


Abstract
There is a pressing need for in silico tools that can aid in the identification of the complete repertoire of protein binding (SLiMs, MoRFs, miniMotifs) and modification (moiety attachment/removal, isomerization, cleavage) motifs. We have created PSSMSearch, an interactive web-based tool for rapid statistical modeling, visualization, discovery and annotation of protein motif specificity determinants to discover novel motifs in a proteome-wide manner. PSSMSearch analyses proteomes for regions with significant similarity to a motif specificity determinant model built from a set of aligned motif-containing peptides. Multiple scoring methods are available to build a position-specific scoring matrix (PSSM) describing the motif specificity determinant model. This model can then be modified by a user to add prior knowledge of specificity determinants through an interactive PSSM heatmap. PSSMSearch includes a statistical framework to calculate the significance of specificity determinant model matches against a proteome of interest. PSSMSearch also includes the SLiMSearch framework’s annotation, motif functional analysis and filtering tools to highlight relevant discriminatory information. Additional tools to annotate statistically significant shared keywords and GO terms, or experimental evidence of interaction with a motif-recognizing protein have been added. Finally, PSSM-based conservation metrics have been created for taxonomic range analyses. The PSSMSearch web server is available at http://slim.ucd.ie/pssmsearch/.
INTRODUCTION
Many functions of a protein are mediated by compact and degenerate motifs that act as binding or post-translational modification sites (1,2). Protein binding motifs, often described as short linear motifs (SLiMs), molecular recognition features (MoRFs) or miniMotifs, promote transient complex formation or mediate regulatory interactions modulating the subcellular localization, half-life, activity or modification state of a protein (2–5). Modification motifs directly encode the modification state of a protein by acting as specificity determinants recognized by enzymes resulting in protein post-translational modification of the motif-containing peptide (1,6). A large number of enzymes have strong preferences for specific motifs and motifs direct a significant proportion of the moiety attachment/removal, isomerization and proteolytic cleavage in the cell (1,2). Protein motifs are a ubiquitous and elusive class of protein module. The human proteome has been estimated to contain tens of thousands of binding motifs and up to a million modification motifs (7). However, to date, only a small fraction of these functional modules has been discovered and subsequently experimentally characterized (1). This can largely be attributed to the experimental and computational difficulty of motif discovery and characterization, and the condition-dependent transient nature of the majority of the interactions mediated by these sites (5,8–10).
Functional motifs often have strict preferences at certain positions where only a single amino acid or a limited subset of physicochemically similar amino acids are allowed. Residues at these positions generally contact the motif-recognizing pocket directly and the preferences reflect the requirement for complementarity with the physicochemical properties and organization of the pocket. Alternatively, solvent-facing positions are usually highly variable, although amino acids that are structurally incompatible with the bound conformation, such as prolines in a motif that binds as a helix, will often be disfavored (2). These site-specific preferences can generally be determined by analyzing the sequence similarity of experimentally characterized instances of peptides known to bind a pocket to create a specificity determinant model of that pocket (1). The model can then be used to screen proteins or proteomes to discover novel protein regions complementary to the binding preferences of the pocket (1,11–14). Two major approaches have been applied to discover novel motifs based on a priori knowledge of motif specificity determinants, regular expressions (RegExs) and position-specific scoring matrices (PSSMs also known as position weight matrices or PWMs) searches (15–17). Both methods have major advantages and disadvantages, and both have been used to successfully discover novel functional motifs (13,18). The simple representations of a RegEx are easy to create, read, edit and search. However, the RegEx approach finds only exact matches and treats all matches as equal. Consequently, when sufficient information is available, PSSMs can be more powerful in motif discovery as a PSSM encodes information on weighted observations of residues at each position and score all matches with a measure of similarity to the set of validated motif-containing peptides. First, this allows peptides to be ranked, and secondly, this allows functional motifs which are highly similar to a well-characterized consensus but do not conform to a RegEx to be discovered. One major drawback of PSSMs over RegExs remains: the fixed length of a PSSM does not allow length flexibility in motif searches. However, this can be solved using multiple PSSMs of different lengths or performing scoring and scanning that can allow for gaps. Several web servers have been created to discover novel motif instances based on a priori motif information (Supplementary Table S1). Many of these servers allow the user to search a protein/proteome with a set of predefined functional motif RegExs (ELM (1), QuasiMotiFinder (19), MiniMotifMiner (20), ScanSite (6)) or PSSMs (ScanProsite (21), ScanSite (6)). Other servers allow the user to search a protein/proteome with user-defined RegExs (SLiMSearch (14), SiRW (22), DoReMi (23) and ScanProsite (21)) or PSSMs (DoReMi (23), MEME (24), PoSSuMSearch (25), MOTIPS (26) and SMALI (27)).
In this article we introduce PSSMSearch, a novel interactive web-based tool that takes a set of known functional motifs, defines the preferences of a motif-recognizing pocket, and uses that information to search for novel regions of the proteome matching the preferences of the pocket. PSSMSearch is a web server for the PSSM-based motif discovery framework developed to discover human and viral docking motifs for the human phosphatase PP2AB56 and validated in Hertz et al. (13). The PSSMSearch framework consists of tools for rapid statistical modeling, visualization, discovery and annotation of protein motif specificity determinants. The framework has several novel and unique features that separate it from the currently available PSSM-based motif discovery tools (See Supplementary Table S1). PSSMSearch includes automated construction of a motif specificity determinant model using one of several available PSSM scoring methods (Table 1) and a custom-made interactive PSSM visualization that permits the manipulation of the created PSSM to add a priori experimental information to the specificity determinant model search. A PSSM-based conservation metric calculates the motif taxonomic range of the motif based on the specificity determinant model. Integration with the SLiMSearch annotation framework adds feature annotation, peptide accessibility and conservation calculations, functional enrichment analysis and advanced filtering of the discovered instances. Finally, two novel motif-recognizing protein data analysis tools give information that can be used to rapidly focus on high confidence novel motifs. Annotation of previously described interactions highlights motifs in proteins with experimental evidence of an interaction between a motif-containing protein and a motif-recognizing protein(s). The calculation of significant shared annotations with a hub protein(s) highlights motifs in proteins with shared function or localization with the analyzed motif-recognizing protein(s).
Table 1. Table of scoring schemes used in the tool.
| Scheme | Description | Reference |
|---|---|---|
| PSI-BLAST | An adapted version of the PSI-BLAST algorithm with corrected background amino acid frequencies. | (28) |
| PSI-BLAST IC | PSSM is obtained identically as from PSI-BLAST method, and then the PSSM scores are adjusted with information content. | (28) |
| MOTIPS | An adapted version of the MOTIPS method to generate PSSM and score peptides. | (26) |
| Log Odds | Binomial statistics of residue frequencies with respect to background amino acid probabilities. | (29) |
| Log Relative Binomial | Log of the binomial cumulative function with over- or under-representation of residues at each position. | |
| Ratio | Amino acid frequencies corrected by background amino acids probabilities. | |
| Frequency | Simple amino acid frequencies. Used only for visualization. | |
| Count | Amino acid counts. Used only for visualization. |
MATERIALS AND METHODS
PSSMSearch is a framework which comprises three major parts: motif specificity determinant model construction; motif discovery and scoring; and motif annotation, filtering and analysis (Figure 1A). First, the motif specificity determinant model construction component creates a PSSM and a motif consensus from an aligned set of peptides or ELM instances, presents the PSSM as a heatmap/logo and allows interactive PSSM manipulation by a user based on a priori knowledge of motif specificity determinants (Figure 1B). Second, the generated PSSM is used in motif scanning and scoring to find PSSM matches and calculate their likelihood. Third, the motif annotation, filtering and analysis framework introduced with the SLiMSearch tool (14) allows the user to construct a high confidence set of putative motif instances for further validation.
Figure 1.

(A) PSSMSearch workflow highlighting the major tools in the framework colored by the component: motif specificity determinant model construction (green); motif discovery and scoring (blue); and motif annotation, filtering and analysis (red). (B) A screenshot of a portion of the output of the interactive PSSM visualization tool of the default PSSMSearch input example, the Calcineurin (PP2B)-docking PxIxIT motif. The calculated PSSM is visualized with a heatmap (with blue to red coloring for enriched to depleted) and logo (Clustal coloring) using aligned PxIxIT peptides retrieved from the ELM database (DOC_PP2B_PxIxI_1). The PSSM was derived using the PSI-BLAST IC method with the default background distribution (human, disorder cut-off 0.5). A priori knowledge of a PxIx[IV] consensus was manually added using the interactivity feature to restrict the residues at the positions 3, 5 and 7 to be proline, isoleucine and isoleucine or valine, respectively. We also prohibited the presence of prolines at the position 4–8. PSSM values at the position 9 were set to zero to allow any residue without preference. (C) Comparison of the distinct properties of different PSSM scoring schemes highlighting the variability in the contribution of different positions in different schemes.
Motif specificity determinant model construction
PSSMSearch constructs a PSSM and a RegEx consensus describing the specificity determinants from a set of aligned user-defined peptides or ELM motif instances. Subsequently, the PSSM is used to find putative novel motif instances by scoring regions of a proteome of interest for similarity to the set of input peptides, and the RegEx consensus is used in conservation score calculations and taxonomic range analysis.
PSSM construction
Numerous statistical and empirical approaches to PSSM creation exist (26,28–29) (Table 1). No PSSM scoring scheme is definable superior in all situations as each scheme has distinct properties (Figure 1B and C). For example, some PSSM scoring schemes, such as Log Odds, weight observed residues in motif positions strongly and will not find peptides that diverge from the consensus. Others, such as PSI-BLAST, will have an increased contribution from the non-consensus flanking residues, as the flanking position of the motif outside of the consensus position include key positive and negative discriminatory information. Some representations encode information about anti-motifs, residues that are negative determinants of binding and down weight such residues. Depending on the search, or the number of instances used to create the PSSM, the user may wish to emphasize the importance of different features of the motif. Consequently, several distinct PSSM construction methods are available in PSSMSearch including the PSI-BLAST (28), MOTIPS (26), Log Odds (29), Log Relative Binomial and Ratio scoring schemes. The PSSM construction is performed based on pre-computed background amino acid probabilities, for supported species and disorder cut-offs, to generate the PSSM based on identical amino acid composition as the PSSM search space. Frequency and counts are also provided to allow a user to observe the raw peptide data. See Supplementary Material for a detailed description of PSSM construction.
PSSM visualization and interactive manipulation
As a result of their complex numeric format, PSSMs are more difficult to understand than RegExs. Furthermore, the integration of a priori experimental knowledge regarding allowed or disallowed amino acids into a PSSM remains complicated. Visualization tools that can represent PSSMs as interactive modifiable human readable sequence logos and heatmaps can overcome both these challenges. A novel PSSM visualization tool, combining a sequence logo and a heatmap has been developed for PSSMSearch (see Figure 1B). The visualization tool allows a user to interactively manipulate the PSSM to add a priori information into a specificity determinant model search (e.g. disallowed amino acids, required amino acids, etc. at each position). The visualization also allows the residues in a column to be grouped and all residues in the group to be scored with either the maximum score or the sum of the scores for the group. This allows the contribution of a physicochemically similar set of residues (e.g. aspartate and glutamate) at a position to be up-weighted. Finally, if required, a given position (column) in the PSSM can be ignored.
PSSM-based RegEx consensus definition
In addition to the PSSM, a RegEx consensus describing the specificity determinants is constructed from the set of aligned user-defined peptides. The motif RegEx consensus defines the significantly enriched residues at every position of the peptide alignment and describes the key affinity and specificity determining residues in the motif. The RegEx consensus is computed based on the position-specific probabilities of amino acid occurrences and corrected by residue frequencies to avoid over- or under-defined residues in consensus representation (see Supplementary Material). This consensus is not used in the search component of the framework and is only used in conservation score calculations and taxonomic range analysis.
Motif discovery and scoring
In the motif discovery and scoring component, the PSSM created by the motif specificity determinant model construction tool (except for Frequency and Counts method) is used to discover matches to the motif specificity determinant model on a proteome-wide level and empirically calculate a background distribution of PSSM scores.
PSSM-based proteome scanning
The scanning of the proteome with the PSSM allows regions of the proteome that are similar to the set of input peptides to be discovered. In theory, the more similar a peptide is to the known experimentally validated functional motifs, the higher the likelihood that the peptide contains an instance of the motif. The target proteome is scanned with the PSSM using a sliding-window-based method. Peptides obtained from scanning are scored with the PSSM. With the exception of the MOTIPS method, the score of a peptide is calculated as the sum of the scores assigned to the observed residue at each position of the peptide based on the PSSM (see Supplementary Material). The MOTIPS PSSM score is calculated differently and the scoring method is adopted from Lam et al. (26). There is a fixed penalty (−3) when the PSSM overhangs the N- and C-termini of the protein. Non-standard amino acids are excluded from scoring.
PSSM score P-value calculation
PSSMSearch empirically calculates the probability of a specificity determinant model match with a given PSSM score at a given position by chance. It uses an observed distribution of PSSM scores of a reversed (default option) or randomly shuffled PSSM against the selected proteome. The background distribution of PSSM scores of the reversed or shuffled PSSM is calculated identically to the PSSM-based proteome scanning as described above. The P-value is then calculated based on this observed distribution of reversed or shuffled PSSM scores (Equation 1). The number of samples used to calculate the background P-value distribution is exactly the same size as the input PSSM search space, thus is approximately equal to the number of amino acids in the proteome search. Peptide matches with a higher PSSM score than any observed in the background model are highlighted with a green bolded PSSM P-value scores. Creating the background P-value distribution against the selected proteome rather than using non-proteome randomly generated peptides removes biases introduced by differing amino acid compositions. The statistic does assume that the reversed or shuffled PSSM will not describe an enriched or depleted sequence feature of the proteome and that there are no dependencies between adjacent residues. The PSSM score P-value and the PSSM score will rank the returned peptides identically. However, the PSSM score is highly variable and correlates with PSSM length, the degeneracy of the motif and the PSSM scoring scheme used. The P-value has no such biases as the P-value defines the likelihood of seeing a given PSSM match with a given PSSM score at a single position. Consequently, the P-value scoring scheme allows an equivalent score cut-off to be applied to all analyses.
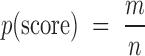 |
Equation 1. PSSM score P-value calculation. m is the number of peptides in the proteome of interest with a PSSM score better than a certain cut-off (score) when scored with a reversed/shuffled PSSM. This cut-off is a maximum score for MOTIPS or a minimum score for other methods. n is the total number of peptides that were used to calculate the background P-value distribution.
Motif annotation, filtering and analysis
PSSMSearch uses the peptide annotation, filtering and analysis framework developed for the SLiMSearch tool to annotate specificity determinant model matches with pertinent biological data, calculate conservation scores, perform functional analyses and filter data. A description of the annotated data including the source of data and calculation of peptide metrics is available in the SLiMSearch paper (14) and provided on the detailed PSSMSearch help page (http://slim.ucd.ie/pssmsearch/help/). Two major novel additions to the peptide annotation, filtering and analysis framework for PSSMSearch are described below.
Interaction, function and co-localization analysis with hub proteins
PSSMSearch allows a user to select a single or a set of motif-recognizing protein(s) as an optional input parameter. This allows PSSMSearch to highlight peptides in proteins with experimental evidence of an interaction between a motif instance-containing protein and the motif-recognizing protein(s), with functional similarity to the motif-recognizing protein(s), or that co-localize with the motif-recognizing protein(s). Experimentally characterized interactions of the selected motif-recognizing protein(s) with the motif instance-containing protein are annotated using information from the HIPPIE database (30). Significant shared annotations between an instance-containing protein and the selected motif-recognizing protein(s) are calculated for each putative motif. This highlights peptides in proteins with shared function or localization with the selected motif-recognizing protein that are unlikely to occur by chance (see Supplementary Material). Both significant shared annotation and interaction information can be used to filter motif instances. For example, by retaining only peptide matches in proteins with experimental evidence of an interaction with the user selected motif-recognizing protein(s).
PSSM-based taxonomic range calculations
Conservation of a motif over a large taxonomic range, the presence of a motif match at the same sequence position in homologs in many divergent species, is a strong positive discriminator of motif function (31–35). PSSMSearch provides a novel extension of the SLiMSearch taxonomic range analysis (14). The PSSMSearch taxonomic range analysis calculates motif taxonomic range based on a searched specificity determinant model using PSSM scoring (PSSM method), matches to a motif consensus (consensus method) or combination of both criteria (combined method). When the PSSM method is used to calculate taxonomic range, the region of an ortholog alignment corresponding to a specificity determinant model match in each protein is scored using the PSSM. The motif is defined as conserved or non-conserved based on the selected conservation P-value cut-off. The RegEx consensus method is the default approach used for the taxonomic range calculations. The RegEx motif consensus is automatically derived from the PSSM as defined above; however, the RegEx consensus can be modified by the user in the input options. PSSM matches can be filtered by taxonomic range (e.g. retaining matches in human conserved outside mammals) or by their presence/absence in a specific species or clade (e.g. retaining matches in human conserved also in Fly or Arthropods). No taxonomic range filtering is applied by default.
Example analysis
The Calcineurin phosphatase holoenzyme (Serine/threonine-protein phosphatase 2B catalytic subunit alpha and Calcineurin subunit B type 1) recruits several substrates with a binding pocket that recognizes the Calcineurin-docking PxIxIT motif (1). Figure 1B and C show specificity determinant models of the Calcineurin (PP2B)-docking PxIxIT- motif binding pocket built using ten experimentally validated PxIxIT motifs from Calcineurin substrates (VTPIISIQE, RLPVIAVND, QVPNIYIQT, PSPRIEITP, KKPKIIITG, KKPKIIITG, GVPRITISD, ESPRIEITS, ESPRIEITS, AIPQIVIDA) (1). This specificity determinant model can be searched against the human proteome and filtered by significant shared annotations to create a list of putative novel PxIxIT motifs (Figure 2). This list includes known PxIxIT containing proteins such as the Nuclear Factor of Activated T-cells (NFAT) proteins and several high scoring novel hits in proteins that are functionally related to the cellular role of Calcineurin. Further, analysis and filtering methods can be used to refine the list of putative motifs.
Figure 2.

Selected regions of the PSSMSearch output showing the results of the PxIxIT motif analysis in Figure 1. The PSSMSearch analysis was performed on the human proteome with taxonomic range method based on PSSM and using the two subunits of the Calcineurin Holoenzyme as the motif-binding partners. Other options were set as default. Instances were filtered based on shared significant annotations with motif-binding partners. (A) Extract of the ranked and filtered instances from the PSSMSearch of the Calcineurin (PP2B)-docking motif PxIxI against the human proteome showing significant shared annotation and previously validated interactions between the motif-containing proteins and the Calcineurin Holoenzyme (i, ii and iii). PSSM P-value scores (iv); Relative local conservation scores for the motif - a tree weighted conservation score that quantifies the conservation of a region compared to the conservation of the surrounding regions (v); Intrinsic Disorder Scores - the mean IUPred disorder score across the peptide match (vi); and warnings showing motifs with attributes or overlapping features that are associated with non-functional matches are also highlighted (vii). (B) Taxonomic range analysis of a PSSM match in Nuclear factor of activated T-cells, cytoplasmic 2 based on the PSSM method. The green dotted box is added to highlight the lack of the lack of the PxIxIT motif in Strongylocentrotus purpuratus and Branchiostoma floridae. (C) ProViz visualization (36) (linked upon clicking a peptide in any PSSMSearch view) of the Nuclear factor of activated T cells, cytoplasmic 2 peptide from Panel B highlighting the lack of conservation of the PxIxIT motif in Strongylocentrotus purpuratus and Branchiostoma floridae in the alignment (Alignment pruned to remove extra species for clarity). A detailed description of the PSSMSearch output, the source of data and the calculation of peptide metrics is provided on the help page (http://slim.ucd.ie/pssmsearch/help/).
Implementation
The PSSMSearch interface is written in JavaScript using the ReactJs (input page, heatmap and logo visualization) and Ember (results, filtering and analysis pages) frameworks. The server side is written as a Python Cornice Web Framework with a GO component for rapid searching and scoring of PSSM matches. A Torque management system is used to queue submitted jobs. Data for peptide annotation is stored in a PostgreSQL database. Results can be downloaded in JSON or tab separated format, or accessed using a job identifier at a later date. Results are stored for two weeks before deletion. A detailed description of PSSMSearch usage and output is provided on the help page (http://slim.ucd.ie/pssmsearch/help/). PSSMSearch has been successfully tested on all major modern browsers.
DISCUSSION
Motif-mediated binding and post-translational modification events are central to a wide range of processes in the cell (1,2). These compact functional modules are key determinants of the subcellular localization, half-life, complex association, activity and modification state of a protein (2,7). Yet, the vast majority of these functional modules remain to be discovered as the field still lacks a set of high quality experimental and in silico methods for high-throughput discovery of peptide motifs (1,7). The PSSMSearch framework will assist biologists designing experiments to discover the remaining functional modules by allowing users with limited bioinformatics skills to discover putative functional protein regions. Compared to previously published web servers (Supplementary Table S1), PSSMSearch is more flexible due to the range of PSSM construction methods provided to build a motif specificity determinant model and the option to encode prior knowledge into the model through an intuitive interactive interface. Additionally, PSSMSearch includes a statistical framework that can rapidly calculate the P-value for each PSSM match based on an empirical PSSM score distribution obtained from reverse or shuffle PSSM scanning. Finally, PSSMSearch uses the powerful SLiMSearch framework (14) for motif annotation, filtering and analysis, which offers more functionality than other servers by integrating biological and evolutionary data into the analysis. However, PSSMSearch significantly extends the functionality of SLiMSearch (14), transitioning to PSSM-based searches from a consensus-based method, annotating motif instances with the significant shared functional annotations with a user-specified motif-binding partner(s) and including a PSSM-based conservation metric for motif taxonomic range calculations.
PSSMSearch currently requires pre-aligned peptides for PSSM construction. Aligning motifs within peptides is a complicated task, especially in situations where the motif has low complexity, the motif has flexible length, the motif is not present in all peptides or there are two or more distinct motif consensuses binding to the same binding partner in the set of input peptides. In future versions of PSSMSearch, we hope to align peptides automatically, removing this responsibility from the user. Numerous multiple sequence alignment tools are available and most can align peptides. Furthermore, powerful tools such as GibbsCluster (37) and MUSI (38) can do so even when multiple specificities are present. The quality of the input alignment has a strong influence on the quality of the PSSM created, and subsequently, the peptides matched. An important issue for many PSSM creation methods is the level of redundancy in the input data. Of the scoring schemes available in PSSMSearch, only the PSI-BLAST scoring schemes down weight the contribution of closely related sequences. This distinct attribute of the scoring scheme is very useful when multiple similar peptides are present in the input dataset. The remaining scoring schemes do not weight peptides and the input alignment needs to be manually cleaned to remove such biases. Finally, PSSMSearch is not currently optimized to deal with alignments of motifs that have flexible length. They can be encoded in a single PSSM when the core can be aligned. However, much of the information of the positional enrichment outside the core is lost. If sufficient information is available, multiple PSSMs can be supplied to PSSMSearch with differing lengths; however, this is not an ideal solution. One of the future goals of the project will be to understand how we can perform searches on gapped PSSMs rapidly and within the current statistical framework.
In conclusion, PSSMSearch was created to improve the understanding of the physiological and pathological processes of the cell at the molecular level. The tool has already successfully characterized novel motifs in the PP2A interactome (13). We believe that PSSMSearch will be an important tool in the quest to tackle the challenging task of characterizing motifs that are central to cell physiology.
DATA AVAILABILITY
PSSMSearch is available at http://slim.ucd.ie/pssmsearch/.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our collaborators and colleagues for their testing of the PSSMSearch tool. We thank Malvika Sharan, Denis Shields, Nicolás Palopoli and Leandro Simonetti for their fruitful discussions and critically reading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Science Foundation Ireland Starting Investigator Research Grant [13/SIRG/2193 to I.K., N.E.D.]; Enterprise Ireland grant to Food for Health Ireland [TC2013001 to J.M.]. Funding for open access charge: Science Foundation Ireland.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gouw M., Michael S., Samano-Sanchez H., Kumar M., Zeke A., Lang B., Bely B., Chemes L.B., Davey N.E., Deng Z. et al. The eukaryotic linear motif resource - 2018 update. Nucleic Acids Res. 2018; 46:D428–D434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Van Roey K., Uyar B., Weatheritt R.J., Dinkel H., Seiler M., Budd A., Gibson T.J., Davey N.E.. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014; 114:6733–6778. [DOI] [PubMed] [Google Scholar]
- 3. Davey N.E., Morgan D.O.. Building a regulatory network with short linear sequence motifs: lessons from the degrons of the Anaphase-Promoting complex. Mol. Cell. 2016; 64:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mészáros B., Kumar M., Gibson T.J., Uyar B., Dosztányi Z.. Degrons in cancer. Sci. Signal. 2017; 10:eaak9982. [DOI] [PubMed] [Google Scholar]
- 5. Van Roey K., Gibson T.J., Davey N.E.. Motif switches: decision-making in cell regulation. Curr. Opin. Struct. Biol. 2012; 22:378–385. [DOI] [PubMed] [Google Scholar]
- 6. Obenauer J.C., Cantley L.C., Yaffe M.B.. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003; 31:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tompa P., Davey N.E., Gibson T.J., Babu M.M.. A million peptide motifs for the molecular biologist. Mol. Cell. 2014; 55:161–169. [DOI] [PubMed] [Google Scholar]
- 8. Gibson T.J. Cell regulation: determined to signal discrete cooperation. Trends Biochem. Sci. 2009; 34:471–482. [DOI] [PubMed] [Google Scholar]
- 9. Gibson T.J., Dinkel H., Van Roey K., Diella F.. Experimental detection of short regulatory motifs in eukaryotic proteins: tips for good practice as well as for bad. Cell Commun. Signal. 2015; 13:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Roey K., Dinkel H., Weatheritt R.J., Gibson T.J., Davey N.E.. The switches.ELM resource: a compendium of conditional regulatory interaction interfaces. Sci. Signal. 2013; 6:rs7. [DOI] [PubMed] [Google Scholar]
- 11. Di Fiore B., Wurzenberger C., Davey N.E., Pines J.. The mitotic checkpoint complex requires an evolutionary conserved cassette to bind and inhibit active APC/C. Mol. Cell. 2016; 64:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gouw M., Sámano-Sánchez H., Van Roey K., Diella F., Gibson T.J., Dinkel H.. Exploring short linear motifs using the ELM database and tools. Curr. Protoc. Bioinformatics. 2017; 58:8.22.21–28.22.35. [DOI] [PubMed] [Google Scholar]
- 13. Hertz E.P.T., Kruse T., Davey N.E., López-Méndez B., Sigurðsson J.O., Montoya G., Olsen J.V., Nilsson J.. A conserved motif provides binding specificity to the PP2A-B56 phosphatase. Mol. Cell. 2016; 63:686–695. [DOI] [PubMed] [Google Scholar]
- 14. Krystkowiak I., Davey N.E.. SLiMSearch: a framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 2017; 45:W464–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aasland R., Abrams C., Ampe C., Ball L.J., Bedford M.T., Cesareni G., Gimona M., Hurley J.H., Jarchau T., Lehto V.P. et al. Normalization of nomenclature for peptide motifs as ligands of modular protein domains. FEBS Lett. 2002; 513:141–144. [DOI] [PubMed] [Google Scholar]
- 16. Boeva V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front. Genet. 2016; 7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000; 16:16–23. [DOI] [PubMed] [Google Scholar]
- 18. Di Fiore B., Davey N.E., Hagting A., Izawa D., Mansfeld J., Gibson T.J., Pines J.. The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev. Cell. 2015; 32:358–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gutman R., Berezin C., Wollman R., Rosenberg Y., Ben-Tal N.. QuasiMotiFinder: protein annotation by searching for evolutionarily conserved motif-like patterns. Nucleic Acids Res. 2005; 33:W255–W261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lyon K.F., Cai X., Young R.J., Mamun A.A., Rajasekaran S., Schiller M.R.. Minimotif Miner 4: a million peptide minimotifs and counting. Nucleic Acids Res. 2018; 46:D465–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Castro E., Sigrist C.J., Gattiker A., Bulliard V., Langendijk-Genevaux P.S., Gasteiger E., Bairoch A., Hulo N.. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006; 34:W362–W365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramu C. SIRW: A web server for the Simple Indexing and Retrieval System that combines sequence motif searches with keyword searches. Nucleic Acids Res. 2003; 31:3771–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horn H., Haslam N., Jensen L.J.. DoReMi: context-based prioritization of linear motif matches. PeerJ. 2014; 2:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beckstette M., Homann R., Giegerich R., Kurtz S.. Fast index based algorithms and software for matching position specific scoring matrices. BMC Bioinformatics. 2006; 7:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lam H.Y., Kim P.M., Mok J., Tonikian R., Sidhu S.S., Turk B.E., Snyder M., Gerstein M.B.. MOTIPS: automated motif analysis for predicting targets of modular protein domains. BMC Bioinformatics. 2010; 11:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L., Wu C., Huang H., Zhang K., Gan J., Li S.S.. Prediction of phosphotyrosine signaling networks using a scoring matrix-assisted ligand identification approach. Nucleic Acids Res. 2008; 36:3263–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Shea J.P., Chou M.F., Quader S.A., Ryan J.K., Church G.M., Schwartz D.. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods. 2013; 10:1211–1212. [DOI] [PubMed] [Google Scholar]
- 30. Alanis-Lobato G., Andrade-Navarro M.A., Schaefer M.H.. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017; 45:D408–D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chica C., Diella F., Gibson T.J.. Evidence for the concerted evolution between short linear protein motifs and their flanking regions. PLoS One. 2009; 4:e6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davey N.E., Cowan J.L., Shields D.C., Gibson T.J., Coldwell M.J., Edwards R.J.. SLiMPrints: conservation-based discovery of functional motif fingerprints in intrinsically disordered protein regions. Nucleic Acids Res. 2012; 40:10628–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davey N.E., Cyert M.S., Moses A.M.. Short linear motifs - ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015; 13:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davey N.E., Shields D.C., Edwards R.J.. Masking residues using context-specific evolutionary conservation significantly improves short linear motif discovery. Bioinformatics. 2009; 25:443–450. [DOI] [PubMed] [Google Scholar]
- 35. Nguyen Ba A.N., Yeh B.J., van Dyk D., Davidson A.R., Andrews B.J., Weiss E.L., Moses A.M.. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012; 5:rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jehl P., Manguy J., Shields D.C., Higgins D.G., Davey N.E.. ProViz-a web-based visualization tool to investigate the functional and evolutionary features of protein sequences. Nucleic Acids Res. 2016; 44:W11–W15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andreatta M., Alvarez B., Nielsen M.. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017; 45:W458–W463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim T., Tyndel M.S., Huang H., Sidhu S.S., Bader G.D., Gfeller D., Kim P.M.. MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets. Nucleic Acids Res. 2012; 40:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PSSMSearch is available at http://slim.ucd.ie/pssmsearch/.