


Abstract
Atomistic molecular dynamics (MD) simulations generate a wealth of information related to the dynamics of proteins. If properly analyzed, this information can lead to new insights regarding protein function and assist wet-lab experiments. Aiming to identify interactions between individual amino acid residues and the role played by each in the context of MD simulations, we present a stand-alone software called gRINN (get Residue Interaction eNergies and Networks). gRINN features graphical user interfaces (GUIs) and a command-line interface for generating and analyzing pairwise residue interaction energies and energy correlations from protein MD simulation trajectories. gRINN utilizes the features of NAMD or GROMACS MD simulation packages and automatizes the steps necessary to extract residue-residue interaction energies from user-supplied simulation trajectories, greatly simplifying the analysis for the end-user. A GUI, including an embedded molecular viewer, is provided for visualization of interaction energy time-series, distributions, an interaction energy matrix, interaction energy correlations and a residue correlation matrix. gRINN additionally offers construction and analysis of Protein Energy Networks, providing residue-based metrics such as degrees, betweenness-centralities, closeness centralities as well as shortest path analysis. gRINN is free and open to all users without login requirement at http://grinn.readthedocs.io.
INTRODUCTION
Atomistic molecular dynamics (MD) simulation is a popular tool for characterizing the dynamic behavior and function of biomolecules, including proteins (1–4). The main output of an MD simulation is a trajectory, typically including several thousands of conformations of a biomolecular system. It is the analysis of this data, not the actual simulation itself, which is the most complicated part of a study involving MD simulations. As such, obtaining comprehendible conclusions from such multidimensional data may be a time-consuming task and require the use of extensive and sophisticated analysis methods and procedures (5).
An interesting output of a MD simulation analysis procedure is the relative importance of individual amino acids—as building blocks—in determining the dynamics and thus the function and activity of a protein (6–8). However, detecting the role of a single residue from high amount of simulation data is far from being straightforward. In order to gain information on residue level, pairwise non-bonded interaction energies between amino acid residues can be calculated either by using a single conformation or an ensemble of conformations (such as one obtained from a MD simulation). Example applications include: identification of stabilizing amino-acids, their interactions in protein structures and construction of Interaction Energy Matrices (IEMs) (9,10), identification of ligand- or mutation-induced changes in amino-acid interactions throughout the protein structure (11,12), construction of interaction energy-based Protein Energy Networks (PENs) to identify residues playing important roles in dynamics (13–17) and stability (18), allosteric pathway identification (13) and calculation of equal-time correlations of pairwise residue interaction energies to identify side-chain dependent dynamic cross-talk between residues (19,20).
Protein Structure Networks (PSNs) denote the application of network theory to protein structures (21–28). PENs can be considered as special types of ‘Protein Structure Networks’ (PSNs) where pair interaction energies between individual residues are used to construct IEMs that represent the ‘strength’ of edges between nodes (residues) in the network structure (13–16,29,30). These strength values can be normalized to represent the ‘weight’ or ‘cost of information transfer’ from one node to another to be used in further network analysis tasks such as shortest path identification. Although there are a variety of online and stand-alone tools for constructing PSNs from single protein structures, such as NAPS (31), WebPSN (32), Bio3D (33), xPyder (34) and RINalyzer (35–37), options for direct PEN construction from structural ensembles or MD simulation snapshots are limited. Two relevant stand-alone programs, PSN-Ensemble (38) and pyInteraph, (39) can construct and/or utilize an IEM to define edge weights, however their use is limited: PSN-Ensemble requires user-supplied IEMs and depends on a MATLAB installation whereas pyInteraph employs a statistical pair-potential for interaction energies which can lead to a reduced accuracy when compared to a force-field based computation in special cases.
Two online research tools construct force-field based IEMs and/or utilize the information contained within. The INTAA web-server offers quick calculation and visualization of pairwise residue interaction energies and construction of an IEM (40). The server, however, operates on single conformations and is therefore not directly suitable for calculations on a conformational ensemble. Another server, Molecular Dynamics Network (MDN) (41) was reported as being able to perform pairwise interaction energy calculations using MD simulation data with subsequent construction and analysis of PENs, however the service seems to be currently unavailable.
Alternatively, popular MD simulation packages feature between-groups interaction energy calculations via decomposing the non-bonded components of the potential energy into interaction elements without evaluating the equations of motion; however, the user still needs to prepare customized scripts for his/her own simulation data. For example, NAMD (42) requires custom configuration files in addition to custom PDB files that identify the interacting atom groups. GROMACS (43), likewise, requires custom index files to perform such a computation. Furthermore, the size of the interaction energy data can be dramatically high for an analysis procedure (on the gigabyte scale), depending on the size of the protein and simulation trajectory. This makes a preliminary filtering out of non-interacting residue pairs in the structure necessary. Finally, the resulting data still needs to be parsed and analyzed further, requiring some additional scripting effort. Hence, the whole workflow can quickly become a cumbersome and a time-consuming task for researchers.
We developed gRINN (get Residue Interaction eNergies and Networks) as an easy-to-use stand-alone software for practical generation and analysis of pairwise residue interaction energy data from protein MD simulation trajectories. gRINN supports simulation trajectories generated by GROMACS or NAMD/CHARMM software. The aforementioned scripting tasks for residue interaction energy calculations with NAMD/GROMACS are completely automated, allowing researchers to focus rather on their actual research question. In addition to residue interaction energies, gRINN can compute correlations between the interaction energy series as well. The resulting data, either in tab-separated or comma-separated format, are stored in a user-specified folder. gRINN offers a rich visualization interface for inspecting the output. Resulting Interaction Energy Matrix (IEM) is used to construct and analyze a PEN in terms of residue-based network metrics such as degrees, betweenness-centralities and closeness-centralities as well as short paths between selected residues in the structure.
MATERIALS AND METHODS
Dependencies
gRINN computes pairwise residue interaction energies by performing repeated calls to the MD simulation executable. gRINN supports NAMD/CHARMM or GROMACS-generated data; hence either NAMD or GROMACS should be pre-installed on the system.
Workflow
A typical workflow of gRINN is presented in Figure 1. gRINN is designated for post-simulation analysis, hence it is targeted for users who have already completed a MD simulation of a protein using either NAMD or GROMACS. Files that describe the protein structure, topology, and an ensemble of structures (trajectory) are required. Depending on the used software, the path of NAMD2 or GMX executable should be provided as well. For NAMD data, it is highly recommended to delete the solvent molecules from all input Protein Structure (PSF), Protein Data Bank (PDB) and DCD files prior the usage of gRINN. Otherwise, a very high amount of computer memory will be required by gRINN for processing the trajectory file, particularly if the user chooses to use multiple CPU cores. Output Folder is specified by the user. This folder must be a path that does not exist prior to gRINN usage. gRINN creates this folder and stores the results in it. For NAMD data, the path of the parameter file should also be provided by the user.
Figure 1.

Overall workflow of gRINN.
In addition to these input files, the user can specify several calculation settings. Two selection strings (Selection 1 and Selection 2), describing the two residue groups between which non-bonded interaction energies are to be computed, can be defined by the user. The atom selection syntax as implemented by ProDy package is used here (44). Note that this setting is only useful when a pairwise interaction energy characterization between specific subsets of the protein structure is desired. In most cases, leaving these two selections at the default value of ‘all’ is advised. Percent cutoff and Filtering distance cutoff settings allow further control over which residue pairs are included in the interaction energy computation. Default values of 60% cutoff and 20 Å filtering distance cutoff imply that only those pairs of residues whose centers-of-mass come closer than 20 Å in at least 60% of trajectory frames will be included in the calculation. Non-bonded cutoff (NAMD) specifies the cutoff distance for non-bonded interaction energy calculations (default value is set to 12 Å). Any interaction beyond this cutoff distance is ignored. Trajectory stride is used to stride over frames included in the simulation trajectory. For example, if there are 1000 frames in the trajectory, specifying a stride value of 10 results in every 10th trajectory being processed by gRINN, yielding a total of 100 frames to be included in the calculation, thereupon reducing the calculation time. Number of processors option specifies the number of processors used in the calculation. By default, this value is one less than the total number of cores available in the system (gRINN reserves one core for progress monitoring purposes). gRINN can also calculate correlations between each interaction energy series as well, which is an optional feature.
The user has two options to compute interaction energies and their correlations using gRINN: the Graphical User Interface (GUI) or the Command-line Interface (CLI). GUI is the default interface for gRINN usage. New Calculation UI provides several GUI elements for specifying the paths of input files and calculation settings. Once they are set, clicking CALCULATE button on this interface starts the calculation. Progress bars on this interface allows the user to track the progress of computation and monitor the estimated time remaining until the calculation is complete. While the New Calculation UI is ideal for analyzing a limited number of simulation data, using the CLI is more efficient for batch processing of simulation trajectories.
The completion time of gRINN depends on the number of trajectory frames analyzed, the number of interacting residue pairs and the number of processors used for computation. Once the calculation is complete, several output files are generated and saved into the specified output folder. These files include the computation log, total, van-der Waals and electrostatic interaction energies’ time series and averages, correlations between these energy times series and a residue correlation (RC) matrix. The RC matrix is constructed using the correlation values (see below). The output folder additionally includes a reference input structure and a trajectory containing the frames used in calculation. Interaction energy and correlation data files are provided in comma-separated values (CSV) format and can be used in a subsequent custom analysis workflow. Alternatively, gRINN offers a View Results UI including several tables and plots for the inspection of output data and construction/analysis of a PEN. Exemplary plots are given in the Results section.
Calculation of pairwise residue interaction energies and interaction energy correlations
The interaction energy between two residues i and j is the sum of the non-bonded interaction energies defined in a force-field. Non-bonded interaction energies are typically considered in van-der Waals and electrostatic terms:
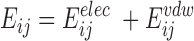 |
(1) |
Here,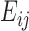 is an array including interaction energies between residues i and j in all trajectory frames included in the calculation. The average interaction energy between residues i and j,
is an array including interaction energies between residues i and j in all trajectory frames included in the calculation. The average interaction energy between residues i and j, 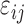 is the average of
is the average of 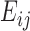 .
.
gRINN does not include a force-field definition of its own. Instead, it performs repeated calls to either NAMD or GROMACS (gmx) executables using specific procedures for extracting residue interaction energy data. Once the interaction energies are computed, correlations between them can be computed as well. For example, correlation between interaction energy series between residues i-j and k-l is computed as such:
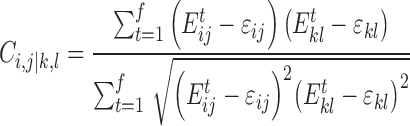 |
(2) |
Here, 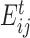 denotes the interaction energy between residues i and j in frame. gRINN computes correlations between all calculated interaction energy series and reports only values above or below 0.4 to eliminate weak correlations and reduce the amount of reported correlation data. In order to employ this interaction correlation matrix (C) to obtain a dynamical correlation measure between pairs of residues, it is projected on the residue space and a residue correlation (RC) matrix is constructed:
denotes the interaction energy between residues i and j in frame. gRINN computes correlations between all calculated interaction energy series and reports only values above or below 0.4 to eliminate weak correlations and reduce the amount of reported correlation data. In order to employ this interaction correlation matrix (C) to obtain a dynamical correlation measure between pairs of residues, it is projected on the residue space and a residue correlation (RC) matrix is constructed:
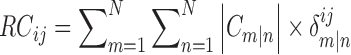 |
(3) |
N is the dimension of the interaction correlation matrix. 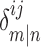 is equal to 1 only if residues i and j are involved in interactions m and n. Otherwise, it is zero. Computing RC matrix this way is based on the mutual occurrence of both residues i and j on the different sides of a correlation interaction. For example, if the correlation between the interaction GLY142–LYS145 and the interaction ILE16–LYS145 is 0.6 and the correlation between the interaction ILE16-LYS223 and the interaction GLY142-ASP191 is −0.5, the residue correlation between ILE16 and GLY142 would be the sum of the absolute values of these two correlation coefficients (1.1)). This summation is performed for all calculated correlations for each residue pair in the structure while constructing the full RC matrix.
is equal to 1 only if residues i and j are involved in interactions m and n. Otherwise, it is zero. Computing RC matrix this way is based on the mutual occurrence of both residues i and j on the different sides of a correlation interaction. For example, if the correlation between the interaction GLY142–LYS145 and the interaction ILE16–LYS145 is 0.6 and the correlation between the interaction ILE16-LYS223 and the interaction GLY142-ASP191 is −0.5, the residue correlation between ILE16 and GLY142 would be the sum of the absolute values of these two correlation coefficients (1.1)). This summation is performed for all calculated correlations for each residue pair in the structure while constructing the full RC matrix.
Protein energy network (PEN) construction and analysis
The term ‘Protein Energy Network’ has been used for the first time by Vijayabaskar and Vishveshwara in a study where they modeled such networks of protein structures using pairwise residue interaction energies from MD simulation trajectories (15). In this method, a network is constructed by taking individual residues as nodes and average interaction energies between each residue pair as the ‘weight’ for the edges that are added between these residue nodes. Once the network is complete, local (node-based) network metrics, such as degree, closeness and betweenness-centralities can be obtained to assess the importance of each residue in terms of protein stability and/or dynamics (45–47).
gRINN constructs a PEN using the information contained within the IEM. Here, each residue in the structure is taken as a node. An edge between two nodes (residues) is added if a non-zero average interaction energy exists between the two respective residues in the IEM. The user can specify a further interaction energy cutoff for edge addition as well. gRINN uses the value of the average IE to determine a ‘weight’ attribute of an edge using the following formula:
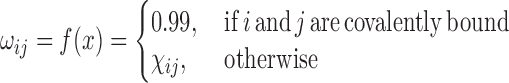 |
(4) |
In the above equation, 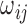 denotes the edge weight between residues i and j.
denotes the edge weight between residues i and j. 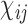 denotes the average interaction energy between residues i and j. gRINN allows the user to dismiss edge addition between covalently-bound residues, if desired.
denotes the average interaction energy between residues i and j. gRINN allows the user to dismiss edge addition between covalently-bound residues, if desired.
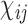 is computed using the following equation:
is computed using the following equation:
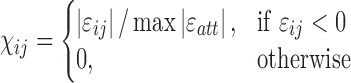 |
(5) |
In this equation, 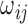 denotes the average interaction energy between residues i and j;
denotes the average interaction energy between residues i and j; 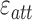 denotes the array of attractive (negative) interaction energies. It should be noted that when using this equation, the more attractive (negative) an interaction is, the higher weight will be assigned to the edge of that specific interaction. In other words, attractive interactions are favored. Repulsive interactions obtain zero weights. Hence, the matrix χ contains values between 0 and 1.
denotes the array of attractive (negative) interaction energies. It should be noted that when using this equation, the more attractive (negative) an interaction is, the higher weight will be assigned to the edge of that specific interaction. In other words, attractive interactions are favored. Repulsive interactions obtain zero weights. Hence, the matrix χ contains values between 0 and 1.
In addition to a weight attribute for each edge, a distance attribute is also calculated by subtracting each weight from 1. For example, if the weight of the edge between residues i and j is 0.2, the corresponding distance attribute would be 0.8. This is performed to extract short paths with preferentially higher edge weights (i.e. with lower distances).
Once a PEN is constructed, a variety of network analysis methods can be used to deduce useful information regarding the role of each individual residue in the protein structure (35,36,47–49). gRINN calculates residue-based local as well as global network metrics and provides shortest path analysis. Residue-based metrics include the degree, betweenness-centrality (BC) and closeness-centrality (CC) of each residue in the structure. Degree denotes the number of edges connected to a respective residue. BC of a residue is a measure of how frequently this residue occurs in all shortest paths between all other residues. BC is computed using the following equation:
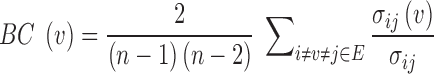 |
(6) |
In this equation, 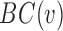 is betweenness-centrality of residue ν, n is the number of residues in the network,
is betweenness-centrality of residue ν, n is the number of residues in the network, 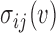 is the number of shortest paths between residue i and j that pass through residue ν and
is the number of shortest paths between residue i and j that pass through residue ν and 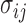 is the total number of shortest paths from i to j.
is the total number of shortest paths from i to j.
Closeness-centrality is a measure of the efficiency of information transfer through a particular residue. CC of a residue is the reciprocal sum of all shortest paths that originate from u to all other n – 1 nodes. Since the sum of the distances is dependent on the number of nodes in a network, the measure is normalized by the sum of all minimum possible distances. CC is computed using the following equation:
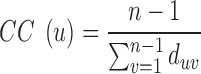 |
(7) |
In this equation, 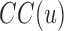 is closeness-centrality of residue u and
is closeness-centrality of residue u and 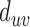 is the shortest path distance from node u to v.
is the shortest path distance from node u to v.
Shortest paths between any residue i and j are identified using the algorithm by Dijkstra (50) using the distance edge property as edge distance in order to favor edges having lower distance values. Betweenness centralities are found using the algorithm by Brandes (51).
RESULTS
Application example: Identifying functional residues in trypsin structure using gRINN
An example application of gRINN is presented on a 50-nanoseconds NAMD simulation data of the bovine pancreatic trypsin enzyme. Bovine pancreatic trypsin is a 243 amino-acid protein and a member of serine protease family enzymes. Like other proteases in this enzyme family, trypsin makes use of a catalytic triad located in the active site of the enzyme (HIS57, ASP102, SER195) to properly position the substrate and perform peptide bond cleavage. In addition to these catalytic residues, residues in the S1 substrate binding pocket (ASP189–SER195, SER214–CYS220, PRO225–TYR228) as well as in L1 (LEU185–LYS188) and L2 (GLN221–LYS224) loops were found to have an active role in determining substrate specificity and/or catalytic activity (52,53).
Details of the MD simulation and input data preparation for gRINN can be found in Supplementary Data. After completing the simulation, solvent molecules were removed from PSF, PDB and DCD files, an equilibrated portion of the trajectory was selected (25–50 ns), conformations were superposed on to the reference (initial) conformation and then saved into a new DCD file using VMD (54). Pairwise residue interaction energies between all possible residue pairs in the structure and their correlations were obtained via gRINN New Calculation UI using the default settings. In this particular application, a total of 10098 unique pairwise residue interaction energy series, each containing 1000 data points (i.e. 1000 trajectory frames), were computed. Total processing time of gRINN was ∼4 h on a workstation with 16 cpu cores.
Following the completion of calculations, View Results UI was used to visualize the data produced by the calculation step. Figure 2 shows several exemplary plots, extracted and shown as they appear in different tab panels of this UI (tables are omitted). By browsing through these tab panels, the user can inspect time-series, distributions and average interaction energies between a selected residue with all other residues (Figure 2A–C), time series of interaction energies between residues taking part in a selected interaction energy correlation pair (Figure 2D) and the corresponding correlation plot (Figure 2E) and finally the IEM (Figure 2F) and the residue correlation (RC) matrices (Figure 2G). The RC matrix is a way of extracting side-chain dynamic correlation between any two residues in the structure. Highest correlations (the darkest data points in Figure 2G) are observed between SER190–ASN223 and THR144–ASN223. The correlation between SER190 and ASN223 implies a side-chain-based dynamic correlation between the S1 binding pocket and the L2 loop. This result can be expected since a concerted motion between the S1 binding pocket and L1/L2 loops was previously found to be important for enzyme specificity (52).
Figure 2.

An overview of output plots as generated by gRINN View Results UI. (A and B) Example single interaction energy time series and distributions, respectively. (C) Average non-zero interaction energies between a selected residue and all other residue in protein structure. (D) Time series of interaction energies between residues taking part in a selected interaction energy correlation pair. (E) Corresponding correlation plot of the plot in D. (F) Interaction Energy Matrix (IEM). (G) Residue Correlation (RC) matrix.
View Results UI provides an additional PEN analysis feature under ‘Network Analysis’ tab panel (Figure 3). Here, a PEN is constructed by taking each residue in the structure as a node and determining edges and edge weights/distances using average interaction energies (see Materials and Methods section for details). Three residue metrics (degree, betweenness-centrality and closeness-centrality) are computed and plotted. The user has the option to include/exclude covalent bonds as edges, specify a cutoff energy value for edge addition and export the network and residue metrics to files.
Figure 3.

A screenshot of the Network Analysis Tab in gRINN View Results UI.
A PEN was constructed for trypsin simulation data by including covalent bonds as edges and adding edges between any two residues whose absolute average interaction energy is equal to or above 1 kcal/mol. Using this PEN, betweenness-centrality (BC) and closeness-centrality (CC) metrics were computed and top 10 residues having the highest BC and CC values are listed in Table 1. The full list of degree, BC and CC values can be found in Supplementary Data. Table 1 also includes a position column to indicate whether the respective residue is in one of the functionally important positions in the structure mentioned above.
Table 1. Top 10 betweenness-centrality and closeness-centrality residues of trypsin given along with the position of residues (52,53).
| Betweenness -centrality (BC) | Closeness-centrality (CC) | ||||
|---|---|---|---|---|---|
| Residue | Position | BC value | Residue | Position | CC value |
| LYS188 | L1 loop | 0.165 | LYS188 | L1 loop | 0.608 |
| ASP189 | S1 binding pocket | 0.099 | ASP194 | S1 binding pocket | 0.606 |
| ASP191 | S1 binding pocket | 0.088 | ASP189 | S1 binding pocket | 0.577 |
| ILE16 | N-terminal residue | 0.086 | ILE16 | N-terminal residue | 0.576 |
| LYS224 | L2 loop | 0.047 | ASP102 | Catalytic aspartate | 0.508 |
| ASP102 | Catalytic aspartate | 0.045 | TYR29 | 0.505 | |
| LYS230 | Near S1 pocket | 0.044 | VAL31 | 0.505 | |
| GLU70 | 0.033 | GLU70 | 0.503 | ||
| ASP165 | 0.029 | SER195 | Catalytic serine | 0.502 | |
| LEU209 | 0.027 | SER54 | 0.501 | ||
Table 1 shows that residues located in the S1 substrate binding site as well as L1 and L2 loops play important roles in information transfer within the enzyme structure. Catalytic ASP102 and catalytic SER195 are also listed among the top 10 BC and CC residues. The fact that these residues are important for determining the catalytic activity (as mentioned above) highlights the utility of the PEN approach as implemented in gRINN.
IMPLEMENTATION
gRINN was developed in Python programming language (version 2.7). In addition to the python core library, several open source Python packages are used. GUI elements are provided by PyQt5. Matplotlib (version 2.0.2) is used to generate two-dimensional line and scatter plots as well as heatmaps (55). ProDy (version 1.9.3) is used for PDB and DCD trajectory manipulations, atom selections and all other general geometric tasks related to protein structure (44). Mdtraj (version 1.9.0) is used to convert GROMACS trajectory file formats to DCD for further processing (56). Open-source PyMol (version 1.8) is used as the molecule viewer in the ‘View Results’ UI. Pexpect (version 4.3.1) is used to interact with the gmx executable. Numpy (version 1.13.3) is used for all matrix operations occurring throughout the computation workflow of gRINN. Pandas (version 0.20.3) is used to store, process and save tabular data. Networkx (v.2.0) is used to construct PENs and calculation of global/local network metrics and short paths.
CONCLUSION
We have developed a stand-alone software, gRINN, for efficient calculation and analysis of amino-acid residue interaction energies from NAMD/CHARMM or GROMACS-generated MD simulations. Extraction of interaction energies from molecular simulation data is considered as a computationally expensive analysis task with enormous amounts of output data. Dealing with such data in the context of whole protein structure requires significant computational expertise. gRINN automatizes the steps necessary for extracting pairwise residue interaction energies using either NAMD or GROMACS executables, thereby greatly simplifying the workflow for the end-user with limited exposure to scripting. gRINN additionally features interaction energy correlation calculations and Protein Energy Network analysis to identify dynamic cross-talk between residues and potentially functional residues in the protein structure.
DATA AVAILABILITY
gRINN is free and open to all users. Executables for Linux and Mac OSX operating systems are available for download at http://grinn.readthedocs.io. We recommend all interested users to follow the tutorial at this website.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank all researchers who have tested gRINN. Special thanks to Metin Yazar, Zekeriya Düzgün, İlayda Bozan, İsmail Hakkı Akgün and Gözde Yalçın for their extensive feedback and help in debugging the software.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Onur Serçinoğlu would like to acknowledge the ÖYP research grant from the Higher Education Council of Turkey. Marmara University Commission of Scientific Research Project, FEN-A-100616-0273, is acknowledged. .
Conflict of interest statement. None declared.
REFERENCES
- 1. Lee E.H., Hsin J., Sotomayor M., Comellas G., Schulten K.. Discovery through the computational microscope. Structure. 2009; 17:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dror R.O., Dirks R.M., Grossman J.P., Xu H.F., Shaw D.E.. Biomolecular Simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012; 41:429–452. [DOI] [PubMed] [Google Scholar]
- 3. Hospital A., Goni J.R., Orozco M., Gelpi J.L.. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem. 2015; 8:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vlachakis D., Bencurova E., Papangelopoulos N., Kossida S.. Current state-of-the-art molecular dynamics methods and applications. Adv. Protein Chem. Struct. Biol. 2014; 94:269–313. [DOI] [PubMed] [Google Scholar]
- 5. Benson N.C., Daggett V.. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B. 2012; 116:8722–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eisenmesser E.Z., Millet O., Labeikovsky W., Korzhnev D.M., Wolf-Watz M., Bosco D.A., Skalicky J.J., Kay L.E., Kern D.. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005; 438:117–121. [DOI] [PubMed] [Google Scholar]
- 7. Agarwal P.K. Role of protein dynamics in reaction rate enhancement by enzymes. J. Am. Chem. Soc. 2005; 127:15248–15256. [DOI] [PubMed] [Google Scholar]
- 8. Narayanan C., Bernard D.N., Bafna K., Gagne D., Chennubhotla C.S., Doucet N., Agarwal P.K.. Conservation of dynamics associated with biological function in an enzyme superfamily. Structure. 2018; 26:426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bendova-Biedermannova L., Hobza P., Vondrasek J.. Identifying stabilizing key residues in proteins using interresidue interaction energy matrix. Proteins. 2008; 72:402–413. [DOI] [PubMed] [Google Scholar]
- 10. Morra G., Genoni A., Colombo G.. Mechanisms of differential allosteric modulation in homologous Proteins: Insights from the analysis of internal dynamics and energetics of PDZ domains. J. Chem. Theory Comput. 2014; 10:5677–5689. [DOI] [PubMed] [Google Scholar]
- 11. Ermakova E., Kurbanov R.. Effect of ligand binding on the dynamics of trypsin. Comparison of different approaches. J. Mol. Graph. Model. 2014; 49:99–109. [DOI] [PubMed] [Google Scholar]
- 12. Kumawat A., Chakrabarty S.. Hidden electrostatic basis of dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E5825–E5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ribeiro A.A., Ortiz V.. Energy propagation and network energetic coupling in proteins. J. Phys. Chem. B. 2015; 119:1835–1846. [DOI] [PubMed] [Google Scholar]
- 14. Ribeiro A.A., Ortiz V.. Determination of signaling pathways in proteins through network Theory: Importance of the topology. J. Chem. Theory Comput. 2014; 10:1762–1769. [DOI] [PubMed] [Google Scholar]
- 15. Vijayabaskar M.S., Vishveshwara S.. Interaction energy based protein structure networks. Biophys. J. 2010; 99:3704–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhattacharyya M., Vishveshwara S.. Probing the allosteric mechanism in pyrrolysyl-tRNA synthetase using energy-weighted network formalism. Biochemistry. 2011; 50:6225–6236. [DOI] [PubMed] [Google Scholar]
- 17. Lu C., Knecht V., Stock G.. Long-Range conformational response of a PDZ domain to ligand binding and release: a molecular dynamics study. J. Chem. Theory Comput. 2016; 12:870–878. [DOI] [PubMed] [Google Scholar]
- 18. Brinda K.V., Vishveshwara S.. A network representation of protein structures: implications for protein stability. Biophys. J. 2005; 89:4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kong Y., Karplus M.. The signaling pathway of rhodopsin. Structure. 2007; 15:611–623. [DOI] [PubMed] [Google Scholar]
- 20. Kong Y., Karplus M.. Signaling pathways of PDZ2 domain: a molecular dynamics interaction correlation analysis. Proteins. 2009; 74:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papaleo E. Integrating atomistic molecular dynamics simulations, experiments, and network analysis to study protein dynamics: strength in unity. Front. Mol. Biosci. 2015; 2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng S., Fu H.L., Cui D.X.. Characteristics analyses and comparisons of the protein structure networks constructed by different methods. Interdiscip. Sci. 2016; 8:65–74. [DOI] [PubMed] [Google Scholar]
- 23. O’Rourke K.F., Gorman S.D., Boehr D.D.. Biophysical and computational methods to analyze amino acid interaction networks in proteins. Comput. Struct. Biotechnol. J. 2016; 14:245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahita J., Sowdhamini R.. Investigating the effect of key mutations on the conformational dynamics of toll-like receptor dimers through molecular dynamics simulations and protein structure networks. Proteins. 2018; 86:475–490. [DOI] [PubMed] [Google Scholar]
- 25. Guzel P., Kurkcuoglu O.. Identification of potential allosteric communication pathways between functional sites of the bacterial ribosome by graph and elastic network models. Biochim. Biophys. Acta. 2017; 1861:3131–3141. [DOI] [PubMed] [Google Scholar]
- 26. Ni D., Song K., Zhang J., Lu S.. Molecular dynamics simulations and dynamic network analysis reveal the allosteric unbinding of monobody to H-Ras triggered by R135K mutation. Int. J. Mol. Sci. 2017; 18:E2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verkhivker G.M. Computational modeling of the Hsp90 interactions with cochaperones and Small-Molecule inhibitors. Methods Mol. Biol. 2018; 1709:253–273. [DOI] [PubMed] [Google Scholar]
- 28. Yao X.Q., Malik R.U., Griggs N.W., Skjaerven L., Traynor J.R., Sivaramakrishnan S., Grant B.J.. Dynamic coupling and allosteric networks in the alpha subunit of heterotrimeric G proteins. J. Biol. Chem. 2016; 291:4742–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhattacharyya M., Ghosh S., Vishveshwara S.. Protein structure and function: looking through the network of side-chain interactions. Curr. Protein Pept. Sci. 2016; 17:4–25. [DOI] [PubMed] [Google Scholar]
- 30. Vijayabaskar M.S., Vishveshwara S.. Comparative analysis of thermophilic and mesophilic proteins using Protein Energy Networks. BMC Bioinformatics. 2010; 11:S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakrabarty B., Parekh N.. NAPS: network analysis of protein structures. Nucleic Acids Res. 2016; 44:W375–W382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seeber M., Felline A., Raimondi F., Mariani S., Fanelli F.. WebPSN: a web server for high-throughput investigation of structural communication in biomacromolecules. Bioinformatics. 2015; 31:779–781. [DOI] [PubMed] [Google Scholar]
- 33. Grant B.J., Rodrigues A.P., ElSawy K.M., McCammon J.A., Caves L.S.. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006; 22:2695–2696. [DOI] [PubMed] [Google Scholar]
- 34. Pasi M., Tiberti M., Arrigoni A., Papaleo E.. xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures. J. Chem. Inf. Model. 2012; 52:1865–1874. [DOI] [PubMed] [Google Scholar]
- 35. Doncheva N.T., Assenov Y., Domingues F.S., Albrecht M.. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012; 7:670–685. [DOI] [PubMed] [Google Scholar]
- 36. Doncheva N.T., Klein K., Domingues F.S., Albrecht M.. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 2011; 36:179–182. [DOI] [PubMed] [Google Scholar]
- 37. Doncheva N.T., Klein K., Morris J.H., Wybrow M., Domingues F.S., Albrecht M.. Integrative visual analysis of protein sequence mutations. BMC Proc. 2014; 8:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhattacharyya M., Bhat C.R., Vishveshwara S.. An automated approach to network features of protein structure ensembles. Protein Sci. 2013; 22:1399–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tiberti M., Invernizzi G., Lambrughi M., Inbar Y., Schreiber G., Papaleo E.. PyInteraph: a framework for the analysis of interaction networks in structural ensembles of proteins. J. Chem. Inf. Model. 2014; 54:1537–1551. [DOI] [PubMed] [Google Scholar]
- 40. Galgonek J., Vymetal J., Jakubec D., Vondrasek J.. Amino Acid Interaction (INTAA) web server. Nucleic Acids Res. 2017; 45:W388–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ribeiro A.A., Ortiz V.. MDN: A Web Portal for Network Analysis of Molecular Dynamics Simulations. Biophys. J. 2015; 109:1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1–2:19–25. [Google Scholar]
- 44. Bakan A., Meireles L.M., Bahar I.. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011; 27:1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. del Sol A., Fujihashi H., Amoros D., Nussinov R.. Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci. 2006; 15:2120–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiong Y., Xia J., Zhang W., Liu J.. Exploiting a reduced set of weighted average features to improve prediction of DNA-binding residues from 3D structures. PLoS One. 2011; 6:e28440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amitai G., Shemesh A., Sitbon E., Shklar M., Netanely D., Venger I., Pietrokovski S.. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004; 344:1135–1146. [DOI] [PubMed] [Google Scholar]
- 48. Bode C., Kovacs I.A., Szalay M.S., Palotai R., Korcsmaros T., Csermely P.. Network analysis of protein dynamics. FEBS Lett. 2007; 581:2776–2782. [DOI] [PubMed] [Google Scholar]
- 49. Taylor N.R. Small world network strategies for studying protein structures and binding. Comput. Struct. Biotechnol. J. 2013; 5:e201302006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dijkstra E.W. A note on two problems in connexion with graphs. Numer. Math. 1959; 1:269–271. [Google Scholar]
- 51. Brandes U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001; 25:163–177. [Google Scholar]
- 52. Ma W., Tang C., Lai L.. Specificity of trypsin and chymotrypsin: loop-motion-controlled dynamic correlation as a determinant. Biophys. J. 2005; 89:1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hedstrom L. Serine protease mechanism and specificity. Chem. Rev. 2002; 102:4501–4524. [DOI] [PubMed] [Google Scholar]
- 54. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 55. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 56. McGibbon R.T., Beauchamp K.A., Harrigan M.P., Klein C., Swails J.M., Hernandez C.X., Schwantes C.R., Wang L.P., Lane T.J., Pande V.S.. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015; 109:1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
gRINN is free and open to all users. Executables for Linux and Mac OSX operating systems are available for download at http://grinn.readthedocs.io. We recommend all interested users to follow the tutorial at this website.