

Abstract
Motivation
Existing approaches to plot association results from genome-wide association studies (GWAS) are in the form of static Manhattan plots and often lack data integration with rich databases on variant regulatory potential as well as population-specific linkage disequilibrium patterns.
Summary
We created an intuitive web module for uploading and efficiently exploring GWAS association results. Interactive plots and sortable tables allow researchers to query genomic regions of interest, facilitating the integration of data on linkage disequilibrium, variant regulatory potential and potential target genes. External links allow for visualization of association results in the UCSC genome browser as well as easy access to publically available databases (e.g. dbSNP and RegulomeDB). Through improved visualization and data integration, LDassoc offers genomic researchers a specialized environment to examine association signals and suggests variants for functional investigation.
Availability and implementation
LDassoc is a free and publically available web tool which can be accessed online at https://analysistools.nci.nih.gov/LDlink/? tab=ldassoc.
1 Introduction
Genome-wide association studies (GWAS) systematically investigate phenotypic associations across select genotyped variants spanning the genome using single nucleotide polymorphism markers. GWAS have led to the discovery of thousands of disease susceptibility loci that have informed knowledge of the genetic architecture of a spectrum of traits and chronic diseases (Chanock, 2014; MacArthur, et al., 2017). Despite GWAS advancements made in discovering germline genetic susceptibility markers associated with a variety of traits, the methods of visualizing and interpreting GWAS association signals remain largely unchanged. Results from GWAS are typically plotted using static Manhattan plots displaying the chromosomal position of genotyped markers across the x-axis and the −log10 of the respective association P-value on the y-axis. While these plots have utility in examining consistency of P-value associations at a genomic locus, they fail to integrate information on linkage disequilibrium (LD) patterns, minor allele frequency (MAF), variant regulatory potential or neighboring genes of interest. A lack of data integration make connecting information from different sources difficult and could result in missed opportunities for discovering novel insights regarding the biology underlying a susceptibility haplotype.
A notable improvement in plotting GWAS association results was implemented in LocusZoom, an online plotting tool that generates regional plots with data on LD patterns, coding variants and nearby genes (Pruim, 2010). Recent improvements to LocusZoom have added interactivity, but so far only for select public datasets. Other plotting tools and packages are available for visualizing GWAS association results (e.g. LocusExplorer and LocusTrack) (Cuellar-Partida, et al., 2015; Dadaev, 2016), but substantial opportunities exist to create an improved interface to upload association results and interactively visualize the data in rich integrative plots and tables. We created LDassoc as a new web module in the LDlink suite of web tools (Machiela and Chanock, 2015) with the goals of developing an intuitive package where GWAS association results could be: (i) easily uploaded, (ii) interactively visualized, (iii) merged with data on LD, MAF frequency, variant regulatory potential and neighboring genes, (iv) filtered and sorted in interactive tables and (v) exported to the University of California Santa Cruz (UCSC) Genome Browser for further integration with data tracks.
2 Implementation
LDassoc is a module in LDlink where researchers can upload space or tab delimited association results from GWAS studies to a secure server for analysis. Required data columns are: chromosome, base pair coordinates (currently GRCh37) and association P-values; although additional columns are permitted in uploaded datasets. File headers are optional and facilitate users when selecting appropriate columns to import into LDassoc. Data uploads are limited to 2Gb in size and generally take under 1 min to upload; with file size and internet connection speed being major determinants of upload time. Uploaded association results are only available for the current user session and are deleted after 4 h.
LDassoc has three options for visualizing regions of association: by gene, by region or by variant. The choice of a gene will require a RefSeq gene name and will zoom in on association results in a window (default is ± 100 Kb) containing the gene. An index variant may be selected as a reference for calculating LD metrics. When no index variant is chosen, the lowest P-value variant is selected as the index variant. Selecting region will enable a researcher to define a genomic window of interest as well as specify an index variant. As with the gene option, if no index variant is entered the lowest P-value variant in the region will be selected as the reference for calculating LD metrics. Finally, using the variant option allows researchers to zoom in on a window around a variant of interest (default is ± 500 Kb). This variant is used as the index variant for calculating LD metrics. Other required LDassoc input is specification of the 1000 genomes super or sub-population of interest (e.g. EAS, CHB) as well as whether the desired LD output is R2 or D′ (default is R2).
Generated results consist of an interactive Manhattan plot, a filterable and sortable table, as well as links to external bioinformatic resources such as visualizing results in the UCSC Genome Browser (Fig. 1). The interactive Manhattan plot displays the index variant in blue and all variants in LD are shaded in red based on their strength of LD. The size of plotted variants is proportional to MAF and numbers annotate RegulomeDB scores (Boyle, 2012). Recombination rate as well as a genome-wide significance line (P-value < 5 × 10−8) are overlaid on the plot. Additional panels show a rug plot of variant density and nearby genes. Interactive tools allow for hovering of additional variant and gene information, zooming, panning, selecting subsets of variants and saving plots for publication. Interactive tables provide intuitive user specified filtering and searching of association results with links to external resources on the variants (e.g. dbSNP, RegulomeDB, HaploReg). Finally, custom links allow for importing of the association data to the UCSC Genome Browser as well as downloading annotated data.
Fig. 1.
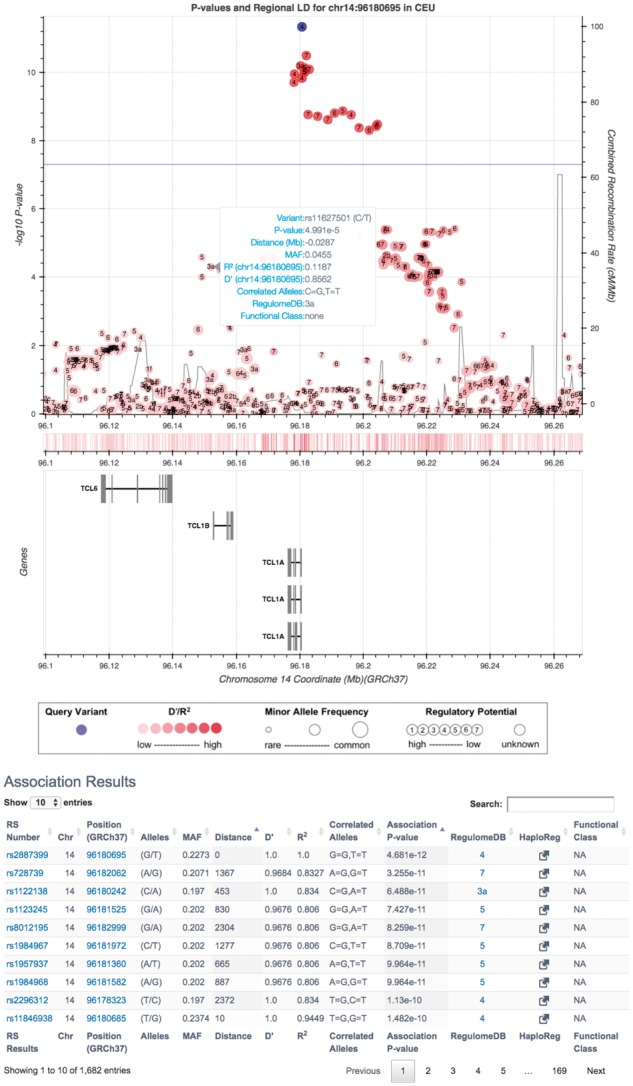
Screen capture of an LDassoc interactive plot and table
LDassoc is written in Python 2.7 and runs on a web-accessible virtual machine with UNIX operating system. Tabix (0.2.5) is used to access phased 1000 genomes genotypes (Phase 3) of variants in the LDassoc query region. LDassoc uses the Bokeh package (0.12.2) to generate interactive plots. The LDassoc page is programed in HTML5 for cross browser and cross platform compatibility. All code for LDassoc is available in our GitHub repository: https://github.com/CBIIT/nci-webtools-dceg-linkage/.
Acknowledgements
Special thanks to Ye Wu and Sue Pan from the Center for Biomedical Informatics and Information Technology (CBIIT) for technical assistance and web development.
Funding
This work was supported by the Intramural Research Program of the US National Institutes of Health; and the Informatics Tool Challenge of the Division of Cancer Epidemiology and Genetics.
Conflict of Interest: none declared.
References
- Boyle A.P. et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock S.J. (2014) Cancer biology: genome-wide association studies In: Stewart B.W., Wild C.P. (eds) World Cancer Report 2014. International Agency for Cancer Research, Lyon, France, pp. 193–202. [Google Scholar]
- Cuellar-Partida G. et al. (2015) LocusTrack: integrated visualization of GWAS results and genomic annotation. Source Code Biol. Med., 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadaev T. et al. (2016) LocusExplorer: a user-friendly tool for integrated visualization of human genetic association data and biological annotations. Bioinformatics, 32, 949–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J. et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela M.J., Chanock S.J. (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics, 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim R.J. et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]