

ABSTRACT
Background
Recent studies have highlighted a critical role for CD40 in the pathogenesis of renal injury and fibrosis. However, little is currently understood about the regulation of CD40 in this setting.
Methods
We use novel Na/K-ATPase cell lines and inhibitors in order to demonstrate the regulatory function of Na/K-ATPase with regards to CD40 expression and function. We utilize 5/6 partial nephrectomy as well as direct infusion of a Na/K-ATPase ligand to demonstrate this mechanism exists in vivo.
Results
We demonstrate that knockdown of the α1 isoform of Na/K-ATPase causes a reduction in CD40 while rescue of the α1 but not the α2 isoform restores CD40 expression in renal epithelial cells. Second, because the major functional difference between α1 and α2 is the ability of α1 to form a functional signaling complex with Src, we examined whether the Na/K-ATPase/Src complex is important for CD40 expression. We show that a gain-of-Src binding α2 mutant restores CD40 expression while loss-of-Src binding α1 reduces CD40 expression. Furthermore, loss of a functional Na/K-ATPase/Src complex also disrupts CD40 signaling. Importantly, we show that use of a specific Na/K-ATPase/Src complex antagonist, pNaKtide, can attenuate cardiotonic steroid (CTS)-induced induction of CD40 expression in vitro.
Conclusions
Because the Na/K-ATPase/Src complex is also a key player in the pathogenesis of renal injury and fibrosis, our new findings suggest that Na/K-ATPase and CD40 may comprise a pro-fibrotic feed-forward loop in the kidney and that pharmacological inhibition of this loop may be useful in the treatment of renal fibrosis.
Keywords: CD40/CD40L, inflammation, Na/K-ATPase, renal fibrosis, Src
INTRODUCTION
Na/K-ATPase is a P-type ATPase capable of transporting sodium and potassium against their concentration gradients [1]. It has been established that four isoforms of the α subunit of Na/K-ATPase exist—of note, the α1 isoform is expressed ubiquitously, while the α2 isoform is predominately found in skeletal muscle [2]. Interestingly, studies from our group and others have demonstrated that in addition to its role as an ion transporter, the α1 isoform can form a functional signaling complex with Src, a non-receptor tyrosine kinase, which is known to be involved in numerous signaling cascades [3]. This Na/K-ATPase/Src signaling complex can be activated by the binding of cardiotonic steroids (CTSs) [4–6]. However, some controversy still exists regarding whether Na/K-ATPase and Src directly interact within this complex [7].
CD40, a type I transmembrane receptor of the toll-like receptor superfamily, was initially discovered as a mediator of humoral immune reactions [8]. However, it is now understood that CD40 is widely expressed, including within renal proximal tubular epithelial cells, and may serve broader functions [9, 10]. Interestingly, we have previously shown that circulating levels of the ligand for CD40, soluble CD40 ligand (sCD40L), are significantly increased in subjects with renal artery stenosis [11]. Follow-up studies have identified circulating CD40 mediators as predictive of changes in renal function in subjects with renal artery stenosis [12] and chronic kidney disease [13]. Furthermore, we have recently demonstrated that functional deletion of CD40 in a strain of rat prone to kidney disease improved renal function and attenuated renal fibrosis [14]. However, little is currently understood about the regulation of CD40 in the setting of renal disease.
Recent studies have established a critical role of the Na/K-ATPase/Src signaling complex in the regulation of cellular signaling pathways important for reactive oxygen species (ROS) generation, inflammation and fibrosis in the kidney [15–17]. Given that both Na/K-ATPase and CD40 play central roles in mediating renal fibrosis and are known to reside in caveolae [18, 19], we speculate that Na/K-ATPase may crosstalk and be critically involved in the regulation of CD40 within renal parenchyma. Demonstration of a relationship between Na/K-ATPase and CD40 would reveal the existence of a profibrotic feed-forward signaling loop and identify a new pharmacological target for the treatment of renal fibrosis.
MATERIALS AND METHODS
Materials
Rabbit anti-CD40 antibody (#13545) was purchased from Abcam (Cambridge, MA, USA). Mouse anti-α2 antibody (AB9094-I) was purchased from EMD Millipore (Billerica, MA, USA). Mouse anti-α1 antibody (α6F) was from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA, USA). Rabbit anti-caveolin-1 antibody (D46G3) was purchased from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-α-tubulin (TU-02), mouse anti-c-Src (B-12) and all horseradish peroxidase–conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) Telocinobufagin (TCB) was purchased from Baoji Herbest Bio-tech (Baoji City, China). pNaKtide was synthesized and purchased from Ohio Peptide (Columbus, OH, USA). Materials for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Dulbecco’s modified Eagle medium (DMEM) with L-glutamine, 4.5 g/L glucose and sodium pyruvate was purchased from Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from Rocky Mountain Biologicals (Missoula, MT, USA). Rabbit anti-phospho-Src (Tyr418; 44-660 G), trypsin ethylenediaminetetraacetic acid (EDTA; 0.25%) and penicillin-streptomycin (10 000 U/mL) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). sCD40L was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Recombinant human TNF-α (300-01A) was purchased from PeproTech (Rocky Hill, NJ, USA).
Cell culture
LLC-PK1 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA; CL-101). For a detailed description of the generation of stable mutant Na/K-ATPase cell lines, please see relevant publications and Table 2 [20–23]. Cells were cultured in DMEM supplemented with 10% FBS with 100 U/mL penicillin and streptomycin.
Table 2.
List of cell lines used
| Cell line | Na/K-ATPase species and isoform | Na/K-ATPase ion-transport function | Ability to form a Na/K-ATPase/ Src complex |
|---|---|---|---|
| LLC-PK1 | Pig α1 | + | + |
| PY-17 | Pig α1 | − | − |
| AAC-19 | Rat α1 | + | + |
| LX-α2 | Rat α2 | + | − |
| LY-α2 | Rat α2 (mutant) | + | + |
| A425P | Rat α1 | + | − |
Animals
Male Sprague Dawley rats were used for all of the studies. All of the animal experimentation described in the article was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals using protocols approved by the University of Toledo, Health Science Campus, Institutional Animal Use and Care Committee. Rats weighing between 250 and 300 g (n ≥ 8 per group) were subjected to sham surgery, 5/6 partial nephrectomy (PNx), or minipump (ALZET model 2004, Durect, Cupertino, CA, USA) infusion of the CTS marinobufagenin (MBG) at 10 µg/kg/day inserted subcutaneously through a flank incision as described previously [24]. The animals were euthanized and tissues were collected for biochemical studies 4 weeks following treatment.
Immunoblot analysis (in vivo)
Renal cortex tissue was isolated from kidneys and homogenized in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (pH 7.0) (Santa Cruz Biotechnology; sc-24948). The proteins, obtained from tissue homogenates, were resolved on an SDS-PAGE using Precast Ready Gels 4–15% Tris-HCl, purchased from Bio-Rad. In all, 60 μg of protein per sample were loaded into each well. The proteins from the gel were electrotransferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in 20 mmol/L Tris-HCl (pH 7.5, 150 mmol/L NaCl and 0.1% Tween 20). Mouse anti-CD40 antibody (Abcam) was used to probe for CD40 and secondary anti-mouse antibody was purchased from Invitrogen (Grand Island, NY, USA).
Immunoblot analysis (in vitro)
Cells were washed with cold phosphate-buffered saline (PBS) and then lysed into radioimmunoprecipitation buffer (0.25% deoxycholate, 1% Nonidet P-40, 1 mM EDTA) with 1× Halt Protease and Phosphatase Inhibitor (Thermo Fisher, #78440). Cell lysates were vortexed and then rotated at 4° for 15 min. Cell lysates were then spun at 15 000 g for 15 min at 4°C and supernatants were transferred to a new tube. Cell lysates were stored at −80°C. Following determination of protein concentration, aliquots containing 30 μg of proteins were subjected to SDS-PAGE under reducing conditions unless otherwise noted. Proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Thermo Fisher, #88518) and then probed using the indicted antibody.
RNA isolation and real-time quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using the RNEasy kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s instructions. RNA was stored at −80°C. The concentration and purity of RNA was assessed using NanoDrop 2000 (Thermo Fisher). RNA was reverse transcribed into cDNA using the Qiagen RT2 First Strand cDNA kit. cDNA was stored at −20°C. qPCR reactions were performed on a Qiagen Rotor Gene Q system using the Qiagen RT2 FAST SYBR Green/ROX kit according to the manufacturer’s instructions.
Primers
Species-specific primers were designed using Primer-BLAST (National Center for Biotechnology Information, Bethesda, MD, USA) and were sent to Integrated DNA Technologies (Coralville, IA, USA) for synthesis. Primer sequences can be found in Table 1. Verification of single product amplification was assessed by analyzing melt curves as well as by running qPCR amplification products on an agarose gel.
Table 1.
List of primer sequences
| Gene name (sus scrofa) | Sequence (forward) | Sequence (reverse) |
|---|---|---|
| MCP-1/CCL2 | TTGCCCAGCCAGA TGCAATTA | ACCCACTTCTGCTT GGGTTC |
| β-actin/ACTB | GACATCCGCAAG GACCTCTA | ACACGGAGTACTT GCGCTCT |
Basal CD40 expression
Cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Once cells reached confluency, either RNA or protein was collected according to the protocols described above.
Effects of TCB on CD40 expression or Src kinase activity
Cells were serum starved upon reaching confluency for 16–24 h. Following serum starvation, appropriate concentrations of TCB were added to the cells for 24 h. For experiments utilizing our novel Na/K-ATPase/Src antagonist, pNaKtide, 1 µM pNaKtide was added to the cells for 30 min prior to any TCB treatments. To assess the effects of TCB on Src kinase activity, serum starved cells were incubated with the appropriate concentrations of TCB for 15 min.
Surface protein biotinylation
Cells were grown in 60-mm dishes until 90–100% confluent. Cells were washed three times with cold PBS containing 1 mM EDTA. Cells were incubated for 1 h with a total of 2.5 mL of sulfo-NHS-SS-biotin solution (Thermo Fisher, #21331), freshly diluted into biotinylation buffer (10 mM triethanolamine, pH 9.0, 250 mM sucrose). Plates were kept on ice, with gentle horizontal shaking. Cells were rinsed three times with PBS-EDTA with 100 mM glycine and then washed twice for 15 min on ice in order to quench any unbound biotin. Plates were then washed twice with cold PBS and cells were lysed in 200 µL of lysis buffer [50 mM Tris-HCl, 5 mM EDTA, 150 mM NaCl, 1%Triton X-100 and protein inhibitor mixture (pH 7.5)]. Cell lysates were vortexed and rotated at 4°C for 15 min. Cell lysates were then spun at 15 000 g for 15 min at 4°C. Following determination of protein concentration, microcentrifuge tubes containing 100 μg of protein, 35 μL of streptavidin agarose beads (Thermo Fisher, #20347) and lysis buffer to a final volume of 500 μL were rotated at 4°C overnight. Microcentrifuge tubes were washed five times with PBS. Laemmli loading buffer (35 μL) was added to each and samples were shaken in a 55°C water bath for 30 min to elute protein. Eluted protein was then subjected to SDS-PAGE, transferred onto a PVDF membrane and probed using the indicated antibody.
sCD40L and TNF-α treatments
The indicated cell type was serum starved for 16–24 h and treated with either 0, 25, 50 or 100 ng/mL sCD40L or 10 ng/mL TNF-α for 24 h. Following treatment, total RNA was collected and monocyte chemoattractant protein-1 (MCP-1/CCL2) gene expression was assessed.
Cellular fractionation
Subcellular fractionation was performed as previously described [25]. Briefly, cell homogenates were obtained in a 500 mM sodium carbonate (pH 11.0) solution. The sucrose content in homogenates was adjusted to 45% using 90% sucrose prepared in MBS (25 mM MES, 0.15 M NaCl, pH 6.5). Samples were placed at the bottom of an ultracentrifuge tube and loaded with 4 mL of 35% sucrose and 4 mL of 5% sucrose (both in MBS containing 250 mM sodium carbonate) and centrifuged at 39 000 rpm for 16–20 h in an SW41 rotor (Beckman Instruments, Fullerton, CA, USA). A total of 11 gradient fractions of 1 mL were collected from the top to the bottom of the centrifuge tube. Fractions 4 and 5 were combined and considered the caveolar-enriched fraction.
Cycloheximide
Cells were grown to 90–100% confluency and then incubated with 10 µg/mL cycloheximide for 24 h.
Statistical analysis
Data presented are the mean standard error (SE) of the mean of at least three independent experiments. Student’s unpaired t-test was used to assess statistically significant differences between two groups. One-way analysis of variance (ANOVA) and post hoc multiple comparisons tests were used when comparing more than two groups. Statistical significance was accepted as P < 0.05. All statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA).
RESULTS
Effects of CTS on CD40 expression
We have previously demonstrated in an in vivo system that chronic infusion of MBG, a CTS and ligand of Na/K-ATPase, causes renal fibrosis [15]. We have also shown that CTSs are increased in animals subjected to 5/6 PNx and that immunization against CTSs in the PNx model can improve renal function and attenuate renal fibrosis [16, 26]. To test whether Na/K-ATPase and CD40 crosstalk in the pathogenesis of renal fibrosis, we assessed kidney cortical expression of CD40 in animals infused with MBG or subjected to PNx. As illustrated in Figure 1A, both PNx and infusion of MBG led to statistically significant increases in CD40 expression in kidney cortex compared with sham-operated controls (P < 0.01). Kidney cortical Na/K-ATPase α1 expression was unaltered (Figure 1B).
FIGURE 1.

CTS causes increased CD40 expression in vivo and in vitro. (A) Kidney cortex homogenate from sham, 5/6 PNx and animals infused with MBG was analyzed by western blot for CD40 expression or (B) Src kinase activity. Blots were normalized using β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control and are expressed as a fraction of the sham control. The data are presented as mean ± SE. *P < 0.05, **P < 0.01 compared with sham. (C) LLC-PK1 cells were serum starved and treated with TCB for 24 h. CD40 expression, following treatment, was analysed by western blot. Results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control. The data are presented as mean ± SE. *P < 0.05, **P < 0.01 compared with control. (D) LLC-PK1 cells were serum starved and treated with TCB for 15 min. Src kinase activity was analysed by western blot. Blots were stripped and reprobed for total Src. Results were normalized using total Src as a loading control and are expressed as a fraction of the control. The data are presented as mean ± SE. *P < 0.05, **P < 0.01 compared with control. (E) TCB-treated LLC-PK1 cells were probed for Na/K-ATPase α1 expression by western blot. Results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control. The data are presented as mean ± SE. *P < 0.05 compared with control.
Next, in order to determine whether Na/K-ATPase is involved in the regulation of CD40 expression, we incubated LLC-PK1 cells, a pig proximal tubule epithelial cell line, for 24 h with TCB, a potent CTS known to be elevated in subjects with chronic kidney disease [27]. Moreover, it has been shown that levels of endogenous ouabain are not significantly altered in animals undergoing PNx, nor in subjects with CKD, which suggests that bufadienolides, such as TCB, may be the more physiologically relevant CTSs to use in our system [28]. By western blotting, we observed a statistically significant increase in CD40 expression with 10 nM of TCB when compared with our control group (Figure 1C; P < 0.01), confirming our in vivo observations with MBG. The effects of lower doses of TCB were also assessed (Supplementary data, Figure S1). Interestingly, our results demonstrated that TCB at both 10 and 100 nM induced increases in Src kinase activity (Figure 1D; P < 0.01).
Furthermore, as shown in Figure 1E, 10 nM TCB induced a statistically significant increase in the expression of α1 in LLC-PK1 cells (P < 0.05). Taken together, our data demonstrate that TCB, at physiologically relevant concentrations, induces the expression of CD40, Src kinase activity and α1 in LLC-PK1 cells.
The α1 isoform of Na/K-ATPase is an important regulator of CD40 expression
In order to further establish a role of the Na/K-ATPase α1 isoform in the regulation of CD40, we measured CD40 expression in LLC-PK1, PY-17 and AAC-19 cells. PY-17 cells are derived from LLC-PK1 and they express <10% of α1 as compared to LLC-PK1 cells (Table 2). AAC-19 cells are PY-17 cells transfected with rat α1 cDNA [20] (Table 2). As expected, while LLC-PK1 and AAC-19 cells expressed comparable amounts of α1, the α1 expression was barely detectable in PY-17 cells (Figure 2A). Interestingly, knockdown of the α1 isoform of Na/K-ATPase caused a statistically significant reduction in CD40 expression in PY-17 cells (P < 0.05). This reduction was fully recovered in AAC-19 cells. In aggregate, these results indicate that α1 is an important regulator of CD40 expression in renal epithelial cells.
FIGURE 2.

Role of Na/K-ATPase α1 in the regulation of CD40. (A) Basal CD40 expression was compared among LLC-PK1, PY-17 and AAC-19 cell lines by western blot. A representative blot showing relative Na/K-ATPase α1, CD40 and α-tubulin expression is shown. CD40 expression results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control (LLC-PK1). The data are presented as mean ± SE. *P < 0.05. (B) Basal CD40 expression was compared in AAC-19 and LX-α2 cell lines by western blot. A representative blot showing relative Na/K-ATPase α1 and α2, CD40 and α-tubulin expression is shown. Membranes were probed initially for α1 and then stripped and reprobed for α2. CD40 expression results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control (AAC-19). The data are presented as mean ± SE. *P < 0.05 compared with AAC-19. (C) Basal Src kinase activity was assessed in LLC-PK1, PY-17 and AAC-19 cell lines by western blot. A representative blot showing relative Src kinase activity and total Src expression is shown. (D) Basal Src kinase activity was assessed in the AAC-19 and LX-α2 cell lines by western blot. A representative blot showing relative Src kinase activity and total Src expression is shown.
To assess whether this regulation is specific to the α1 isoform of Na/K-ATPase, we measured CD40 expression in LX-α2 cells. LX-α2 cells are PY-17 cells transfected with rat α2 expressing vectors [21] (Table 2). As such, only the α2 isoform of Na/K-ATPase is detectable in LX-α2 cells. This is illustrated in the top two panels of Figure 2B. Interestingly, in contrast to AAC-19 cells, expression of α2 did not restore the expression of CD40 in LX-α2 cells. Because ouabain-sensitive ATPase activity in LX-α2 cells was comparable to that in AAC-19 cells [23], these findings suggest that the regulatory effect of α1 Na/K-ATPase on CD40 expression is unlikely, due to changes in ion pumping capacity.
We also compared the basal Src kinase activity in these cell lines. As illustrated in Figure 2C and 2D, PY-17 and LX-α2 cells had greater levels of Src kinase activity compared with their respective controls. These findings are in accordance with what has been previously reported in the literature [20, 21].
A functional Na/K-ATPase/Src complex is critical for regulation of CD40 expression
A major difference between the α1 and α2 isoforms of Na/K-ATPase is their ability to interact and regulate Src [23]. Because our results suggest that alterations in ion transport function alone are insufficient to explain the differences in CD40 expression among different cell lines, we tested the hypothesis that Na/K-ATPase/Src signaling complex might play a role in the regulation of CD40. We have previously shown that knockdown of the α1 isoform of Na/K-ATPase inhibits α1 Na/K-ATPase-mediated signal transduction in PY-17 cells. In contrast, expression of rat α1 fully restores this signaling mechanism in AAC-19 cells [20]. As depicted in Figure 3A, we found that TCB failed to stimulate CD40 expression in PY-17 cells and also failed to induce increases in Src kinase activity. On the other hand, both 100 nM and 1 μM TCB caused a statistically significant increase in CD40 expression in AAC-19 cells (Figure 3B; P < 0.05 and P < 0.01, respectively). Moreover, 1 μM TCB also caused a statistically significant increase in Src kinase activity (P < 0.05). Please note that because rat α1 is much less sensitive to TCB than that of pig α1, higher concentrations of TCB were used in this experiment [29].
FIGURE 3.

CTS mediates changes in CD40 expression via a Na/K-ATPase/Src-dependent mechanism. (A) PY-17 cells were serum starved and treated with TCB for 24 h to assess for CD40 expression or 15 min to assess for Src kinase activity. Results were normalized using α-tubulin or stripped and reprobed for total Src for use as a loading control. Results are expressed as a fraction of the control. The data are presented as mean ± SE. (B) AAC-19 cells were serum starved and treated with TCB for 24 h to assess for CD40 expression or 15 min to assess for Src kinase activity. Results were normalized using α-tubulin or stripped and reprobed for total Src for use as a loading control. Results are expressed as a fraction of the control. The data are presented as mean ± SE. *P < 0.05, **P < 0.01 compared with control. (C) LLC-PK1 cells were serum starved and treated with TCB with or without 1 µM pNaKtide pretreatment (30 min prior to addition of TCB) for either 24 h or 15 min. Results were normalized using α-tubulin or stripped and reprobed for total Src for use as a loading control. Results are expressed as a fraction of the control. The data are presented as mean ± SE. *P < 0.05 compared with control.
The above experiments suggest that signaling through the α1 Na/K-ATPase/Src complex may be important for regulation of CD40 expression. To further test this hypothesis, we determined the effect of pNaKtide, a specific inhibitor of the α1 Na/K-ATPase/Src complex [30, 31], on TCB-induced CD40 expression in LLC-PK1 cells. As depicted in Figure 3C, pNaKtide pretreatment inhibited TCB-induced increases in both CD40 expression and Src kinase activity.
To substantiate the above findings, we measured CD40 expression in a unique cell line that expresses a mutant α2 isoform of Na/K-ATPase with a gain-of-Src binding capability. Our prior studies have identified two putative Src binding sites in α1 Na/K-ATPase, sites that are completely conserved in mammalian α1. In contrast, the lack of these binding sites are also conserved in mammalian α2 Na/K-ATPase (publication under review). Substitution of four amino acids in α2 Na/K-ATPase into the corresponding α1 sequence resulted in a mutant α2 that possesses two α1-like Src binding sites. Transfection of PY-17 cells with this mutant α2 generated a stable cell line, named LY-α2, which responds to ouabain stimulation (publication under review) (Table 2). Our results, shown in Figure 4A, indicated that gain-of-α2 Src binding capabilities was sufficient to restore the expression of CD40 to a level similar to that in AAC-19 cells.
FIGURE 4.

A functional Na/K-ATPase/Src complex is critical for CD40 expression. (A) Basal CD40 expression was compared among the AAC-19, LX-α2 and LY-α2 cell lines by western blot. A representative blot showing relative Na/K-ATPase α1 and α2, CD40 and α-tubulin is shown. Membrane was initially probed for α1 and then stripped and reprobed for α2. CD40 expression results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control (AAC-19). The data are presented as mean ± SE. *P < 0.05. (B) Basal CD40 expression was compared between AAC-19 and A425P cell lines by western blot. A representative blot showing relative Na/K-ATPase α1 and α2, CD40 and α-tubulin is shown. CD40 expression results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control (AAC-19). The data are presented as mean ± SE. **P < 0.01 compared with AAC-19.
To further verify the importance of Src binding, we next measured CD40 expression in a novel loss-of-Src binding α1 Na/K-ATPase cell line. This cell line was generated by introducing a single amino acid substitution at alanine 425 (A425P), which has been shown to effectively disrupt the interaction between the α1 isoform of Na/K-ATPase and Src, abolishing ouabain-induced signal transduction [23] (Table 2). When we compared the basal CD40 expression in this mutant cell line with AAC-19 cells, we observed that there was a statistically significant reduction in CD40 expression (Figure 4B; P < 0.01). Importantly, the α1 expression in this cell line was comparable to the α1 expression in AAC-19 cells.
Na/K-ATPase regulates surface CD40 expression and function
Because CD40 is a transmembrane receptor [8], we reasoned that overall changes in CD40 expression may not be of functional relevance if surface expression of CD40 remained unaltered in our various mutant cell lines. To address this, we utilized a surface protein biotinylation assay to specifically assess surface expression of CD40. Much like what was observed with regards to total levels of CD40, our results show that LX-α2 cells expressed statistically significant lower amounts of surface CD40 compared with AAC-19 and LY-α2 cells (Figure 5; P < 0.05). To ensure that cytosolic proteins were not biotinylated, we tested for expression of HSP60 and ERK1/2 in both surface and total preparations. No significant amounts of HSP60 or ERK1/2 were found in biotinylated preparations (Supplementary data, Figure S2).
FIGURE 5.

The Na/K-ATPase/Src complex regulates surface CD40 expression. AAC-19, LX-α2 and LY-α2 cells underwent a surface biotinylation procedure as described in the ‘Materials and Methods’ section. A representative western blot showing relative surface CD40 (100 µg) and total CD40 (30 µg) expression is shown. Surface CD40 results are presented as mean ± SE. *P < 0.05.
Next, because we observed differences in surface CD40, we assessed whether disruption of the Na/K-ATPase/Src complex affected downstream CD40 signaling pathways. It has been previously established that activation of CD40 in proximal tubule cells leads to an increase in MCP-1, a pro-inflammatory molecule [10]. In order to test whether disruption of the Na/K-ATPase/Src complex reduces CD40 signaling–induced MCP-1 expression, we activated CD40 signaling pathways in LLC-PK1 cells using sCD40L. As illustrated in Figure 6A, a statistically significant increase in MCP-1 expression was observed with 100 ng/mL of sCD40L by real-time qPCR (P < 0.01). In contrast, in PY-17 cells, which we have shown to have reduced basal CD40 expression, there was no statistically significant change in MCP-1 expression (Figure 6B). In AAC-19 cells, 100 ng/mL sCD40L caused a statistically significant increase in MCP-1 messenger RNA (mRNA) expression, although the magnitude of the increase was less than what was observed in LLC-PK1 cells (Figure 6C; P < 0.01). As shown in Figure 6D, our pump-functional, Na/K-ATPase/Src signaling deficient, LX-α2 cells had no change in MCP-1 expression following sCD40L treatment, which mirrors what was observed in PY-17 cells (Figure 6B). Finally, in our α2 gain-of-function mutant cell line, LY-α2, the effects of sCD40L on MCP-1 expression were restored (Figure 6E; P < 0.01). Finally, to rule out the possibility that the lack of response in sCD40L treated PY-17 and LX-α2 cells was due to a general defect in MCP-1, we incubated all cell lines with 10 ng/mL TNF-α for 24 h. By comparing the ΔΔCT of control versus TNF-α-treated cells, our results demonstrate that although not statistically significant, there was a trend towards LLC-PK1 cells having the largest increase in MCP-1 mRNA expression following incubation with TNF-α (Figure 6F). Moreover, PY-17, AAC-19 and LY-α2 cells all had similar levels of induction in MCP-1 mRNA expression (Figure 6F). Somewhat surprisingly, we observed that although LX-α2 still responded to TNF-α by increasing MCP-1 mRNA expression (by about two cycles), the magnitude of increase was statistically significantly lower as compared with the other tested cell lines (Figure 6F). This suggests that the lack of response to sCD40L in LX-α2 but not PY-17 cells may be due in part to a general defect in MCP-1 regulation.
FIGURE 6.

Disruption of the Na/K-ATPase/Src complex affects CD40 signaling pathways. (A) LLC-PK1, (B) PY-17, (C) AAC-19, (D) LX-α2 and (E) LY-α2 cells were serum starved and treated with sCD40L (0, 10, 50 and 100 ng/mL) for 24 h. Change in MCP-1 expression was measured by real-time qPCR. Results were normalized using levels of β-actin mRNA as a loading control and expressed as a fraction of the control using the comparative CT method (2−ΔΔCT). The data are presented as mean ± SE. **P < 0.01 compared with control. (F) LLC-PK1, PY-17, AAC-19, LX-α2 and LY-α2 were serum starved and treated with 10 ng/mL TNF-α for 24 h. Change in MCP-1 expression was measured by real-time qPCR. Results were normalized using levels of β-actin mRNA as a loading control and are expressed using the ΔΔCT method.
Both Na/K-ATPase and CD40 are known to reside in caveolae [18, 19]. We therefore tested whether disruption of the Na/K-ATPase/Src signaling complex affected the cellular localization of these receptors. We followed a previously established protocol of bicarbonate extraction and sucrose density centrifugation to fractionate cell lysates [32]. We observed that caveolin-1 (Figure 7A), a structural protein of caveolae, resided in low-density fraction 4/5 in AAC-19 and LX-α2 cells. α2 Na/K-ATPase (Figure 7B) and Src (Figure 7C) migrated away from fraction 4/5 in LX-α2 cells but not in AAC-19 cells, as previously reported (publication under review). Interestingly, CD40 remained concentrated in low-density fraction 4/5 in both AAC-19 and LX-a2 cells (Figure 7D).
FIGURE 7.

Disruption of the Na/K-ATPase/Src complex does not alter CD40 localization. Cell lysates from AAC-19 and Lx-α2 cells were collected and subjected to fractionation by sucrose gradient centrifugation as described in the ‘Materials and Methods’ section. An equal volume was loaded for fractions 4/5–11. Cellular localization of (A) caveolin-1, (B) Na/K-ATPase α1 or α2, (C) Src and (D) CD40 was analysed by western blot. A western blot representative of at least three independent experiments is shown.
Na/K-ATPase regulates CD40 expression at the posttranscriptional level
To further characterize the relationship between the Na/K-ATPase/Src signaling complex and CD40, we assessed the basal CD40 mRNA expression in our different cell lines. Interestingly, in contrast to what was seen at the protein level, we observed no statistically significant differences in baseline CD40 mRNA expression (Figure 8A and B). Moreover, TCB incubation was unable to induce changes in LLC-PK1 mRNA expression (Figure 8C). Therefore this suggests that the Na/K-ATPase/Src signaling complex regulates CD40 expression at the posttranscriptional level. Thus we treated AAC-19 cells and LX-α2 with 10 µg/mL cycloheximide for 24 h and compared the effects of protein synthesis inhibition on basal CD40 expression between these two cell lines. We observed that 10 µg/mL cycloheximide induced a statistically similar degree of degradation of CD40 in both AAC-19 and LX-α2 cells (Figure 8D).
FIGURE 8.

Na/K-ATPase/Src complex regulates CD40 expression at the posttranscriptional level. Relative (2−ΔΔCT) basal CD40 mRNA levels were compared between (A) AAC-19 and PY-17 cells or (B) LY-α2 and LX-α2 cells. Results were normalized using β-actin mRNA as a loading control. The data are presented as mean ± SE. (C) LLC-PK1 cells were serum starved and treated with either 10 or 100 nM TCB for 24 h. The change in CD40 mRNA expression was analyzed by real-time qPCR. Results were normalized using levels of β-actin mRNA as a loading control and expressed as a fraction of the control using the comparative CT method (2−ΔΔCT). (D) AAC-19 and LX-α2 were treated with 10 µg/mL cycloheximide for 24 h. The change in CD40 protein expression was analyzed by western blotting. Results were normalized using α-tubulin as a loading control and are expressed as a fraction of the control. The data are presented as mean ± SE. **P < 0.01 compared with control.
DISCUSSION
We report the identification of a novel Na/K-ATPase α1–mediated CD40 regulatory pathway. In particular, we identify the α1 Na/K-ATPase/Src complex as an integral part of this regulatory mechanism, as disruption or blockade of the Na/K-ATPase/Src complex leads to dysregulated basal CD40 and inhibition of CTS-mediated increases in CD40 expression. We further demonstrate that disruption of the Na/K-ATPase/Src complex causes a reduction in surface CD40 expression and affects downstream CD40 signaling pathways.
Interestingly, our data supports the hypothesis that it is not necessarily the absolute degree of Src kinase activity that is critical in regulation of CD40 per se, as we observed that in both PY-17 and LX-a2 cells there was increased basal Src kinase activity but reduced basal expression of CD40. Instead, we hypothesize that the functional status of the Na/K-ATPase/Src signaling complex is the critical factor in the regulation of CD40 expression and function. A similar phenomenon has been previously described in the literature as greater levels of Src kinase activity, in the setting of a disrupted Na/K-ATPase/Src signaling complex, were paradoxically associated with diminished cellular proliferation and cell spreading, two processes known to be regulated by Src [23].
Somewhat surprisingly, we were unable to observe any statistically significant changes in the expression of CD40 at the mRNA level with any of our manipulated Na/K-ATPase cell lines or with the use of TCB as a Na/K-ATPase/Src signaling agonist (Figure 8). This suggests a posttranscriptional regulatory mechanism. Moreover, the effects of cycloheximide on CD40 expression in AAC-19 and LX-α2 cells were similar. We have previously demonstrated that CTS-induced increases in α1 were mediated through the mammalian target of rapamycin (mTOR) pathway, a pathway that is known to be involved in the regulation of mRNA translation [33]. Whether the Na/K-ATPase/Src complex regulates CD40 expression in a similar fashion by affecting the rate of protein translation is the subject of future studies.
CD40 lacks intrinsic kinase domains and requires both caveolae and Src family kinases to mediate signaling [9, 18, 34–37]. We have previously demonstrated that the Na/K-ATPase/Src complex plays a critical role in caveolin-1, an important structural component of caveolae trafficking [32]. In accordance with previous publications, we observed that in LX-α2 cells, lack of a functional Na/K-ATPase/Src complex led to changes in the localization of Na/K-ATPase α2 and Src from caveolae into higher-density fractions. Interestingly, CD40 remained localized in caveolar fractions in both AAC-19 and LX-α2 cells. Therefore, although the Na/K-ATPase/Src complex is critical in the regulation of CD40 expression and function, disruption of this complex does not affect CD40 localization. On the other hand, our results show that CD40 signaling pathways are disrupted in LX-α2 cells, as sCD40L failed to stimulate an induction in MCP-1. We hypothesize that this may be due in part to the fact that, in these cells, surface CD40 expression was decreased and a subset of Src was no longer located within caveolae. Loss of this caveolar Src may disrupt the ability of CD40 to interact with Src and activate downstream signaling cascades. To this end, it has been previously demonstrated that Na/K-ATPase interacts with and facilitates signaling of another intrinsic kinase-less receptor in caveolae, scavenger receptor CD36, in a similar fashion [38, 39]. However, it is important to note that LX-α2 cells may have generalized defects in MCP-1 regulation. Therefore, MCP-1 may not be the most appropriate marker for CD40 signaling in this particular cell line.
CD40 has been implicated in the pathogenesis of renal fibrosis. Previous studies from our group and others have demonstrated that plasminogen activation inhibitor-1 (PAI-1) as well as the Src family kinase Lyn are both involved in CD40-mediated renal fibrosis [14, 40, 41]. Interestingly, it has been demonstrated that in vascular smooth muscle cells, ROS play an important role in the regulation of CD40 and that activation of CD40 leads to subsequent ROS generation [42, 43]. Moreover, recent studies have begun to unravel a complex relationship between the Na/K-ATPase/Src complex, ROS and CTS. Specifically, it has been demonstrated that CTS-induced activation of the Na/K-ATPase/Src complex is mediated in part by ROS signaling and that ROS can activate Na/K-ATPase/Src signaling cascades directly [44–46]. Given the large body of literature implicating ROS as a key player in the pathogenesis of renal fibrosis [47–49], we speculate that ROS may be a potential driver in a Na/K-ATPase/CD40 profibrotic feed-forward loop (Figure 9). Importantly, it has been shown that pNaKtide is capable of attenuating Na/K-ATPase/Src-mediated ROS signaling in an obesity model in vivo [50]. Moreover, we show here that pNaKtide can block CTS-induced increases in CD40 expression in vitro. Critically, the in vivo utility of pNaKtide in suppressing the Na/K-ATPase/CD40 signaling axis and its potential effect on renal fibrosis still needs to be evaluated.
FIGURE 9.
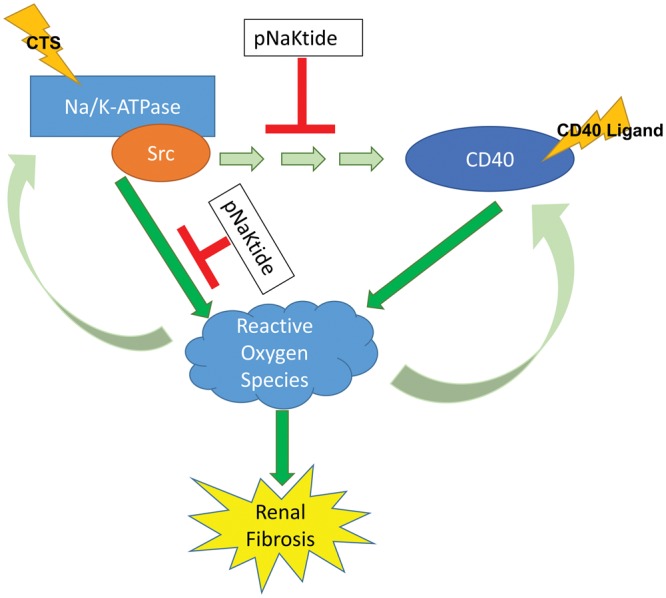
Na/K-ATPase/CD40 feed-forward signaling loop.
In short, we have uncovered a novel CD40 regulatory pathway. Our results have identified a critical role for the α1 isoform of Na/K-ATPase in the regulation of CD40 expression and function. It should be noted that a number of specific details regarding the precise mechanism of this regulatory process remain unanswered. Yet, given the known importance of both of these signaling molecules in the pathogenesis of renal fibrosis, our results provide evidence of a profibrotic feed-forward signaling loop between Na/K-ATPase and CD40. Future endeavors will evaluate the therapeutic potential of pharmacological blockade of the Na/K-ATPase/CD40 signaling dyad for renal fibrosis.
AUTHORS’ CONTRIBUTIONS
J.X.X., X.C., J.Z., H.Y., F.K.K. and S.Z. performed the experiments. D.M., J.I.S., J.T. and S.T.H. contributed to the conception and design of the study. J.X.X., DK, J.L.S., J.T. and S.T.H. analyzed and interpreted the data. J.X.X. and S.T.H. drafted the manuscript. All authors contributed to the revision and final approval of the manuscript.
FUNDING
The authors gratefully acknowledge support from the American Heart Association (13POST16860035 to S.T.H., 12SDG12050473 to D.J.K.), the National Institutes of Health (grant no. 1RO1HL105649 to J.T.; 1RO1HL137004 to S.T.H, J.T. and D.J.K.), the David and Helen Boone Foundation Research Fund (D.J.K.), Early Career Development Award from the Central Society for Clinical and Translational Research (D.J.K.) and the University of Toledo Women and Philanthropy Genetic Analysis Instrumentation Center (D.J.K, S.T.H.).
We would like to thank Dr Zijian Xie for providing us with the mutant Na/K-ATPase cell lines.
SUPPLEMENTARY DATA
Supplementary data are available at ndt online.
CONFLICT OF INTEREST STATEMENT
None declared. The results presented in this article have not been published previously in whole or part, except in abstract format.
Supplementary Material
REFERENCES
- 1. Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 1957; 23: 394–401 [DOI] [PubMed] [Google Scholar]
- 2. Lingrel JB. Na, K-ATPase: isoform structure, function, and expression. J Bioenerg Biomembr 1992; 24: 263–270 [DOI] [PubMed] [Google Scholar]
- 3. Tian J, Cai T, Yuan Z. et al. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell 2006; 17: 317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu J, Tian J, Haas M. et al. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 2000; 275: 27838–27844 [DOI] [PubMed] [Google Scholar]
- 5. Nguyen AN, Wallace DP, Blanco G.. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J Am Soc Nephrol 2007; 18: 46–57 [DOI] [PubMed] [Google Scholar]
- 6. Li J, Zelenin S, Aperia A. et al. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J Am Soc Nephrol 2006; 17: 1848–1857 [DOI] [PubMed] [Google Scholar]
- 7. Yosef E, Katz A, Peleg Y. et al. Do Src kinase and caveolin interact directly with Na,K-ATPase? J Biol Chem 2016; 291: 11736–11750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Kooten C, Banchereau J.. CD40-CD40 ligand. J Leukoc Biol 2000; 67: 2–17 [DOI] [PubMed] [Google Scholar]
- 9. Schonbeck U, Libby P.. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci 2001; 58: 4–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H, Nord EP.. CD40 ligation stimulates MCP-1 and IL-8 production, TRAF6 recruitment, and MAPK activation in proximal tubule cells. Am J Physiol Renal Physiol 2002; 282: F1020–F1033 [DOI] [PubMed] [Google Scholar]
- 11. Haller S, Adlakha S, Reed G. et al. Platelet activation in patients with atherosclerotic renal artery stenosis undergoing stent revascularization. Clin J Am Soc Nephrol 2011; 6: 2185–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haller ST, Kalra PA, Ritchie JP. et al. Effect of CD40 and sCD40L on renal function and survival in patients with renal artery stenosis. Hypertension 2013; 61: 894–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie JX, Alderson H, Ritchie J. et al. Circulating CD40 and sCD40L predict changes in renal function in subjects with chronic kidney disease. Sci Rep 2017; 7: 7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haller ST, Kumarasamy S, Folt DA. et al. Targeted disruption of Cd40 in a genetically hypertensive rat model attenuates renal fibrosis and proteinuria, independent of blood pressure. Kidney Int 2017; 91:365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fedorova LV, Raju V, El-Okdi N. et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol 2009; 296: F922–F934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haller ST, Drummond CA, Yan Y. et al. Passive immunization against marinobufagenin attenuates renal fibrosis and improves renal function in experimental renal disease. Am J Hypertens 2014; 27: 603–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tian J, Shidyak A, Periyasamy SM. et al. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension 2009; 54: 1313–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Nord EP.. Functional caveolae are a prerequisite for CD40 signaling in human renal proximal tubule cells. Am J Physiol Renal Physiol 2004; 286: F711–F719 [DOI] [PubMed] [Google Scholar]
- 19. Liang M, Tian J, Liu L. et al. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem 2007; 282: 10585–10593 [DOI] [PubMed] [Google Scholar]
- 20. Liang M, Cai T, Tian J. et al. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J Biol Chem 2006; 281: 19709–19719 [DOI] [PubMed] [Google Scholar]
- 21. Xie J, Ye Q, Cui X. et al. Expression of rat Na-K-ATPase α2 enables ion pumping but not ouabain-induced signaling in α1-deficient porcine renal epithelial cells. Am J Physiol Cell Physiol 2015; 309: C373–C382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lai F, Madan N, Ye Q. et al. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. J Biol Chem 2013; 288: 13295–13304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banerjee M, Duan Q, Xie Z.. SH2 ligand-like effects of second cytosolic domain of Na/K-ATPase alpha1 subunit on Src kinase. PLoS One 2015; 10: e0142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haller ST, Yan Y, Drummond CA. et al. Rapamycin attenuates cardiac fibrosis in experimental uremic cardiomyopathy by reducing marinobufagenin levels and inhibiting downstream pro-fibrotic signaling. J Am Heart Assoc 2016; 5: e004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H, Haas M, Liang M. et al. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 2004; 279: 17250–17259 [DOI] [PubMed] [Google Scholar]
- 26. Kennedy DJ, Vetteth S, Periyasamy SM. et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006; 47: 488–495 [DOI] [PubMed] [Google Scholar]
- 27. Komiyama Y, Dong XH, Nishimura N. et al. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem 2005; 38: 36–45 [DOI] [PubMed] [Google Scholar]
- 28. Kolmakova EV, Haller ST, Kennedy DJ. et al. Endogenous cardiotonic steroids in chronic renal failure. Nephrol Dial Transplant 2011; 26: 2912–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loreaux EL, Kaul B, Lorenz JN. et al. Ouabain-sensitive α1 Na, K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol 2008; 19: 1947–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z, Cai T, Tian J. et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem 2009; 284: 21066–21076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J, Tian J, Chaudhry M. et al. Attenuation of Na/K-ATPase mediated oxidant amplification with pnaktide ameliorates experimental uremic cardiomyopathy. Sci Rep 2016; 6: 34592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai T, Wang H, Chen Y. et al. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 2008; 182: 1153–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian J, Li X, Liang M. et al. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J Biol Chem 2009; 284: 14921–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vidalain PO, Azocar O, Servet-Delprat C. et al. CD40 signaling in human dendritic cells is initiated within membrane rafts. Embo J 2000; 19: 3304–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brenner B, Koppenhoefer U, Lepple-Wienhues A. et al. The CD40 ligand directly activates T-lymphocytes via tyrosine phosphorylation dependent PKC activation. Biochem Biophys Res Commun 1997; 239: 11–17 [DOI] [PubMed] [Google Scholar]
- 36. Neron S, Suck G, Ma XZ. et al. B cell proliferation following CD40 stimulation results in the expression and activation of Src protein tyrosine kinase. Int Immunol 2006; 18: 375–387 [DOI] [PubMed] [Google Scholar]
- 37. Mukundan L, Milhorn DM, Matta B. et al. CD40-mediated activation of vascular smooth muscle cell chemokine production through a Src-initiated, MAPK-dependent pathway. Cell Signal 2004; 16: 375–384 [DOI] [PubMed] [Google Scholar]
- 38. Kennedy DJ, Chen Y, Huang W. et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension 2013; 61: 216–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Y, Kennedy DJ, Ramakrishnan DP. et al. Oxidized LDL-bound CD36 recruits an Na+/K+-ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci Signal 2015; 8: ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kairaitis L, Wang Y, Zheng L. et al. Blockade of CD40-CD40 ligand protects against renal injury in chronic proteinuric renal disease. Kidney Int 2003; 64: 1265–1272 [DOI] [PubMed] [Google Scholar]
- 41. Pontrelli P, Ursi M, Ranieri E. et al. CD40L proinflammatory and profibrotic effects on proximal tubular epithelial cells: role of NF-κB and lyn. J Am Soc Nephrol 2006; 17: 627–636 [DOI] [PubMed] [Google Scholar]
- 42. Souza HP, Frediani D, Cobra AL. et al. Angiotensin II modulates CD40 expression in vascular smooth muscle cells. Clin Sci 2009; 116: 423–431 [DOI] [PubMed] [Google Scholar]
- 43. Laxmanan S, Datta D, Geehan C. et al. CD40: a mediator of pro- and anti-inflammatory signals in renal tubular epithelial cells. J Am Soc Nephrol 2005; 16: 2714–2723 [DOI] [PubMed] [Google Scholar]
- 44. Yan Y, Shapiro AP, Haller S. et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem 2013; 288: 34249–34258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y, Ye Q, Liu C. et al. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic Biol Med 2014; 71: 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elkareh J, Kennedy DJ, Yashaswi B. et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007; 49: 215–224 [DOI] [PubMed] [Google Scholar]
- 47. Sedeek M, Nasrallah R, Touyz RM. et al. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 2013; 24: 1512–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ha H, Lee HB.. Reactive oxygen species and matrix remodeling in diabetic kidney. J Am Soc Nephrol 2003; 14: S246–S249 [DOI] [PubMed] [Google Scholar]
- 49. Barnes JL, Gorin Y.. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int 2011; 79: 944–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sodhi K, Maxwell K, Yan Y. et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv 2015; 1: e1500781. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.