


Abstract
ArDock (ardock.ibcp.fr) is a structural bioinformatics web server for the prediction and the visualization of potential interaction regions at protein surfaces. ArDock ranks the surface residues of a protein according to their tendency to form interfaces in a set of predefined docking experiments between the query protein and a set of arbitrary protein probes. The ArDock methodology is derived from large scale cross-docking studies where it was observed that randomly chosen proteins tend to dock in a non-random way at protein surfaces. The method predicts interaction site of the protein, or alternate interfaces in the case of proteins with multiple interaction modes. The server takes a protein structure as input and computes a score for each surface residue. Its output focuses on the interactive visualization of results and on interoperability with other services.
INTRODUCTION
As fundamental elements of life, proteins perform their biological functions through molecular interactions. Functional interactions are regulated by protein availability and localization, while new functions can emerge through transient or permanent interactions. Over the past decades, tremendous efforts have been made to experimentally identify the molecular properties and functions of individual proteins and complexes (1,2). However, the identification of protein–protein complexes, let alone their structural characterization, remains a challenging task that is difficult to address accurately with high-throughput methods. These limitations can be addressed with computational methods for studying protein–protein interactions. Among the computational approaches, docking methods aim at predicting protein complexes starting from the individual structure of the proteins. ‘Cross-docking’ studies are large scale docking experiments where the subunits of biological dimers are separated and their structures all docked against each other (3). These studies led to the conclusion that it was difficult to separate biological and non-biological docking complexes by current computational means (4). Nevertheless, the proteins assembled by cross-docking tend to ‘arbitrarily’ dock their cognate and non-cognate partners at similar regions of their surface (5,6). As a consequence, arbitrary docking was acknowledged as a viable tool to predict protein binding sites at the surface of a protein. Computational methods based on arbitrary docking are now being developed to study proteins and predict their binding sites (7–9).
Meanwhile, experimental protein–protein complexes have been extensively characterized in terms of physicochemical, geometrical and sequence-related properties (10,11). Data from these studies can be analyzed to derive various features that can be mapped onto residues in order to build binding-region predictors. Frequently used features include sequence information (amino acid conservation across homologs, sequence profiles or residue propensity), side-chain physicochemical properties (hydrophobicity, electrostatic potential) or geometrical properties (accessible surface area, planarity). Predictors can be applied at the single residue level, or extended to clusters of adjacent surface residues (12). Protein-binding region predictors frequently combine different kinds of features. A first group of predictors based on machine-learning algorithms, includes the following web services: Cons-PPISP (13), PROFISIS (14), PresConst, SPPIDER (15) and PredUs (16). Another class of predictors such as SHARP2 (17), WHISCY (18) or PINUP (19) uses scoring function maximization. A third class which comprises the JET2 (20) and ProMate (21) servers uses clustering approaches. They all provide limited interaction and visualization capabilities to the user. The new ArDock web server proposed here is an original addition to this set of tools on two levels: methodologically, the information provided by arbitrary docking calculations is an addition to currently used features; in terms of the user interface, the ArDock server provides rich and interactive visualization of binding site predictions.
MATERIALS AND METHODS
Docking of arbitrary peptide probes
The protein submitted to the ArDock server is docked against 25 molecular probes. Each probe is docked independently by the Hex software (22). The 10 best computed protein-probe conformations, as scored by the Hex energy function, are used to count the number of times that each surface residue of the protein is found at a protein-probe interface. The protein residues are considered to form the interface when their solvent accessibility changes from the initial to the probe-bound conformation. The solvent accessibility of the amino acids is computed with NACCESS (23). The surface amino acids are finally assigned a normalized score:
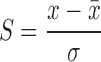 |
Where x is the number of protein-probe interface in which the amino acid is found, 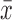 is the averaged value of x over all the surface amino acids and σ the estimated standard deviation. The set of small molecular probes was culled from the Nh3D (24) database. The probes cover a range of structures at the Topology level of the CATH structural classification database (25). Details of the culling procedure can be found in an earlier publication (5).
is the averaged value of x over all the surface amino acids and σ the estimated standard deviation. The set of small molecular probes was culled from the Nh3D (24) database. The probes cover a range of structures at the Topology level of the CATH structural classification database (25). Details of the culling procedure can be found in an earlier publication (5).
Input
A protein structure file complying with the Protein Data Bank (26) format can be submitted to the server. Only natural amino acids are currently supported, any other molecular component will be discarded. The submitted coordinates are processed in the navigator by two JavaScript components: a parser (https://github.com/glaunay/pdb-lib) and the NGL (27,28) webGL renderer. This allows the user to interactively select amino acid chains before their analysis by the server. It is important to note that different combinations of protein chains can lead to distinct results because the ArDock method is sensitive to the overall shape of the protein. Additionally, different protein structures can be submitted to the same session. We plan to extend the treatment to nucleic acids and to modified protein residues.
Client interface
The interface has a tabular scaffold. A top-level tab is created upon PDB file submission. Multimeric proteins can be split into subcomplexes (see Figure 1) and each subcomplex will be open in a dedicated sub-tab for further inspection. The top-right necklace menu gives access to the sequence representation of the protein. The sequence of each protein chain is displayed in a dedicated draggable window. This is a scrollable one-letter code representation of the protein sequence in fasta numbering. Clicking on a given amino acid will highlight its position in the three-dimensional (3D) representation of the protein (see Figure 1).
Figure 1.

(A) The ArDock user interface allows the manipulation of different proteins and set of protein chains, each opened in distinct tab. (B) Protein chains can be selected or deleted before being analyzed by the server. (C) The amino acid sequence of each chain can be visualized, and individual amino acids highlighted in the structure representation (here an aspartic acid colored in red). (D) Ribbon, ball-and-stick and surface representations are available. The two icons on the right-end side allow to modify the background color and to take a snapshot of the structure.
Server implementation
The protein coordinates with the desired set of chains are sent to the server backend, on which 25 docking simulations are performed in parallel by our NodeJS cluster scheduler (https://github.com/glaunay/nslurm). The docking software is the version 8.0.0 of the HeX program (hex.loria.fr/). Computations use the shape complementarity scoring function with a ligand–receptor maximum range of 40Å and scan steps of 0.75Å, the complete list of parameters can be found in a previous publication (5). All the tasks required by the ArDock procedure are organized in an asynchronous pipeline. When a task is finished, its results are streamed in the pipeline but can also be captured immediately by the application server. Thus, full completion of the pipeline is not required to begin visualization and any relevant intermediate results can be passed to the web client. The communication between the server and the client is two ways and is built upon the socket.io framework (http://socket.io/). This allows for a real-time update of the web client every time a protein probe docking experiment is completed on the server. The protein molecular representation is continuously colored as probes are being docked at its surface.
DISCUSSION
Visualizing binding regions
When the server analysis of the protein surface is completed, results are displayed on the molecular surface and in an interactive data table (see Figure 2). Each line of the table refers to a particular amino acid. The three left-hand columns correspond to the amino acid type, number and chain. The two right-hand columns correspond to the amino acid raw and normalized scores. While the initial order of the lines respects the amino acid sequence, the user can reorder the table by clicking on a column header. If the user clicks on a line in the data table, the structure of the corresponding amino acid will be highlighted in the molecular structure. By default, the amino acids with the highest scores (those most susceptible to form protein–protein interface) are colored in red. A button at the bottom of the table triggers the downloading of the results as a tabulated file. Additionally, results can also be downloaded as a PDB format file with raw scores in the B-factor field.
Figure 2.

ArDock results display: (A) individual amino acid scores are presented in the left-hand table. This table has searchable and sortable functionalities. (B) The table content can be downloaded in a tabulated format. (C) An upper center console provides save and resume functionalities as buttons. Buttons (left to right): communication of the results to the ENDscript server, download of the results as a PDB file and copying of the job-specific key for later reload. (D) In order to easily inspect an amino acid in the structure, the whole structure transparency is increased and only the inspected amino acid remains fully opaque (in this case, threonine 105 of chain A).
Resuming analysis and interoperability
Upon job submission, a center console dedicated to data saving and export appears. The right-end button of the console displays a personal key that allows future access to the job results. If the user is forced to quit at this stage, the computations will be carried out and remain accessible. To resume the analysis the user will paste his personal key into the restore procedure accessible from the welcome page. The client will then fetch all relevant data from the server and restore the proper 3D rendering and the tabular results.
The left-hand button of the console will establish a communication with the ENDscript (29) server (http://endscript.ibcp.fr). The structure file annotated with the ArDock result will be passed to the ENDscript server and a dedicated tab will open in the browser. The ENDscript server will provide the user with additional tools to study protein sequences and structures. Hence, the user can automatically generate a multiple sequence alignment from the query, which is colored according to residue conservation and adorned with secondary structure elements of each homologous protein of known structure. Potential interaction regions identified by ArDock are shown with colored bars at the bottom of sequence blocks. The user can also generate an interactive 3D PyMOL representation of the query, whose main chain is depicted as a tube whose radius is proportional to the differences in Cα between the query and all homologous proteins of known structures. Potential interaction areas will be highlighted anew with colored meshes.
Prediction performances
The predictive performance of arbitrary docking was explored in our previous studies (5,30). Here, we present the performance of the ArDock server on the protein benchmark created for the critical assessment of on-line resources for the prediction of protein interface residue (31). The dataset is made of 90 target proteins crystal complexes, which were obtained from the Protein Docking Benchmark Set 4.0 (32) the following way : (i) excluding complexes for which the receptor (i.e. longer chain) is shorter than 50 or longer than 600 residues; (ii) discarding complexes with more than two chains, and interfaces smaller than 20 residues; (iii) interface residues were defined using a 5Å distance cutoff. Accessible residues (relative accessibility greater than 5%) were classified as interface or non-interface depending on their arbitrary docking score. By varying the cutoff, we were able to compute a receiver operating characteristic (ROC) curve and the area under the curve (AUC). For a given cutoff, we collected the number of true positives (TP: interface residues predicted as such), true negatives (TN: non-interface residues predicted as such), false positives (FP: non-interface residues predicted as interface) and false negatives (FN: interface residues predicted as non-interfaces). We then computed the usual performance indicators: accuracy (ACC), precision or positive predictive value (PPV), sensitivity or true positive rate (TPR), specificity (SPC), false positive rate (FPR) and Matthew’s correlation coefficient (MCC):
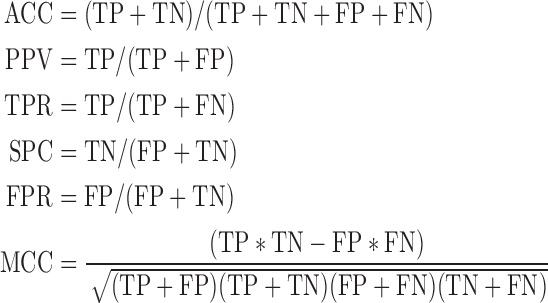 |
Following the original benchmark study, we present the indicators computed with the score threshold that maximizes the MCC value, see Table1. We also computed the number of predicted interfaces by ArDock with TPR > 15%, in line with the work of Ripoche et al. (20): it is equal to 88.9%. The corresponding AUC is 0.664. The statistics were computed over the BM90C benchmark (31) composed of 22 076 exposed residues in 90 proteins.
Table 1. Comparison of the performance of the ArDock server and 11 web services for the prediction of protein interface residues.
| MCC | TPR | FPR | SPC | PPV | ACC | |
|---|---|---|---|---|---|---|
| Pseudo-meta | 0.481 | 0.692 | 0.094 | 0.905 | 0.417 | 0.887 |
| PredUs | 0.383 | 0.701 | 0.156 | 0.843 | 0.302 | 0.831 |
| eFindSitePPI | 0.375 | 0.396 | 0.045 | 0.954 | 0.459 | 0.905 |
| cons-PPISP | 0.247 | 0.279 | 0.052 | 0.947 | 0.338 | 0.888 |
| ArDock | 0.189 | 0.595 | 0.319 | 0.682 | 0.206 | 0.671 |
| SPPIDER | 0.173 | 0.340 | 0.125 | 0.875 | 0.208 | 0.827 |
| ProMate | 0.165 | 0.526 | 0.295 | 0.704 | 0.210 | 0.684 |
| WHISCY | 0.164 | 0.130 | 0.025 | 0.975 | 0.334 | 0.900 |
| PIER | 0.118 | 0.066 | 0.012 | 0.987 | 0.342 | 0.906 |
| VORFFIP | 0.117 | 0.531 | 0.401 | 0.598 | 0.337 | 0.579 |
| PSIVER | 0.103 | 0.645 | 0.463 | 0.536 | 0.118 | 0.546 |
| InterProSurf | 0.100 | 0.435 | 0.291 | 0.709 | 0.163 | 0.677 |
Data for the other web services were taken from Maheshwari et al. (31).
According to the benchmark and classification of Maheshwari et al. (31), the ArDock server achieves the best performances among the web servers based on residue features (group I) and physicochemical and structural features (group III). Aside from one meta-predictor, ArDock is only outperformed by two template-based methods (16,31) and a machine learning method (13). In contrast to these methods which use sequence statistics or supervised classification, ArDock does not require any training or prior knowledge. More importantly, ArDock identifies interface residues using a structural information which is not taken into account by any other available method. The information provided by ArDock is hence orthogonal to the one provided by other comparable web services. This makes of ArDock an accurate tool on its own but also a significant addition to the current set of on-line resources for the detection of protein interaction site.
Experimental interface detection examples
In addition, to these global performance indicators, the use of ArDock can be illustrated on specific examples. Here, two complexes were taken from the aforementioned benchmark and only one partner in each complex was processed by the ArDock server. We now present the accuracy of ArDock at detecting the interaction surface of these partners that form the interface in their corresponding complexes. The first case is the human cell division control protein 42 (CDC42). This protein is a small GTPase of the Rho family. In the crystal structure of the benchmark (PDB code: 1GRN) a monomer of CDC42 forms a complex with a domain of its specific GTPase-activating protein. Their surface of interaction is 2332.2Å2 for a total of 65 interchain residue contacts. A set of 22 residues from CDC42 participates in the experimental interface. As it can be seen in Figure 3, ArDock analyzed the surface of the CDC42 monomer and predicted 21 of these 22 residues as interacting residues.
Figure 3.

ArDock interface prediction of the human cell division control protein 42 (PDB code: 1GRN). Amino acids that are part of the experimental interface are colored in green if they are also predicted as interface residue by ArDock. Amino acids that part of the experimental are colored in orange if they were not predicted as interface residue by ArDock.
The second case is a hydrolase/inhibitor complex. The enzyme is a yellow meal worm α-amylase. The experimental structure (PDB code: 1TMQ) displays an interface of 2401.0Å2. A total of 32 enzyme residues participates in 70 interchain residue contacts. The enzyme monomer was processed by the server (see Figure 4). ArDock successfully predicted 29 surface residues of the enzyme out of the 32 that form the experimental interface.
Figure 4.

ArDock interface prediction of a glycosylase, the yellow meal worm α-amylase (PDB code: 1TMQ). In this case, the experimental interface is larger and a similar color code is used. The experimental interface residues are colored in green if they are also predicted as interface residue by ArDock and colored in orange if they were not predicted as interface residue by ArDock.
CONCLUSION
We present the ArDock server. Working from a PDB input file, this web resource detects protein surface residues likely to be involved in protein–protein interactions. The server uses an approach based solely on physical properties that helps to identify biologically relevant protein interfaces. ArDock does not perform explicit clustering of surface residues to predict interaction patches, unlike other effective prediction methods (33). ArDock will benefit from the future addition of a clustering procedure and the integration of additional residue properties. In its current state, the ArDock web service targets a large audience. It uses a friendly interactive interface for the visualization of annotated protein structures. It is built on an asynchronous pipeline on the server side and has a modular organization of the software on both the client and server sides. This architecture will support future extensions to include the calculation and interactive visualization of additional amino acid properties (physicochemical or sequence-based).
ACKNOWLEDGEMENTS
The authors thank R. Lavery for his advice at the origin of the web service initiative and for his help with the manuscript. We thank M.Brylinski for providing the BM90 dataset. We thank Alexis Michon and Samuel Bosquin (UMS 3760, Institut de Biologie et Chimie des Protéines, Lyon, France) for helpful discussions, technical assistance and hardware support.
FUNDING
European Union’s Horizon 2020 Framework Programme for Research and Innovation (Human Brain Project SGA1) [720270]. Funding for open access charge: European Union’s Horizon 2020 Framework Programme for Research and Innovation (Human Brain Project SGA1) [720270].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shoemaker B.A., Panchenko A.R.. Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comput. Biol. 2007; 3:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petschnigg J., Snider J., Stagljar I.. Interactive proteomics research technologies: recent applications and advances. Curr. Opin. Biotechnol. 2011; 22:50–58. [DOI] [PubMed] [Google Scholar]
- 3. Wass M.N., Fuentes G., Pons C., Pazos F., Valencia A.. Towards the prediction of protein interaction partners using physical docking. Mol. Syst. Biol. 2011; 7:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sacquin-Mora S., Carbone A., Lavery R.. Identification of protein interaction partners and protein-protein interaction sites. J. Mol. Biol. 2008; 382:1276–1289. [DOI] [PubMed] [Google Scholar]
- 5. Martin J., Lavery R.. Arbitrary protein–protein docking targets biologically relevant interfaces. BMC Biophys. 2012; 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fernandez-Recio J., Totrov M., Abagyan R.. Identification of protein-protein interaction sites from docking energy landscapes. J. Mol. Biol. 2004; 335:843–865. [DOI] [PubMed] [Google Scholar]
- 7. Lopes A., Sacquin-Mora S., Dimitrova V., Laine E., Ponty Y., Carbone A.. Protein-protein interactions in a crowded environment: an analysis via cross-docking simulations and evolutionary information. PLoS Comput. Biol. 2013; 9:e1003369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vamparys L., Laurent B., Carbone A., Sacquin-Mora S.. Great interactions: how binding incorrect partners can teach us about protein recognition and function. Proteins. 2016; 84:1408–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laine E., Carbone A.. Protein social behavior makes a stronger signal for partner identification than surface geometry. Proteins. 2017; 85:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keskin O., Gursoy A., Ma B., Nussinov R.. Principles of protein-protein interactions: what are the preferred ways for proteins to interact. Chem. Rev. 2008; 108:1225–1244. [DOI] [PubMed] [Google Scholar]
- 11. Jones S., Thornton J.M.. Principles of protein-protein interactions. Proc. Natl Acad. Sci. U.S.A. 1996; 93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keskin O., Tuncbag N., Gursoy A.. Predicting protein-protein interactions from the molecular to the proteome level. Chem. Rev. 2016; 116:4884–4909. [DOI] [PubMed] [Google Scholar]
- 13. Chen H., Zhou H.X.. Prediction of interface residues in protein-protein complexes by a consensus neural network method: test against NMR data. Proteins. 2005; 61:21–35. [DOI] [PubMed] [Google Scholar]
- 14. Ofran Y., Rost B.. ISIS: interaction sites identified from sequence. Bioinformatics. 2007; 23:e13–e16. [DOI] [PubMed] [Google Scholar]
- 15. Porollo A., Meller J.. Prediction-based fingerprints of protein-protein interactions. Proteins. 2007; 66:630–645. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Q.C., Deng L., Fisher M., Guan J., Honig B., Petrey D.. PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 2011; 39:W283–W287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murakami Y., Jones S.. SHARP2: protein-protein interaction predictions using patch analysis. Bioinformatics. 2006; 22:1794–1795. [DOI] [PubMed] [Google Scholar]
- 18. de Vries S.J., van Dijk A.D.J., Bonvin A.M.J.J.. WHISCY: what information does surface conservation yield? Application to data-driven docking. Proteins. 2006; 63:479–489. [DOI] [PubMed] [Google Scholar]
- 19. Liang S., Zhang C., Liu S., Zhou Y.. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006; 34:3698–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ripoche H., Laine E., Ceres N., Carbone A.. JET2 Viewer: a database of predicted multiple, possibly overlapping, protein–protein interaction sites for PDB structures. Nucleic Acids Res. 2017; 45:D236–D242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neuvirth H., Raz R., Schreiber G.. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 2004; 338:181–199. [DOI] [PubMed] [Google Scholar]
- 22. Ghoorah A.W., Devignes M.D., Smal-Tabbone M., Ritchie D.W.. Protein docking using case-based reasoning. Proteins. 2013; 81:2150–2158. [DOI] [PubMed] [Google Scholar]
- 23. Hubbard S.J., Thornton J.M.. NACCESS, Computer Program, Department of Biochemistry and Molecular Biology, University College London. 2001; 1st edn. [Google Scholar]
- 24. Thiruv B., Quon G., Saldanha S.A., Steipe B.. Nh3D: a reference dataset of non-homologous protein structures. BMC Struct. Biol. 2005; 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dawson N.L., Lewis T.E., Das S., Lees J.G., Lee D., Ashford P., Orengo C.A., Sillitoe I.. CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017; 45:D289–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rose P.W., Prli A., Altunkaya A., Bi C., Bradley A.R., Christie C.H., Costanzo L.D., Duarte J.M., Dutta S., Feng Z. et al. . The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017; 45:D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rose A.S., Hildebrand P.W.. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prli A., Rose P.W.. Web-based molecular graphics for large complexes. Proceedings of the 21st International Conference on Web3D Technology. 2016; NY: ACM; 185–186. [Google Scholar]
- 29. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin J. Benchmarking protein-protein interface predictions: why you should care about protein size: Size Bias in Predictions. Proteins. 2014; 82:1444–1452. [DOI] [PubMed] [Google Scholar]
- 31. Maheshwari S., Brylinski M.. Predicting protein interface residues using easily accessible on-line resources. Brief. Bioinform. 2015; 16:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Howook H., Vreven T., Janin J., Weng Z.. Protein-protein docking benchmark version 4.0. Proteins. 2010; 78:3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laine E., Carbone A.. Local geometry and evolutionary conservation of protein surfaces reveal the multiple recognition patches in protein-protein interactions. PLoS Comput. Biol. 2015; 11:e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]