

Abstract
Liver disease is a leading cause of HIV-related mortality. Hepatitis C virus (HCV)–related fibrogenesis is accelerated in the setting of HIV coinfection, yet the mechanisms underlying this aggressive pathogenesis are unclear. We identified formalin-fixed paraffin-embedded liver tissue for HIV-infected patients, HCV-infected patients, HIV/HCV-coinfected patients, and controls at Duke University Medical Center. De-identified sections were stained for markers against the wound repair Hedgehog (Hh) pathway, resident T-lymphocytes, and immune activation and cellular aging. HIV infection was independently associated with Hh activation and markers of immune dysregulation in the liver tissue.
Keywords: fibrogenesis, GLI, Hedgehog, hepatitis C virus, human immunodeficiency virus, patched, pathogenesis, Sonic Hedgehog, wound repair
Liver disease is a leading cause of HIV-related morbidity and mortality [1]. In the United States, chronic hepatitis C virus (HCV) infection is the leading cause of liver disease and related mortality in HIV infection [1]. HCV is a metabolically active virus that, in the setting of HIV coinfection, results in accelerated liver disease pathogenesis [2]. The mechanisms identified as potentially contributing to this more severe disease pathogenesis include increased oxidative stress and downstream induction of profibrotic pathways by cytokines including TGFß-1 and activation of hepatic stellate cells (HSCs) via the Toll-like receptor 4, cysteine-X-cysteine receptor 4, and chemokine motif receptor 5 receptors [3, 4].
Fibrosis is a wound-healing response to tissue (epithelial) injury. The regenerative responses of wound-healing differ depending on the severity and chronicity of liver injury. Although initially beneficial, the repair process can become pathogenic if it continues aberrantly, leading to considerable tissue remodeling and scar. The fetal morphogen hedgehog (Hh) signaling pathway has been implicated in multiple metabolic diseases, including nonalcoholic steatohepatitis, obesity, and diabetes [5–7]. The Hh pathway has also been implicated in HCV mono-infection, which induces hepatic steatosis and insulin resistance [8], and now for the first time the Hh pathway has been implicated in HIV-associated nephropathy [9].
The Hh pathway, originally identified in Drosophilia, is a highly conserved signaling pathway that orchestrates multiple aspects of organogenesis and tissue remodeling [10]. Hh pathway activation typically enhances the growth and viability of Hh-responsive cells, whereas abrogating Hh signal transduction usually triggers apoptosis, conferring a selective advantage for cell types capable of responding to Hh [11]. Hh-ligands upregulate expression of chemokines and receptors in immature liver epithelial cells, thus facilitating hepatic recruitment and retention of particular types of immune cells [12]. Specifically, activated NKT cells are recruited, and by avoiding apoptosis, they accumulate in liver tissue, in turn enhancing liver disease progression. Gli2, a Hh pathway transcription factor, is also reported to regulate transcription of TGFß-1, a key profibrogenic cytokine implicated in HIV/HCV-related fibrogenesis [13].
Using liver tissue specimens from HIV-infected patients, HCV-infected patients, HIV/HCV-coinfected patients, and controls, we assessed for markers of the Hh pathway, T-lymphocyte cellular subpopulations, cellular aging and senescence, and fibrogenesis. We proposed testing the hypothesis that Hh signaling is aberrantly activated in the liver tissue of HIV-infected patients with HCV and that this increased Hh activity correlates with increased liver NK T-cell populations and increased markers of fibrosis.
METHODS
Study Population
Formalin-fixed paraffin-embedded (FFPE) liver tissue sections were identified for HIV-infected patients, HCV-infected patients, HIV/HCV-coinfected patients, and de-identified controls from the Department of Pathology at Duke University Medical Center, in accordance with National Institutes of Health (NIH) and institutional guidelines for human subjects research. All HIV/HCV-coinfected patients with available liver biopsy specimens who did not have any exclusion criteria were included. HCV-monoinfected patients were then identified by matching to HIV/HCV-coinfected patients by age (±5 years) at the time of liver biopsy, sex, and METAVIR stage (0–1 and 2–4) of liver fibrosis on the biopsy. Due to the small number, all HIV-monoinfected patients with available liver biopsy who did not have any exclusion criteria were included. Reasons provided on the requisition form for liver biopsy for the HIV-monoinfected patients included abnormal liver enzymes and steatosis noted on imaging (n = 4), ill-defined lesions on imaging (liver tissue was normal), abnormal liver enzymes off of antiretrovirals, and assessment for cirrhosis (no underlying liver injury etiology provided and liver tissue was normal). Active or recent alcohol use, evidence of nonalcoholic steatohepatitis (NASH) on biopsy with Brunt stage 2 or greater fibrosis, and active HBV infection (+HBsAg) were excluded. Control liver tissue is acquired from localized colorectal cancer tumor metastasis resection as part of a research tissue biorepository protocol at Duke. Histologically normal tissue that is >1 cm from the tumor mass is collected for future use. HIV viral suppression was defined as RNA <200 copies/mL.
Immunohistochemistry
De-identified FFPE liver tissue sections were stained with antibodies against (1) key components of the Hh pathway, including Sonic Hedgehog (SHH) ligand, glioblastoma 2 (GLI2) transcription factor, and patched (PTCH1) receptor to assess liver repair; (2) resident T-lymphocytes (CD3, CD8, CD56, CD28), CXCL16 (NKT cell cytokine); and (3) markers of immune activation and cellular aging (CD57 and p16INK4a). In diseased liver tissue, CD56 may also be expressed by reactive bile ducts. Please see Supplementary Table 1 for commercial suppliers of the antibodies used. Staining for the myofibroblast marker alpha-smooth muscle actin (αSMA) was also performed. In addition, isotype/negative controls were completed for each antibody to ensure that positive staining is due to the antibody binding of the desired antigen and not to some general unspecific binding of the immunoglobulin to the tissue.
Morphometric analysis was completed with MetaView software (Universal Imaging Corp, Downington, PA), and morphometric quantification was done with 50 randomly chosen, 20x fields per section. Immunoreactive cells were quantified using similar approaches for each of these stains; the numbers of cells with stained nuclei (GLI2) and stained membranes (immune markers, cytokines) were counted in 20 random, 20x fields per section.
Statistical Analysis
The Student t test and analysis of variance were used to compare histologic changes across the study groups, with significance set at a P value of <.05. To test for independent associations for each immunohistochemistry marker, multivariate linear regression models were built and adjusted for independent variables: sex, race, age, METAVIR fibrosis stage, METAVIR inflammation score, steatosis, HIV infection, HCV infection, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and diabetes. An interaction term was included to assess the effect of HIV/HCV coinfection when compared with monoinfection. If the HIV/HCV coinfection interaction term is reported as significant, it suggests that HIV/HCV coinfection has a unique impact on the staining of the marker of interest beyond that of HIV or HCV monoinfection. Controls were not included in the regression models due to the lack of clinical data. Additional multivariate linear regression models were used to determine if HIV viral load suppression, in addition to the variables mentioned above, was associated with any of the markers in a subset of patients with either HIV or HIV/HCV. All P values were adjusted using the Benjamini-Hochberg procedure for multiple comparisons.
RESULTS
Overall, 66 liver specimens were identified: 5 controls, 7 HIV-infected patients, 27 HCV-infected patients, and 27 HIV/HCV-coinfected patients. Two HCV-infected subjects did not have adequate tissue on staining and were therefore removed from the analysis. The controls were predominantly female (80%), and the mean age at the time of liver biopsy was 52 years (±10.7). Race was only available for 1 of the controls, who was white. The final analysis cohort (n = 59) with demographic data was 44% female and 54% black, with a mean age at the time of the liver biopsy (SD) of 46 (8.7) years (Supplementary Table 2). Thirty-four percent of all patients with HCV infection had severe liver fibrosis (METAVIR stage 3 or 4), and the majority (92%) had genotype-1 infection. For all patients with HIV infection, the median CD4 (SD) was 651 (386) cells/mm3, and 85% were on antiretrovirals at the time of liver biopsy. Ten patients (17%) had diabetes.
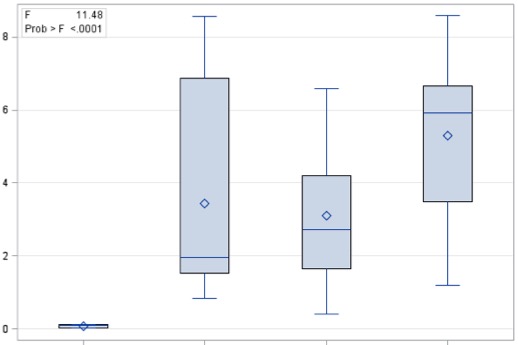
The 3 infected cohorts (HIV, HCV, HIV/HCV) had greater expression of markers of tissue repair and fibrogenesis (GLI2 P < .0001, Shh P < .0001, PTCH1 P = .0214, αSMA P < .0001), T-cell populations (CD3 P < .0001), NK-like cells (CD56 P < .0001), NKT cell recruitment cytokine (CXCL16 P < .0001), and markers of cellular senescence/aging (CD57 P = .0003 and p16INK4aP < .0001) than controls (Figures 1 and 2; Supplementary Figure 1). We observed increased staining for the Hh pathway transcription factor GLI2 and ligand SHH in all 3 virally infected cohorts (Figure 1A and B). A representative immunohistochemistry section to show staining of GLI2 and SHH across all cohorts is provided in Figure 1C. Expression of tissue markers was greatest in HIV/HCV coinfection for PTCH1 (P = .016), CD3 (P = .028), CD57 (P = .017), CXCL16 (P = .0002), and p16ink4a (P = .096). Increased T-cell activation marker (CD57) staining was also observed for all 3 virally infected cohorts, with the greatest staining in patients with HIV/HCV coinfection (Figure 2A). Staining for the αSMA fibrosis marker, shown in Figure 2B, is increased in HIV-monoinfected patients as compared with controls and is greatest for patients with HCV and HIV/HCV infection. A representative immunohistochemistry section to show staining for CD57, NKT cell marker (CD56), and αSMA across all cohorts is provided in Figure 2C and D. In addition, several markers were associated with elevated liver enzymes, including PTCH1 (r = .411, P = .0017), CD56 (r = .421, P = .0011), CD57 (r = .297, P = .025) with AST, and CD56 (r = .286, P = .031) and CD28 (r = .264, P = .047) with ALT.
Figure 1.

Hedgehog (Hh) activation markers in hepatic parenchyma of healthy and virally infected patients. Viral infection is associated with activation of the Hh pathway (GLI2 transcription factor and Sonic Hedgehog [SHH] ligand). Staining of liver tissue sections from controls (Con), patients with HIV infection (HIV), HCV infection (HCV), and HIV/HCV coinfection (HIV/HCV). Box and whisker plots of (A) GLI2 marker staining and (B) SHH marker staining across the 4 patient cohorts. C, Representative immunohistochemistry section with ductular and immune cell staining for GLI2 (brown) and ductular cell staining for SHH (green).
Figure 2.

Immune activation and fibrosis markers in hepatic parenchyma of healthy and virally infected patients. Viral infection is associated with immune activation (CD57), NKT cell inflammation (CD56) and fibrogenesis (αSMA). Staining of liver tissue sections from controls (Con), patients with HIV infection (HIV), HCV infection (HCV), and HIV/HCV coinfection (HIV/HCV). Box and whisker plots of (A) CD57 T-cell terminal differentiation marker staining and (B) αSMA fibrosis marker staining across the 4 patient cohorts. C, Representative immunohistochemistry section with immune cell staining for CD57 (brown). D, Representative immunohistochemistry section with immune and ductular cell staining for CD56 (brown) and ductular and stromal (hepatic stellate cells, myofibroblasts) cell staining for αSMA (green).
After adjusting for clinical and demographic variables, the multivariate models identified METAVIR stage, HIV infection, HCV infection, and the HIV/HCV coinfection interaction term as the primary independent predictors (P < .05) of increased marker staining (Supplementary Table 3). As expected based on prior studies, METAVIR fibrosis stage was independently associated with Hh activation markers, increased T-cell markers (activity and senescence), and increased αSMA staining. HIV infection was independently associated with Hh activation markers, increased T-cell markers (activity, recruitment, and senescence), and increased cellular aging markers. HCV infection was independently associated with Hh activation markers, increased T-cell markers (activity, recruitment, and senescence), and increased cellular aging and αSMA markers. HIV/HCV coinfection had an additive impact, above and beyond that of HIV or HCV alone, on levels of staining for SHH, CD3, CD8, CD56, and p16ink4a and trended toward significance (P < .1) for CD57 and CXCL16. In the subset of patients with either HIV or HIV/HCV, HIV viral load suppression was not associated with any of the markers.
DISCUSSION
HIV/HCV coinfection accelerates progressive liver fibrosis, yet the mechanisms remain poorly understood. The data, presented here for the first time, support a role for Hh activity in HIV-related liver disease. Furthermore, the evidence of Hh activation even in HIV-infected patients without HCV coinfection suggests a potential role for ongoing liver fibrosis after HCV eradication or a risk of fibrosis in the absence of HCV infection. There is an increasing concern for progressive fibrogenesis after HCV eradication, with a recent study suggesting that a subset of HIV-infected patients will not normalize liver enzymes after HCV eradication [14]. It is critical to gain a more comprehensive understanding of the active profibrogenic pathways in HIV/HCV coinfection and to develop serologic biomarkers of fibrogenic risk with the long-term goal of targeting these pathways for novel therapeutics.
The evidence for a role of Hh activation in human liver disease includes nonalcoholic steatohepatitis (NASH), hepatitis B and C virus infections, schistosomiasis, and hepatocellular carcinoma [12, 15]. In animal models of NASH, Hh-responsive HSCs undergo epithelial-to-mesenchymal transition into myofibroblasts, which acquire a profibrogenic phenotype [10]. A similar transition was recently reported in animal models of HIV-associated nephropathy, for the first time implicating Hh signaling in HIV-associated kidney fibrosis [9]. Hh ligands also upregulate expression of chemokines and receptors in immature liver epithelial cells, thus facilitating hepatic recruitment and retention of immune cells known to play a key role in fibrogenesis [12]. Here we confirm, in the setting of 2 chronic viral infections, that the pathologic stage of fibrosis on liver biopsy is independently associated with activation of the Hh pathway (GLI2, SHH, and PTCH). Our data also indicate that fibrosis is independently associated with T-cell populations in the liver, including NKT cells and CXCL16, a cytokine associated with NKT cell recruitment and retention in liver tissue. Lastly, we show that pathologic fibrosis stage is independently associated with T-cell terminal differentiation, a marker of chronic immune activation and senescence of the T cells in liver tissue.
We report for the first time that liver tissue from patients with HIV infection exhibits markers of liver tissue repair (Hh signaling), T-cell populations including NKT cells, and markers of cellular aging and senescence, independent of HCV coinfection. In addition, we have confirmed prior work that HCV infection is independently associated with liver tissue repair, T-cell recruitment, and markers of fibrogenesis. The additive effect of HIV and HCV coinfection was evident for a majority of the markers, suggesting that patients with HIV/HCV coinfection have a greater wound healing response to tissue injury and more severe immune dysregulation, which would be expected to translate to a greater risk of developing fibrosis.
Limitations of the current study include the use of preexisting clinical samples, which resulted in limited numbers of patients; thus these preliminary data will need to be confirmed in a larger study. In addition, the cross-sectional and retrospective nature of the study and access only to fixed formalin tissue did not allow for more extensive investigations into mechanism; thus we cannot comment on causation. Although we excluded any HIV-monoinfected patients with grossly abnormal liver tissue, there is likely selection bias in those who underwent liver biopsy in the first place because liver biopsies in HIV monoinfection are uncommon in clinical practice, and they were primarily done for elevated liver enzymes and/or abnormal imaging. To limit the potential impact of nonalcoholic steatohepatitis, we excluded HIV-infected patients with significant fibrosis, which limited our ability to assess the relationship of Hh signaling and immune activation with fibrosis in the HIV-infected cohort. Similarly, we failed to show that HIV/HCV coinfection has independently associated with the fibrogenesis marker αSMA. This was due to our study design, which matched HCV-infected and HIV/HCV-coinfected patients by severity of pathologic fibrosis stage. Further studies will be conducted to address these potential limitations.
In conclusion, this is the first study to assess the role of the Hh signaling pathway in HIV-associated liver disease. Compared with controls, and independent of HCV coinfection, patients with HIV infection had increased Hh pathway activity, increased markers of T-cell-mediated inflammation, chronic activation, and cellular aging. The majority of these markers were elevated by HIV/HCV coinfection compared with either HIV or HCV monoinfection. These data suggest that HIV accentuates HCV-related fibrogenesis pathways, specifically the canonical Hh pathway. Furthermore, the increased T-cell presence in HIV/HCV-coinfected liver tissue, increased staining for NKT cells, and increased cellular aging markers support a role for immune dysfunction that may be promoted by differential constraint on the Hh pathway noted across the different viral infection cohorts. Additional studies are needed to confirm our findings and further elucidate the causal relationship of Hh pathway activation and T-cell recruitment, retention, and exhaustion.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Prior presentations. This work was presented in part at the European Association for the Study of Liver Disease Meeting; April 2015; Vienna, Austria.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases K23-AI096913 and Duke Center for AIDS Research (AI064518) to S.N.; Department of Veteran Affairs (Ralph H Johnson VAMC, Charleston) and MUSC GI Division Start-Up Funds (MUCU-2220300-35876-6125-00) to W.S.
Potential conflicts of interest. Dr. Diehl reports receiving consulting fees from Pfizer, Novartis, Boehringer Ingelheim, Allergan, Celgene, and Lumena; participating in clinical trials with Allergan, Gilead Sciences, Conatus, Galmed, NGM Biopharmaceuticals, Bristol-Myers Squibb, Madrigal, Galectin Therapeutics, Exalenz Biosciences, Shire, Intercept, Immuron, Boehringer Ingelheim, and Genfit; receiving grant support from and participating in a clinical trial with Immuron; receiving grant support for research collaborations from Metabolon, Prometheus, and Celgene; and holding a pending patent application for “Development of Novel Therapeutics to Treat Non-Alcoholic Steatohepatitis (NASH).” Dr. Naggie reports research support in the form of clinical trials or grant funding from AbbVie, Gilead, Janssen, Bristol Meyers Squibb, Tacere, and Merck.
References
- 1. Smith CJ, Ryom L, Weber R, et al. ; D:A:D Study Group Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet 2014; 384:241–8. [DOI] [PubMed] [Google Scholar]
- 2. Thein HH, Yi Q, Dore GJ, Krahn MD. Natural history of hepatitis C virus infection in HIV-infected individuals and the impact of HIV in the era of highly active antiretroviral therapy: a meta-analysis. AIDS 2008; 22:1979–91. [DOI] [PubMed] [Google Scholar]
- 3. Lin W, Weinberg EM, Chung RT. Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J Infect Dis 2013; 207(Suppl 1):S13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Salloum S, Holmes JA, Jindal R, et al. . Exposure to human immunodeficiency virus/hepatitis C virus in hepatic and stellate cell lines reveals cooperative profibrotic transcriptional activation between viruses and cell types. Hepatology 2016; 64:1951–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guy CD, Suzuki A, Zdanowicz M, et al. ; NASH CRN Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012; 55:1711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braune J, Weyer U, Matz-Soja M, et al. . Hedgehog signalling in myeloid cells impacts on body weight, adipose tissue inflammation and glucose metabolism. Diabetologia 2017; 60:889–99. [DOI] [PubMed] [Google Scholar]
- 7. Desbois AC, Cacoub P. Diabetes mellitus, insulin resistance and hepatitis C virus infection: a contemporary review. World J Gastroenterol 2017; 23:1697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi SS, Bradrick S, Qiang G, et al. . Up-regulation of Hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology 2011; 54:1580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lan X, Wen H, Cheng K, et al. . Hedgehog pathway plays a vital role in HIV-induced epithelial-mesenchymal transition of podocyte. Exp Cell Res 2017; 352:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi SS, Omenetti A, Syn WK, Diehl AM. The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol 2011; 43:238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004; 432:324–31. [DOI] [PubMed] [Google Scholar]
- 12. Syn WK, Witek RP, Curbishley SM, et al. . Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur J Immunol 2009; 39:1879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Furler RL, Uittenbogaart CH. GLI2 regulates TGF-β1 in human CD4+ T cells: implications in cancer and HIV pathogenesis. PLoS One 2012; 7:e40874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hadigan CM, Howard L, Sheehan J, et al. . Persistent aminotransferase elevation following HCV clearance in adults with and without HIV. Paper presented at: 18th International Workshop on Co-morbidities and Adverse Drug Reactions in HIV; 12–13 September 2016; New York, NY. [Google Scholar]
- 15. Pereira Tde A, Witek RP, Syn WK, et al. . Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab Invest 2010; 90:1690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.