

Abstract
Summary
NGLview is a Jupyter/IPython widget to interactively view molecular structures as well as trajectories from molecular dynamics simulations. Fast and scalable molecular graphics are provided through the NGL Viewer. The widget supports showing data from the file-system, online data bases and from objects of many popular analysis libraries including mdanalysis, mdtraj, pytraj, rdkit and more.
Availability and implementation
The source code is freely available under the MIT license at https://github.com/arose/nglview. Python packages are available from PyPI and bioconda. NGLview uses Python on the server-side and JavaScript on the client. The integration with Jupyter is done through the ipywidgets package. The NGL Viewer is embedded client-side to provide WebGL accelerated molecular graphics.
1 Introduction
Interactive computing is a crucial strategy to tackle any scientific data heavy problem. It allows an exploratory process where each computation yields results that guide the next step, help verify previous steps and ultimately build knowledge. Jupyter Notebooks (http://jupyter.org/) are interactive computing environments for creating, sharing and editing documents that contain executable code, rich media and text. They are embraced by the research community for analyzing and sharing data (Grüning et al., 2017; Williams et al., 2017). By providing inlined output visualizations and explanatory text, code and data are put into context.
Performing analyses over large sets of molecular structures (like the Protein Data Bank archive) or trajectories from molecular dynamics simulations are challenging but common tasks in structural bioinformatics and computational biophysics. Traditionally, standalone visualization packages like VMD (Humphrey et al., 1996), Chimera (Pettersen et al., 2004) or PyMOL (http://pymol.org) are used to integrate molecular graphics and computations on molecular data. However, while very powerful and rich (via plugins), those tools are rather monolithic. Integrating computation and visualization in Jupyter notebooks offers a more modular and easier to contribute to approach. Several Python libraries are available to do computations on molecular structures and trajectories, for example biopython (Cock et al., 2009), mdanalysis (Michaud-Agrawal et al., 2011), mdtraj (McGibbon et al., 2015) and pytraj (https://github.com/Amber-MD/pytraj). By integrating computations and interactive visualization, Jupyter notebooks enable exploratory analysis of molecular data. Here, we present NGLview, a widget that provides interactive molecular graphics within Jupyter notebooks. Related tools that integrate molecular graphics in Jupyter notebooks include 3dmol.js (Rego and Koes, 2015), chemview (https://github.com/gabrielelanaro/chemview) and pv (https://github.com/biasmv/pv). The focus of NGLview is in offering a broad set of features via an API and the integration of many third-party libraries for computations on molecular data.
2 NGLview
NGLview is a Python package that provides a visualization widget for Jupyter/IPython notebooks. It allows interactive viewing of molecular structures as well as trajectories from molecular dynamics simulations (Fig. 1). To offer fast and scalable molecular graphics the NGL Viewer is embedded in the widget to provide WebGL accelerated molecular graphics (Rose and Hildebrand, 2015; Rose et al., 2016). Coordinates are sent frame by frame to the notebook allowing viewing of large trajectories.
Fig. 1.
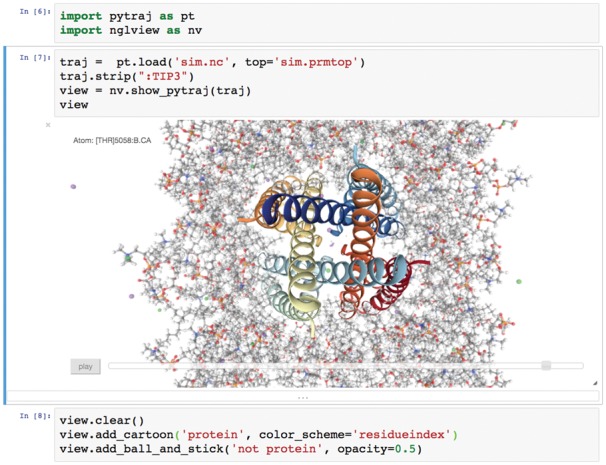
Example Jupyter notebook session. A topology and a trajectory are loaded with pytraj and then rendered with NGLview, showing a cartoon representation for the protein and a ball and stick representation for everything else
2.1 Python API
The main object of NGLview is the NGLWidget class. An instance of the widget can either be constructed directly [NGLWidget()] or via one of the convenience methods to quickly load data from another library [e.g. show_pytraj(pytraj_object)], the file system or an online resource [e.g. show_pdbid(’1crn’)]. Each loaded object (structure, trajectory, volume) is displayed by any number of representations, for example cartoon or spacefill for molecular structures. Upon loading a set of default representations is displayed, but methods exist to add, change and remove representations. The API contains many more methods to customize the visualization and interact with it. Examples include, creating screenshots and download/use them within the notebook, synchronizing the orientation of the view in multiple widgets within a single notebook or getting information about a picked/clicked atom for use within the notebook. The full API documentation is available online (http://nglviewer.org/nglview/latest/api.html).
2.2 Supported data sources and extendability
NGLview can read structures and/or trajectories from the data classes in a growing number of libraries, currently including ase (Hjorth Larsen et al., 2017), biopython, cctbx (Grosse-Kunstleve et al., 2002), HTMD (Doerr et al., 2016), mdanalysis, mdtraj, ParmEd (https://github.com/ParmEd/ParmEd), pyrosetta (Chaudhury et al., 2010), pytraj and rdkit (http://www.rdkit.org/). Moreover, structures can be downloaded from the Protein Data Bank or loaded from the file system.
Users can extend NGLview’s data reading support. Although NGLview supports a large number of popular data sources (library classes, online databases) it is straightforward to add custom adaptors for structure or trajectory data. Any class that implements the Structure or Trajectory interface from base_adaptor can be passed to the NGLWidget constructor to create a view for that data.
2.3 Example use cases
NGLview is used by pytraj and pdb4amber as part of the AmberTools suite http://ambermd.org/, which describes in a number of tutorials how to use NGLview and pytraj within Jupyter notebooks. Tutorials include setting up simulations, using Jupyter notebook remotely, basic visualization of molecules, visualize trajectories, including running simulations. Similar tasks can be performed with other analysis libraries (e.g. HTMD, mdanalysis or mdtraj) as well.
The Molecular Projection Explorer, molPX (https://github.com/markovmodel/molPX), makes use of NGLview to provide interactive visualization of projected coordinates of molecular dynamics trajectories within Jupyter notebooks. The packages connect matplotlib plots (e.g. projected FESs and transition networks) bi-directionally with an NGLWidget to make corresponding 3D structures easily accessible.
The protein-protein interaction docking analysis package, pida (https://github.com/jharman25/pida), streamlines analysis and visualization of protein-protein interaction results from various docking servers. It uses NGLview to visualize individual docking solutions and color it, for example, by a heatmap of aggregated atomic contacts.
3 Conclusion
NGLview is a versatile tool for molecular visualization within Jupyter notebooks. It provides a simple but powerful API. The adoption by the structural bioinformatics and computational biophysics community shows the need for and usefulness of rich media visualizations alongside computations in notebooks.
Acknowledgement
The authors would like to thank S.K. Burley for useful discussions as well as all contributors to the NGLview and NGL projects (http://nglviewer.org).
Funding
The RCSB PDB was jointly funded by the NSF, the NIH and the US DoE [NSF DBI-1338415; PI: SK Burley]. H.N. was supported by NIH Grant GM103297, ‘The Center for HIV RNA Studies.’
Conflict of Interest: none declared.
References
- Chaudhury S. et al. (2010) PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics, 26, 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P.J.A. et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr S. et al. (2016) HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput., 12, 1845–1852. [DOI] [PubMed] [Google Scholar]
- Grosse-Kunstleve R.W. et al. (2002) The computational crystallography toolbox: crystallographic algorithms in a reusable software framework. J. Appl. Cryst., 35, 126–136. [Google Scholar]
- Grüning B.A. et al. (2017) Jupyter and Galaxy: easing entry barriers into complex data analyses for biomedical researchers. PLoS Comput. Biol., 13, e1005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorth Larsen A. et al. (2017) The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter, 29, 273002. [DOI] [PubMed] [Google Scholar]
- Humphrey W. et al. (1996) VMD: visual molecular dynamics. J. Mol. Graph., 14, 33–38. [DOI] [PubMed] [Google Scholar]
- McGibbon R.T. et al. (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J., 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N. et al. (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem., 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F. et al. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem., 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rego N., Koes D. (2015) 3Dmol.js: molecular visualization with WebGL. Bioinformatics, 31, 1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose A.S., Hildebrand P.W. (2015) NGL Viewer: a web application for molecular visualization. Nucleic Acids Res., 43, W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose A.S. et al. (2016) Web-based molecular graphics for large complexes. Proc. 21st Int. Conf. Web3D Technol. Web3D, 16, 185–186. [Google Scholar]
- Williams E. et al. (2017) Image data resource: a bioimage data integration and publication platform. Nat. Methods, 14, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]