

Abstract
Summary
Short reads sequencing technology has been used for more than a decade now. However, the analysis of RNAseq and ChIPseq data is still computational demanding and the simple access to raw data does not guarantee results reproducibility between laboratories. To address these two aspects, we developed SeqBox, a cheap, efficient and reproducible RNAseq/ChIPseq hardware/software solution based on NUC6I7KYK mini-PC (an Intel consumer game computer with a fast processor and a high performance SSD disk), and Docker container platform. In SeqBox the analysis of RNAseq and ChIPseq data is supported by a friendly GUI. This allows access to fast and reproducible analysis also to scientists with/without scripting experience.
Availability and implementation
Docker container images, docker4seq package and the GUI are available at http://www.bioinformatica.unito.it/reproducibile.bioinformatics.html.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Whole transcriptome sequencing (WTS) and ChIPseq made obsolete the corresponding array hybridization based technologies. Short reads sequencing technology has been used for more than a decade now, and experience shows that the main bottleneck in sequencing workflows is the time spent in analyzing and interpreting data (Wong et al. 2017).
The primary analysis of the data, i.e. mapping short read sequences on the reference genome, is still computationally demanding and requires computer performances that are not commonly available in laptops. In particular, WTS requires a significant amount of RAM and multicores processors. The needs of high performance computing infrastructure for the analysis of sequencing data has brought to the development of cloud based analysis tools, e.g. Illumina BaseSpace (https://basespace.illumina.com/home/index), Galaxy (https://usegalaxy.org/), etc. However, cloud based solutions suffer of some criticalities, e.g. data uploading speed, limited storage space and significant computing and data transfer costs. Moreover, although all available data analysis platforms guarantee a certain level of reproducibility, typically storing the version of the software being used, tracking changes in the system libraries, which might lead to sneaky reproducibility issues, is not provided.
To combine reproducible data generation with cost effective but efficient hardware we have developed SeqBox, a software/hardware ecosystem providing the most common analyses of RNAseq and ChIPseq data (i.e. genomic mapping, experimental power evaluation, differential expression, transcription factors/histone-marks peaks identification, etc.) on a consumer game computer (Fig. 1A).
Fig. 1.
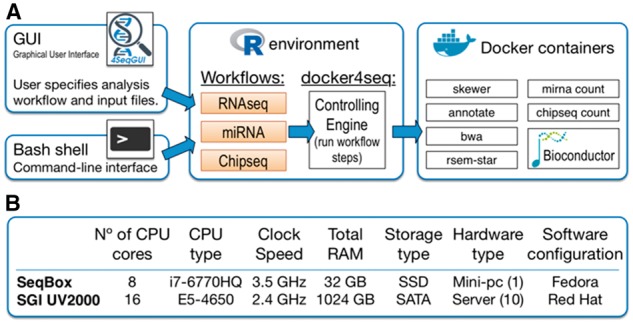
(A) SeqBox framework: depicting the structure of SeqBox and its functionalities from a user point of view. The analysis flows from left to right. (B) Characteristics of the hardware used to evaluate the SeqBox performances
2 Materials and methods
The SeqBox ecosystem (Fig. 1A) is the union of SeqBox software and SeqBox target hardware.
A user can access the system either through a Java-based graphical interface (GUI, see Supplementary Material), or through R console (see Supplementary Material). Independently of the access type, the user can exploit three different workflows: RNAseq, miRNA and ChipSeq, which are managed by a Controlling Engine written in R (Fig. 1A). The functions that realize the workflows are either standard analysis algorithms or a set of supporting functions that have been developed and included in Bioconductor packages (e.g. DESeq2, ChIPpeakAnno). The algorithms used for sequencing data analysis include STAR (Dobin et al., 2013) for RNAseq genomic mapping, DESeq2 (Love et al., 2014) for differential expression analysis, BWA (Li and Durbin, 2009) for ChIPseq genomic mapping, MACS (Zhang et al., 2008), and SICER (Xu et al., 2014) for ChIP peaks detection (see Supplementary Material). All of them are encapsulated into Docker images.
A Docker image is a lightweight, stand-alone, executable package that includes everything needed to run a specific software. A runtime instance of an image, called container, runs completely isolated from the host environment except for user-specified host files. The advantage of using Docker images is that the whole environment is fixed, the images are available in the Docker repository, and the identity of the images is the only element needed to reproduce the results. The execution of the Docker images, implementing the workflow chosen by the user, is done by docker4seq, a R package which embeds a set of functions providing the running parameters to the mapping and counting engine.
SeqBox provides six Docker images: (i) skewer.2017.01, which uses skewer (Jiang et al., 2014) for adapter trimming; (ii) rsemstar.2017.01, which uses STAR (Dobin et al., 2013) to map short reads mapping on the reference genome and RSEM (Li and Dewey, 2011) for gene and isoform-level quantification; (iii) annotate.2017.01, which is used to associate RSEM output id with gene symbols; (iv) mirnaseq.2017.01, which implements the miRNAseq analysis workflow described in (Cordero, et al., 2012); (v) r332.2017.01, which allows differential expression analysis via Bioconductor package DESeq2; (vi) chipseq.2017.01, which uses BWA (Li and Durbin, 2009) to map short reads on the reference genome, MACS (Zhang et al., 2008) to detect transcription factors binding sites, and SICER (Xu et al., 2014) to define histone-marks.
The GUI provides a graphical access to the docker4seq functions allowing the use of the tools to biologists without scripting experience.
SeqBox hardware: The parameters setting of the algorithms (in terms of memory size versus number of assigned cores) is optimized for an execution on the game computer NUC617KYK (Fig. 1A), which is based on an Intel Core I7, featuring 4 cores running up to eight threads that share a common memory of 32 Gb and a SSD disk of 256 GB.
3 Results
We benchmarked SeqBox with respect to a high-end server (SGI UV2000, Fig. 1B). The performance comparison was done for the three workflows (see Supplementary Figs S1–S3). In brief, we compared the workflows using increasing amounts of reads (see Supplementary Material) on SeqBox, using eight threads, and on the SGI server increasing the number of threads from 8 to 160 (Supplementary Figs S1–S3). Parallelization provided by the SGI server did not improve very much the overall performances in the RNAseq workflow (Supplementary Fig. S1). SeqBox significantly outperformed the server, because of the presence of a SSD with high I/O performance which can cope with the limited parallelism of SeqBox. In the case of miRNA and ChIPseq workflows the parallelization is only available for the reads mapping procedures. The limited parallelization of these two workflows combined with the higher I/O performances of the SSD with respect to the SATA array makes SeqBox extremely effective even with very high number of reads to be processed (Supplementary Figs S2 and S3).
4 Conclusion
The majority of the algorithms used in the considered bioinformatics workflows is strongly I/O bound and exhibits a limited exploitation of parallelism. Our experiments show that a combination of a consumer computer with a fast storage is able to over-perform a high-end server. The integration of Docker technology within a mini-PC consumer computer such as Intel NUC6I7KYK provides therefore, to small biology laboratories, a solution for Next Generation Sequence (NGS) analysis which is cheap, efficient and reproducible.
Funding
This work has been supported by the EPIGEN FLAG PROJECT.
Conflict of Interest: none declared.
Supplementary Material
References
- Cordero F. et al. (2012) Optimizing a massive parallel sequencing workflow for quantitative miRNA expression analysis. PLoS One, 7, e31630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A. et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. et al. (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics, 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. et al. (2017) Applied Reconfigurable Computing. In 13th International Symposium ARC 2017, Delft, The Netherlands, April 3–7, 2017, Lecture Notes in Computer Science, volume 10216.
- Xu S. et al. (2014) Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol., 1150, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.