

Abstract
Summary
bioBakery is a meta’omic analysis environment and collection of individual software tools with the capacity to process raw shotgun sequencing data into actionable microbial community feature profiles, summary reports, and publication-ready figures. It includes a collection of pre-configured analysis modules also joined into workflows for reproducibility.
Availability and implementation
bioBakery (http://huttenhower.sph.harvard.edu/biobakery) is publicly available for local installation as individual modules and as a virtual machine image. Each individual module has been developed to perform a particular task (e.g. quantitative taxonomic profiling or statistical analysis), and they are provided with source code, tutorials, demonstration data, and validation results; the bioBakery virtual image includes the entire suite of modules and their dependencies pre-installed. Images are available for both Amazon EC2 and Google Compute Engine. All software is open source under the MIT license. bioBakery is actively maintained with a support group at biobakery-users@googlegroups.com and new tools being added upon their release.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The bioBakery suite is a collection of computational tools for quantitative microbial community analysis based on meta’omic (shotgun metagenome or metatranscriptome) sequencing data. It includes individual tools, workflows for executing them reproducibly, and a pre-built virtual environment that abrogates the burden of identifying and installing those tools and their dependencies. A full list of tools in the suite is included in the Supplementary Material.
bioBakery implements complete ‘fire-and-forget’ analysis workflows for sample quality control, profiling and visualization, reducing the time users spend actively directing computations while ensuring workflow accuracy and completeness. The workflows perform dependency-driven, scalable analysis and produce a set of validated data products and standardized summary reports. Each can be executed with a single command and enables seamless distribution of tasks locally or across a grid computing environment.
2 The bioBakery suite
The tool suite is composed of software in three categories: (i) composition analysis, (ii) statistical analysis, and (iii) infrastructure and utilities (Supplementary Fig. S1). Composition analysis tools take shotgun-sequencing data as input to quantify the presence and abundance of microbial features, e.g. species, strains, gene families, and metabolic pathways. The products from these tools are data tables, which can then be processed by statistical analysis or visualization tools to identify significant associations (among microbial features or with sample metadata). Infrastructure and utilities tools support common meta’omic analyses in part through dependency-driven, reproducible workflows (http://huttenhower.sph.harvard.edu/biobakery_workflows) built with AnADAMA2 (http://huttenhower.sph.harvard.edu/anadama2).
3 bioBakery workflows
bioBakery workflows provide simple and reproducible execution of the many complex steps in processing meta’omic sequencing data. bioBakery includes a collection of data processing and visualization workflows that, starting from raw shotgun sequencing reads (metagenomic or metatranscriptomic), efficiently processes data through read-level quality control, taxonomic, and functional profiling steps. Workflows also exist to process raw 16S RNA gene sequencing data. The outputs of the data processing workflow are then forwarded to a visualization workflow for generating data reports and publication-ready figures (Supplementary Fig. S2). For detailed workflow diagrams, default workflow settings, and instructions on how to customize workflow parameters, refer to the bioBakery workflows user manual.
3.1 Metagenomic application example
The following two commands execute integrated workflows that process raw shotgun metagenomic sequencing reads (Fig. 1):
Fig. 1.
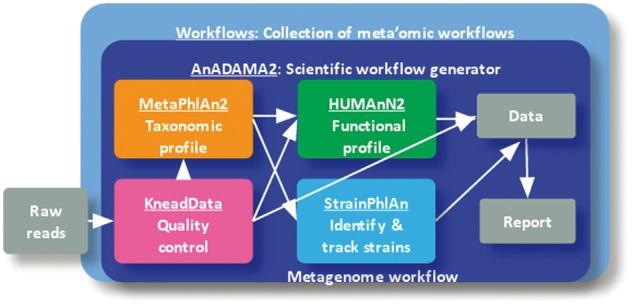
The default metagenome workflow incorporates several individual tools that together process raw sequences into a set of data products, reports and visualizations
$ biobakery_workflows wmgx –input <fastq_folder> –output <data_folder>
$ biobakery_workflows wmgx_vis –input <data_folder> –output <vis_folder> –project-name <project>
In the first command, the input is a directory containing shotgun sequencing data (e.g. gzip-compressed fastq files) and the output is a directory where the data products are written (e.g. feature abundance tables). In the second command, the input is the directory of data products created by the first command and the output is a directory where the visualizations are written. See the bioBakery workflows tutorial for demo data sets including raw input files, data products and visualizations.
4 bioBakery homebrew packages
The bioBakery tool suite can be installed locally on MacOS with the Homebrew package manager (http://brew.sh/), and on Linux with the Linuxbrew package manager (http://linuxbrew.sh/). Linuxbrew does not require root permissions, thus making it ideal for a single user installing tools in a grid computing environment that does not have a container platform available. The bioBakery Homebrew formulas are available at https://github.com/biobakery/homebrew-biobakery. For detailed instructions on how to install the bioBakery Homebrew formulas, see the section on “installing bioBakery” in the Supplementary Material.
5 bioBakery virtual machine
The bioBakery virtual machine (VM) is a Vagrant (https://www.vagrantup.com/) box with VirtualBox (https://www.virtualbox.org/) as the provider, currently running Ubuntu 16.04. All releases are hosted by Vagrant at https://app.vagrantup.com/biobakery/boxes/biobakery. See the Supplementary Material for detailed instructions on installing and running the bioBakery VM. The bioBakery VM is ideally suited for analyzing small data sets or for learning how to run tools in the suite with their corresponding tutorials. The recommended resources for running the VM are 12 GB of RAM [of which 8 GB (tunable default) is allocated to the VM] and 16 GB of available disk space. Users lacking these resources can forego the VM installation and fetch individual bioBakery tools using the Homebrew formulas or Docker images (detailed in Supplementary Material).
6 Scaling up bioBakery
For large data sets, a grid or cloud computing environment is recommended with the tool suite installed using Homebrew or Docker images. If using Google Compute Engine (GCE), a public Google Cloud image is available for bioBakery through the bioBakery Google Cloud Bucket. There are two steps to start running with the bioBakery Google Cloud image: (i) create your own image from the public image and (ii) create your own VM instance from your new image. If using Amazon EC2, use the public bioBakery Amazon Machine Image (AMI) when creating your instance. The minimal recommended configuration (sufficient for running all bioBakery demos) is the machine type ‘n1-standard-2’ for GCE and ‘t2.large’ for Amazon EC2 (each providing ∼8 GB of RAM and 2 CPU cores).
7 Conclusion
bioBakery provides a complete meta’omic analysis environment with simple-to-use, reproducible workflows that efficiently process raw data into analysis reports containing publication-ready figures.
Supplementary Material
Acknowledgements
We would like to thank many lab members over several years for contributions to bioBakery components, including Bahar Sayoldin, Afrah Shafquat, Cesar Arze, Timothy Tickle, Gholamali Rahnavard, George Weingart, Himel Mallick, Jason Lloyd-Price, Melanie Schirmer, Tommi Vatanen, Emma Schwager, Boyu Ren, Jim Kaminski and Liu Cao.
Funding
This work has been supported by the following: National Institutes of Health NIDDK [U54DE023798] (C.H.), JDRF 3-SRA-2016-141-Q-R (C.H.), Sloan Foundation [4406J0B] (Jessica Green), National Institutes of Health NHGRI [R01HG005220] (Rafael Irizzary), NSF DBI-1053486 (C.H.), National Institutes of Health NHGRI [R01HG005969] (C.H.), ARO W911NF-11-1-0473 (C.H.).
Conflict of Interest: none declared.
References
- Abubucker S. et al. (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol., 8, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniela B. et al. (2015) A reproducible approach to high-throughput biological data acquisition and integration. PeerJ., 3, e791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duy T.T. et al. (2017) Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res., 27, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski J. et al. (2015) High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput. Biol., 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M.G.I. et al. (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol., 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan X.C. et al. (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol., 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. (2011) Metagenomic biomarker discovery and explanation. Genome Biol., 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods, 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. (2013) PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun., 4, 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M. et al. (2016) Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods, 13, 435–438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.