can quickly explore the spatial distribution of somatic mutations, the mutational spectrum along with systematic decomposition by known signatures and the associations of regional mutation density with many functional elements in the genome, all of which provide deep insights into the processes that are attributable to the mutations. We believe that Mutalisk can be used for quality control of mutation calls, because false positive calls frequently show distinctive spectra from true calls, therefore are easily identified from Mutalisk. As validated using reference datasets mentioned above, the results from Mutalisk are consistent with published findings and thus are held here to be highly accurate (16,17,23). We have also shown that compared to two other mutational signature decomposition tools, Mutalisk decomposition results are the most similar to the results presented by Nik-Zainal et al. (23), which are based on the original mutational signature decomposition method. All Mutalisk results, including the elegant vector graphics (Supplementary Figures S2–11) and annotated text files (Supplementary Table S5), are downloadable for further private downstream analysis and for users’ manuscripts. Of note, reference epigenome datasets are provided from a multitude of cancer and normal cell lines. Therefore, by matching the cancer types of the user-provided vcf files and the reference dataset, more accurate association analyses are possible. Multiple vcf files can be simultaneously uploaded and analyzed in parallel at once (Supplementary Figures S1 and 2). At last, Mutalisk allows biologists and clinicians to easily analyze somatic mutations without intricate, technical and/or preparatory procedures. Therefore, we believe that Mutalisk will facilitate the elucidation of the diversity of mutational processes underlying the development and clonal evolution of cancer cells.
AVAILABILITY
Mutalisk is freely available at http://mutalisk.org without login requirements.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Center, Korea [NCC-1610450, NCC-1710370 and NCC-1711280 to D.H.]; Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI); Ministry of Health & Welfare, Republic of Korea [HI16C2387 to Y.S.J. and D.H.]; Korean Federation of Science and Technology Societies (KOFST) [162S-1-3-1673 to H.C., in part]. Funding for open access charge: National Cancer Center, Korea [NCC-1610450, NCC-1710370 and NCC-1711280 to D.H.]; Korea Health Technology R&D Project through the KHIDI; Ministry of Health & Welfare, Republic of Korea [HI16C2387 to Y.S.J. and D.H.]; Korean Federation of Science and Technology Societies (KOFST) [162S-1-3-1673 to H.C., in part].
Conflict of interest statement. None declared.
REFERENCES
- 1. Stratton M.R., Campbell P.J., Futreal P.A.. The cancer genome. Nature. 2009; 458:719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reddy E.P., Reynolds R.K., Santos E., Barbacid M.. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature. 1982; 300:149–152. [DOI] [PubMed] [Google Scholar]
- 3. Tabin C.J., Bradley S.M., Bargmann C.I., Weinberg R.A., Papageorge A.G., Scolnick E.M., Dhar R., Lowy D.R., Chang E.H.. Mechanism of activation of a human oncogene. Nature. 1982; 300:143–149. [DOI] [PubMed] [Google Scholar]
- 4. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. . Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 5. Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W. et al. . Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–954. [DOI] [PubMed] [Google Scholar]
- 6. Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L. et al. . An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., Yan H., Gazdar A., Powell S.M., Riggins G.J. et al. . High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304:554. [DOI] [PubMed] [Google Scholar]
- 8. Lee J.K., Lee J., Kim S., Kim S., Youk J., Park S., An Y., Keam B., Kim D.W., Heo D.S. et al. . Clonal history and genetic predictors of transformation into Small-Cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. 2017; 35:3065–3074. [DOI] [PubMed] [Google Scholar]
- 9. Ju Y.S., Martincorena I., Gerstung M., Petljak M., Alexandrov L.B., Rahbari R., Wedge D.C., Davies H.R., Ramakrishna M., Fullam A. et al. . Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017; 543:714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al. . Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. et al. . Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T. et al. . Mutational signatures associated with tobacco smoking in human cancer. Science. 2016; 354:618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G. et al. . An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013; 45:970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kazanov M.D., Roberts S.A., Polak P., Stamatoyannopoulos J., Klimczak L.J., Gordenin D.A., Sunyaev S.R.. APOBEC-Induced cancer mutations are uniquely enriched in Early-Replicating, Gene-Dense, and active chromatin regions. Cell Rep. 2015; 13:1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al. . Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pleasance E.D., Cheetham R.K., Stephens P.J., McBride D.J., Humphray S.J., Greenman C.D., Varela I., Lin M.L., Ordonez G.R., Bignell G.R. et al. . A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010; 463:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schuster-Bockler B., Lehner B.. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012; 488:504–507. [DOI] [PubMed] [Google Scholar]
- 18. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R.. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013; 3:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C.. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goncearenco A., Rager S.L., Li M., Sang Q.X., Rogozin I.B., Panchenko A.R.. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017; 45:W514–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gel B., Serra E.. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017; 33:3088–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. et al. . Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rahbari R., Wuster A., Lindsay S.J., Hardwick R.J., Alexandrov L.B., Turki S.A., Dominiczak A., Morris A., Porteous D., Smith B. et al. . Timing, rates and spectra of human germline mutation. Nat. Genet. 2016; 48:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polak P., Kim J., Braunstein L.Z., Karlic R., Haradhavala N.J., Tiao G., Rosebrock D., Livitz D., Kubler K., Mouw K.W. et al. . A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017; 49:1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.