


Abstract
T cell receptors (TCRs), along with antibodies, are responsible for specific antigen recognition in the adaptive immune response, and millions of unique TCRs are estimated to be present in each individual. Understanding the structural basis of TCR targeting has implications in vaccine design, autoimmunity, as well as T cell therapies for cancer. Given advances in deep sequencing leading to immune repertoire-level TCR sequence data, fast and accurate modeling methods are needed to elucidate shared and unique 3D structural features of these molecules which lead to their antigen targeting and cross-reactivity. We developed a new algorithm in the program Rosetta to model TCRs from sequence, and implemented this functionality in a web server, TCRmodel. This web server provides an easy to use interface, and models are generated quickly that users can investigate in the browser and download. Benchmarking of this method using a set of nonredundant recently released TCR crystal structures shows that models are accurate and compare favorably to models from another available modeling method. This server enables the community to obtain insights into TCRs of interest, and can be combined with methods to model and design TCR recognition of antigens. The TCRmodel server is available at: http://tcrmodel.ibbr.umd.edu/.
INTRODUCTION
The adaptive immune system is responsible for specific molecular recognition of a vast array of foreign antigens. In humans, mice, and other species, cellular adaptive immune recognition is performed by millions of unique T cell receptors (TCRs), which are generated somatically in each individual. Much of the diversity of TCRs is within the complementarity determining region (CDR) loops, which engage antigenic peptides presented by major histocompatibility complex (MHC) proteins, as well as a range of other non-peptide antigens presented by MHC-like proteins.
TCRs are critically important in the effective immune clearance of viruses and pathogens, while TCR autoreactivity is associated with a range of autoimmune diseases including diabetes (1) and multiple sclerosis (2), and studies have demonstrated that TCRs have major potential as cell-based (3,4) and soluble therapeutics (5) for cancer. Many studies have generated sets of TCR sequences, representing tumor-infiltrating T cells targeting neoantigens (6), and large-scale studies of TCR sequences targeting viruses and bacterial pathogens (7–9). While sequence-based insights have been made based on several of these datasets (8,9), high resolution 3D structural information on TCRs can provide critical insights into the basis of their antigen targeting and specificity (7,10), and experimental structure determination of more than a small set of these TCRs is not feasible due to time and resources involved. Accurate structural modeling of TCR structures would provide the opportunity to view, analyze, and compare the structures of TCRs of interest.
To provide the means to easily and accurately model TCR structures from sequence, we have developed a TCR modeling algorithm in the powerful modeling framework, Rosetta (11), and have implemented its functionality in a new web server, TCRmodel. With a goal of producing TCR models quickly with a simple interface, this enables the use of a powerful modeling method by the broader research community, such as immunologists, biologists, or clinicians and researchers focused on cancer and autoimmune diseases.
MATERIALS AND METHODS
Template library assembly
To provide templates for modeling of TCRs, a library of nonredundant TCR structures was constructed as follows. The amino acid sequences of the α and β variable domains (Vα and Vβ) of the A6 TCR were used to search for other α and β chains in the Protein Data Bank (PDB) (12) using BLAST (13). Approximately 90 nonredundant Vα and Vβ structures were collected manually among top BLAST hits, and a structure-based multiple sequence alignment was obtained using the algorithm MAMMOTH-MULT (14). Hidden Markov models were constructed from each multiple sequence alignment, which was then used to search all PDB sequences using HMMER (15), to identify all Vα and Vβ structures in the PDB. To provide consistent residue numbering and identification of CDR loop termini, the program ANARCI (16) was used to number TCR chains. Structures with resolution worse than 3.2 Å were removed, commensurate with the resolution cutoff used for structure selection for previously reported protein-protein docking and TCR-peptide-MHC docking benchmarks (3.25 Å) (17,18). Additionally, structures with missing CDR loop residues were removed, and redundant structures, identified by matching CDR loop sequences, were also removed (highest resolution representatives were retained), resulting in ∼150 structures out of over 500 from the original set from the PDB; most reductions were due to redundancies due to multiple chains in the asymmetric unit, or separately solved structures containing the same TCR.
Modeling protocol
The algorithm to model TCRs from sequence was written as a new protocol in the program Rosetta (11), and reflects the general scheme used by template-based antibody structure modeling methods, such as RosettaAntibody (19), with several distinctions. A brief description of the TCRmodel protocol follows:
Input α and β chain amino acid sequences are parsed using regular expressions to identify CDR1, CDR2 and CDR3 loops and framework regions within the variable regions. In this context, the CDR2 loop definition was extended to include the HV4 loop, as canonical CDR2 loop C-termini are highly structurally variable. Top matching CDR and framework templates are identified from the template library using the BLOSUM62 scoring function (20).
Grafting of CDR loop stem residues onto framework regions is performed, entailing superposition of residue backbone atoms for CDR N- and C-termini onto corresponding framework residues (three residue overlap); in the case of one structural template matching both CDR1 and CDR2 loops (TRAV or TRBV germline gene template match), only the CDR3 loop is grafted.
Orientation of modeled Vα and Vβ structures is performed based on a Vα/Vβ template (identified using framework sequence similarity).
The modeled variable domain structure is then minimized using constrained all-atom refinement in Rosetta, to reduce clashes and unfavorable energies from backbone and side chain conformations in the grafted model.
Further minimization of the CDR3 loops (available as an option on the TCRmodel server) is performed using the kinematic closure loop modeling refinement protocol (21), using the recently described REF15 function (22) to perform loop sampling and select a top model among a set of 100 candidate refined loop models.
In addition to its web server implementation, this algorithm is available as source code as part of the Rosetta software suite (www.rosettacommons.org), in a new protocol and application (‘tcr’).
Web server implementation
The implementation of the Rosetta TCR modeling protocol as a web server was performed in python using the web framework, Flask (flask.pocoo.org). Protein structure visualization is performed using a custom designed interface in JavaScript using the PV protein viewer (biasmv.github.io/pv/). Default Rosetta execution (without loop refinement) is performed locally on the web server, while loop refinement is performed on a Linux computing cluster. Germline gene amino acid sequences for TRAV and TRBV were obtained from the IMGT database (23).
RESULTS
Input and library coverage
The TCRmodel server provides three options to generate TCR structural models from sequence, two of which are shown Figure 1. The first option is that users may enter amino acid sequences containing TCR α and β variable domains (Figure 1A). Upon sequence entry, the server will automatically display if there is a match to any TRAV or TRBV germline gene. The second input option allows the user to specify TCR TRAV/TRAJ and TRBV/TRBJ germline genes and CDR3 amino acid sequences, from which the server will generate full Vα and Vβ amino acid sequences (Figure 1B). This option allows the direct entry of TCR sequence data in this format found in the literature (e.g. (7)) as well as public databases including VDJdb (24). To enable modeling of multiple TCRs, a third input option is available to users, where files containing sequences of one or more TCRs in FASTA format can be submitted for batch processing.
Figure 1.

TCRmodel server input. Two main options are available to users for TCR input: (A) input of TCR amino acid sequences, and (B) input of germline gene and CDR3 amino acid sequences, which the server uses to generate TCR amino acid sequences. For TCR sequence input, germline gene is automatically detected in the input sequence and displayed (blue font in (A)).
Given that many TCRs share germline gene and amino acid sequences, which include the CDR1, CDR2, and HV4 loops, we assessed the coverage of human germline genes (TRAV and TRBV) among structural templates (Supplementary Tables S1 and S2). Over half of TRAV and TRBV genes were represented among TCR structures: 27 out of 45 TRAV genes, and 26 out of 48 TRBV genes. Furthermore, as representation of germline genes in mature immune repertoires is not uniform, we calculated the percent of unique human α and β sequences from VDJdb (24) with structural matches to germline genes. Out of ∼5000 unique α sequences and 13 000 unique β sequences, structural coverage was approximately 81% and 72% respectively; this increase in percent coverage versus number of genes is likely because characterized TCRs with solved X-ray structures represent more frequently utilized germline genes, versus a random uniform sampling. This supports the direct identification of TCR germline gene structure matches, which is assessed in the template identification stage of the protocol, avoiding the need for CDR1 and CDR2 loop grafting in many cases.
Upon user submission, the TCRmodel server will immediately show the alignment of input sequences to identified TCR templates, followed by display of full results upon model completion. The modeling results page features an interactive model structure viewer, a link to download model coordinates, as well as template alignments and sequence information.
Benchmarking and predictive performance
To benchmark the performance of the TCRmodel server, we assembled a set of test cases from recently released TCR structures from the PDB that are nonredundant (containing matching CDR sequences or CDR point mutants) with previously released TCR structures (Supplementary Table S3). This benchmark represents a variety of germline genes, CDR3 lengths and CDR3 sequences, encompassing structures released since 2016. For these cases, TCRmodel processing time was found to be less than one minute on average by default, while refinement of CDR3 loops (an option that can be selected on the input page) increased job running time to approximately 10 minutes on average (detailed results by test case are shown in Supplementary Table S4). The default TCRmodel performance compares favorably with a previously released TCR modeling web server, LYRA (25), where average modeling time was found to be over two minutes on average (Supplementary Table S4).
Predictive performance of TCRmodel was then compared with LYRA, excluding all recently determined structures as templates. This benchmarking approach, where recently determined structures were modeled using older structures as templates, was also used in another study focused on template-based modeling of protein complexes (26). It was not necessary to explicitly exclude benchmark structures templates for LYRA modeling, given that the LYRA template database did not appear to include these recent entries. Overall, predictive performance in TCRmodel was comparable or superior to LYRA (Figure 2A, Supplementary Tables S5 and S6). Part of this difference in performance may be due to template selection, as TCRmodel and LYRA selected different templates in their modeling procedures (Supplementary Table S7). Based on overall RMSDs, default TCRmodel (no CDR3 refinement) gave better predictions for 73% of test cases (16 out of 22 cases). It should be noted that one TCR test case (PDB code: 5WJO) caused an execution failure in LYRA, and was excluded from comparison.
Figure 2.

Predictive performance of TCRmodel on a benchmark of recently determined TCR structures. (A) TCRs were modeled using structures from prior to 2016 as templates, and performance was compared with LYRA, another TCR modeling server (25), run with the same template restrictions. Backbone atom root mean square distances (RMSDs) between models and crystal structures were computed for full TCR models (Global), framework residues (FW), and individual CDR loops. Statistically significant lower RMSDs for TCRmodel (P < 0.05, two-tailed t-test) were observed for framework, CDR2α, CDR1β, and CDR2β. CDR3 loop refinement led to significantly improved RMSDs for CDR3α and CDR3β loops. (B) Performance comparison on the TCR benchmark, excluding templates from variable domains with >90% sequence identity to the modeled TCRs. Statistically significant lower RMSDs for TCRmodel (P < 0.05, two-tailed t-test) were observed for full models (Global), framework, CDR1α, CDR2α, CDR1β, and CDR2β. As in (A), CDR3 loop refinement in TCRmodel led to significantly improved RMSDs for CDR3α and CDR3β loops. Figure generated using the ggplot2 package in R (r-project.org).
It is evident that relatively lower TCRmodel performance for some test cases was due to CDR3 inaccuracies; the three cases with the highest overall RMSDs from TCRmodel (5EUO, 5KS9, 5TEZ), all had outlier CDR3α RMSDs over 6 Å (Figure 2A, Supplementary Table S5). However, CDR3 refinement improved the RMSDs of each of these loops, and overall led to a significant improvement in CDR3 predictive performance over template-based models (P < 0.05 for both CDR3 loops, based on two-tailed t-test). To highlight structural improvement from CDR3 loop refinement, Figure 3 shows the refined structure of the CDR3β loop for one test case (5NMD), in comparison with the initial (template-based) model and X-ray structure. During refinement, the loop backbone RMSD improves from 3.16 to 1.38 Å, and several key side chains are positioned more accurately.
Figure 3.
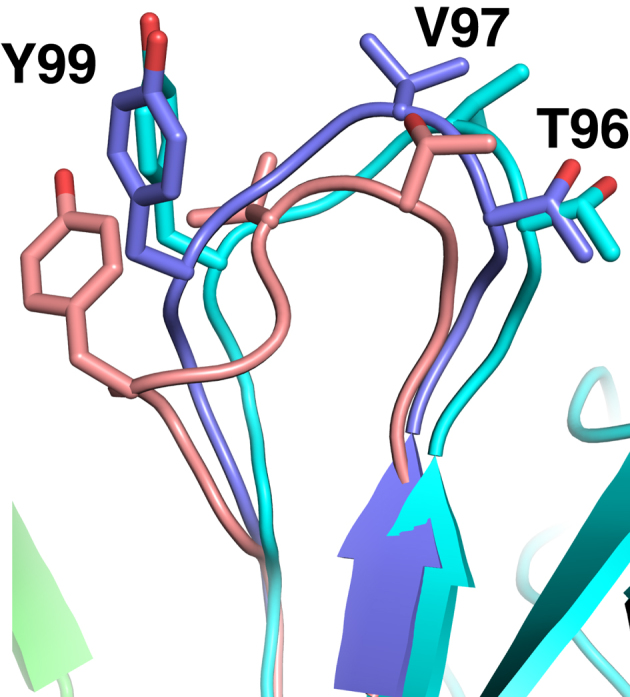
CDR3 loop improvement from refinement. The initial CDR3β loop model from TCRmodel (pink) is compared with the refined loop model (slate) and X-ray structure of the 868 TCR (β chain cyan, α chain green) (31). Backbone RMSD for the initial loop CDR3β model to the crystallographic structure is 3.16 Å, which improves to 1.38 Å upon refinement. For clarity and context, non-CDR3 regions of the TCR are shown only for the X-ray structure. Selected residues at the CDR3β loop apex are shown as sticks and labeled. Figure generated in PyMOL (Schrödinger, LLC).
We also assessed the predictive performance on this set of benchmark cases using a different restriction on template selection, disallowing any templates from variable domains with 90% or greater sequence identity with the target TCR, in accordance with algorithm benchmarking by the developers of LYRA (26). For LYRA, these templates were excluded manually by PDB code during job submission. In this scenario, TCRmodel performance was also superior to LYRA (Figure 2B), with significant improvements in total model RMSDs as well as germline CDRs; 90% of test cases (18 out of 20) had better RMSDs for TCRmodel. TCRmodel performance was superior in some cases compared with the initial benchmarking (Figure 2A), likely due to the inclusion of more recently solved template structures (2016–present). Two test cases were excluded from this analysis (5ISZ, 5NMD), due to incorrect number of residues in LYRA models for these cases, which prevented RMSD calculations. Finally, to confirm the performance of TCRmodel using a larger set of cases, we tested it on a set of over 100 nonredundant TCRs that pre-dated the benchmark set (Supplementary Table S8), again using the 90% template identity cutoff. These results (Supplementary Figure S1) show RMSDs that are comparable to benchmarking using the smaller set of more recently determined TCR structures.
Case study: modeling a tumor-specific TCR, 3995
To demonstrate the capacity of TCRmodel to enable a 3D structural view of TCRs of interest, we modeled a recently described TCR (‘3995’) which was isolated from tumor infiltrating lymphocytes of a patient with metastatic colon cancer (6), and displays cytolytic activity against cells displaying the KRASG12D neoantigen presented by HLA-C*08:02 MHC. As this TCR has no reported experimentally determined structure, this model, shown in Figure 4, provides the first view of its putative 3D structure and features of its CDR loops. TCRmodel identified germline CDR structural templates for both chains of this TCR (PDB codes 4OZI and 4JRY, for α and β chains respectively), and the CDR3 loop template alignments and models (Figure 4B) provide intriguing clues regarding the basis of its tumor antigen engagement. Interestingly, both CDR3 apexes include a number of exposed polar and charged residues, including a ‘DMD’ set of residues in the CDR3α loop that corresponds to ‘DMN’ residues in the CDR3α sequence of the template, which is from the unbound X-ray structure of an alloreactive TCR that targets a different tumor antigen presented by the HLA-A2 MHC (27). The 3995 TCR is included as an example for TCR sequence input and results on the TCRmodel server.
Figure 4.

Model of the KRASG12D neoantigen-specific TCR, 3995. The full 3995 TCR model (containing TCR variable domains) is shown in (A), with α chain colored green, β chain cyan and CDR3 loops in dotted box. (B) Template alignments of model (‘Input’) CDR3α and CDR3β loops from the TCRmodel results page are shown, along with close-up view of modeled CDR3 loops, with loop apex residues shown as sticks. Selected CDR3α loop residues are labeled. Figure structures generated in PyMOL (Schrödinger, LLC).
DISCUSSION
By providing the community with the means to generate high resolution TCR structural models from sequence, TCRmodel complements recent public databases that provide TCR sequences (24,28), providing the means to extend the insights from these TCR sequence datasets into structural space. As was recently illustrated in the context of antibody modeling, the combination of immune repertoire sequencing data with structural modeling can be highly informative (29). One future development would be the direct linking of such sequence database sets to TCRmodel, to provide direct ‘one click’ input into TCRmodel for structural modeling. Additionally, integration of TCR predictive docking methods to peptide-MHC (pMHC) complexes would enable greater insights for TCRs with known pMHC targets. As we previously developed a TCR-pMHC docking algorithm in Rosetta (18), such a modeling pipeline should be possible to implement. Of note, this TCR docking protocol was previously utilized to investigate potential shared targeting features of TCRs that engage an epitope from human cytomelagovirus (30), using TCR models as input for docking.
A major emphasis of the TCRmodel server will be developments and enhancements to address the needs and feedback of users. Additionally, we will regularly update the library of TCR structures on a monthly basis to add new templates from the PDB.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Johnathan Guest and Kunal Kundu (IBBR) for useful suggestions, and Bong-Hyun Kim (Intrexon Corporation) for assistance with the web server interface. We also thank Zhiping Weng (University of Massachusetts Medical School) and Brian Baker (University of Notre Dame) for helpful discussions during initial web server development. We are grateful to the Maryland Advanced Research Computing Center (MARCC) for computing resources to conduct algorithm development and benchmarking, as well as Christian Presley and the Institute for Bioscience and Biotechnology Research (IBBR) computing facility for resources and assistance with server implementation and web hosting.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Startup funding from the University of Maryland (to B.G.P.). Funding for open access charge: Startup funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Pugliese A. Autoreactive T cells in type 1 diabetes. J. Clin. Invest. 2017; 127:2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lang H.L., Jacobsen H., Ikemizu S., Andersson C., Harlos K., Madsen L., Hjorth P., Sondergaard L., Svejgaard A., Wucherpfennig K. et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002; 3:940–943. [DOI] [PubMed] [Google Scholar]
- 3. Tran E., Robbins P.F., Lu Y.C., Prickett T.D., Gartner J.J., Jia L., Pasetto A., Zheng Z., Ray S., Groh E.M. et al. T-Cell transfer therapy targeting mutant KRAS in Cancer. N. Engl. J. Med. 2016; 375:2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hinrichs C.S., Rosenberg S.A.. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2014; 257:56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liddy N., Bossi G., Adams K.J., Lissina A., Mahon T.M., Hassan N.J., Gavarret J., Bianchi F.C., Pumphrey N.J., Ladell K. et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012; 18:980–987. [DOI] [PubMed] [Google Scholar]
- 6. Tran E., Ahmadzadeh M., Lu Y.C., Gros A., Turcotte S., Robbins P.F., Gartner J.J., Zheng Z., Li Y.F., Ray S. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015; 350:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen G., Yang X., Ko A., Sun X., Gao M., Zhang Y., Shi A., Mariuzza R.A., Weng N.P.. Sequence and structural analyses reveal distinct and highly diverse human CD8+ TCR Repertoires to immunodominant viral antigens. Cell Rep. 2017; 19:569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glanville J., Huang H., Nau A., Hatton O., Wagar L.E., Rubelt F., Ji X., Han A., Krams S.M., Pettus C. et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017; 547:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017; 547:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin Y., Li Y., Mariuzza R.A.. Structural basis for self-recognition by autoimmune T-cell receptors. Immunol. Rev. 2012; 250:32–48. [DOI] [PubMed] [Google Scholar]
- 11. Leaver-Fay A., Tyka M., Lewis S.M., Lange O.F., Thompson J., Jacak R., Kaufman K., Renfrew P.D., Smith C.A., Sheffler W. et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011; 487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rose P.W., Beran B., Bi C., Bluhm W.F., Dimitropoulos D., Goodsell D.S., Prlic A., Quesada M., Quinn G.B., Westbrook J.D. et al. The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res. 2011; 39:D392–D401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lupyan D., Leo-Macias A., Ortiz A.R.. A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics. 2005; 21:3255–3263. [DOI] [PubMed] [Google Scholar]
- 15. Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunbar J., Deane C.M.. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. 2016; 32:298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vreven T., Moal I.H., Vangone A., Pierce B.G., Kastritis P.L., Torchala M., Chaleil R., Jimenez-Garcia B., Bates P.A., Fernandez-Recio J. et al. Updates to the integrated Protein-Protein interaction Benchmarks: Docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol. 2015; 427:3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pierce B.G., Weng Z.. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013; 22:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sivasubramanian A., Sircar A., Chaudhury S., Gray J.J.. Toward high-resolution homology modeling of antibody Fv regions and application to antibody-antigen docking. Proteins. 2009; 74:497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henikoff S., Henikoff J.G.. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stein A., Kortemme T.. Improvements to robotics-inspired conformational sampling in rosetta. PLoS One. 2013; 8:e63090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alford R.F., Leaver-Fay A., Jeliazkov J.R., O’Meara M.J., DiMaio F.P., Park H., Shapovalov M.V., Renfrew P.D., Mulligan V.K., Kappel K. et al. The Rosetta All-Atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017; 13:3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lefranc M.P., Giudicelli V., Ginestoux C., Jabado-Michaloud J., Folch G., Bellahcene F., Wu Y., Gemrot E., Brochet X., Lane J. et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009; 37:D1006–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shugay M., Bagaev D.V., Zvyagin I.V., Vroomans R.M., Crawford J.C., Dolton G., Komech E.A., Sycheva A.L., Koneva A.E., Egorov E.S. et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018; 46:D419–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klausen M.S., Anderson M.V., Jespersen M.C., Nielsen M., Marcatili P.. LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res. 2015; 43:W349–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kundrotas P.J., Zhu Z., Janin J., Vakser I.A.. Templates are available to model nearly all complexes of structurally characterized proteins. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simpson A.A., Mohammed F., Salim M., Tranter A., Rickinson A.B., Stauss H.J., Moss P.A., Steven N.M., Willcox B.E.. Structural and energetic evidence for highly peptide-specific tumor antigen targeting via allo-MHC restriction. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21176–21181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tickotsky N., Sagiv T., Prilusky J., Shifrut E., Friedman N.. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. 2017; 33:2924–2929. [DOI] [PubMed] [Google Scholar]
- 29. DeKosky B.J., Lungu O.I., Park D., Johnson E.L., Charab W., Chrysostomou C., Kuroda D., Ellington A.D., Ippolito G.C., Gray J.J. et al. Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E2636–E2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang X., Gao M., Chen G., Pierce B.G., Lu J., Weng N.P., Mariuzza R.A.. Structural basis for clonal diversity of the public T Cell response to a dominant human cytomegalovirus epitope. J. Biol. Chem. 2015; 290:29106–29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole D.K., Fuller A., Dolton G., Zervoudi E., Legut M., Miles K., Blanchfield L., Madura F., Holland C.J., Bulek A.M. et al. Dual molecular mechanisms govern escape at immunodominant HLA A2-Restricted HIV epitope. Front. Immunol. 2017; 8:1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.