


Abstract
With the rapid accumulation of high-throughput microRNA (miRNA) expression profile, the up-to-date resource for analyzing the functional and disease associations of miRNAs is increasingly demanded. We here describe the updated server TAM 2.0 for miRNA set enrichment analysis. Through manual curation of over 9000 papers, a more than two-fold growth of reference miRNA sets has been achieved in comparison with previous TAM, which covers 9945 and 1584 newly collected miRNA-disease and miRNA-function associations, respectively. Moreover, TAM 2.0 allows users not only to test the functional and disease annotations of miRNAs by overrepresentation analysis, but also to compare the input de-regulated miRNAs with those de-regulated in other disease conditions via correlation analysis. Finally, the functions for miRNA set query and result visualization are also enabled in the TAM 2.0 server to facilitate the community. The TAM 2.0 web server is freely accessible at http://www.scse.hebut.edu.cn/tam/ or http://www.lirmed.com/tam2/.
INTRODUCTION
MicroRNAs (miRNAs) are endogenous small RNAs that play versatile regulatory roles in cell proliferation, development and metabolism by targeting other genes (1). Analyzing the involvement of miRNAs becomes increasingly important not only for understanding the molecular mechanisms of physiology and pathology but also for discovering novel clinical biomarkers and therapeutic targets of complex diseases like cancer and cardiovascular diseases (2–5). With high-throughput transcriptome profiling techniques like microarray and RNA-seq, one could easily obtain a list of de-regulated miRNAs. Methods to interpret the functions of these de-regulated miRNAs are therefore demanded. Enrichment analysis has been widely applied to interpret de-regulated coding genes, proteins or microbiota (6–8). Similarly, miRNA set enrichment analysis, which compares the query miRNA list with the reference miRNA sets to infer functional and disease associations, has been increasingly required in the pipeline of high-throughput miRNA assays (9).
Intuitively, the usefulness of an enrichment analysis tool heavily depends on the quality and coverage of its reference miRNA sets. Because the annotations of miRNAs are less comprehensive than those of coding genes, one solution would be to deduce the function of miRNAs from the functions of their target genes. Indeed, several available tools implemented such target gene-based miRNA enrichment analysis, e.g. miRWalk 2.0 (10,11) and DIANA-miRPath v3.0 (12). Nevertheless, due to the promiscuity of miRNA targeting, one miRNA could be related to hundreds of genes and therefore to many weakly associated functional terms, which finally results in unsatisfactory quality of miRNA sets and biased analysis results (9,13). The alternative choice is to directly compile the miRNA sets through manually curation. Manually curated annotations suffer from the publication bias (e.g. cancer is much more intensively investigated than others) and inaccuracy (e.g. some molecular phenotype could be artefact resulted from experiment design). Nevertheless, it is still a well-accepted way to obtain miRNA-disease associations (14,15). In 2010, we established a miRNA set enrichment tool TAM (16), which was based on our miRNA-disease association database HMDD (14) and literature reading. After two rounds of updates, the TAM tool covered 362 miRNA sets, including 43 miRNA-function sets and 183 miRNA-disease sets, and became one of the most popular miRNA set enrichment analysis tool. However, since the manual literature reading is labor-intensive, the available tools often prominently shared their curated miRNA sets albeit their statistical methods would differ. The miSEA server (http://www.baskent.edu.tr/∼hogul/misea/) implemented distinct statistical method than TAM, but its reference miRNA sets largely overlapped with those of TAM, with noticeable exception of ∼200 newly curated miRNA-disease sets that provided important adding value (17). The miEAA server (http://www.ccb.uni-saarland.de/mieaa_tool/) was the most up-to-date tool with significantly improved functionality than TAM (18). Nevertheless, the reference miRNA sets of miEAA were enriched by target-derived functional annotations while the manually curated, miRNA-oriented annotations were not significantly improved (e.g., for miRNA-disease sets, miEAA used HMDD annotations only).
To overcome the challenge in the curated reference miRNA sets, we performed extensive manual literature reading and established the updated TAM 2.0 server. From over 9000 papers, we compiled 547 miRNA-disease and 158 miRNA-function sets, which resulted in the most comprehensive manually curated miRNA sets to our best knowledge. Moreover, we also performed in-depth curation of miRNA-disease associations by discriminating the disease promoting (or disease activated) miRNAs and disease suppressing (or disease inhibited) miRNAs. This curation strategy enables a new function of TAM 2.0, which compares the positive/negative correlation between de-regulated miRNAs between different conditions. Such correlation would provide more useful information for biomedical application (e.g. negative correlation would imply a protective effect against disease). Finally, to facilitate users, the miRNA set query function and graphical visualization of results were also added to our updated TAM 2.0 web server.
METHODS
Collection of miRNA sets
TAM 2.0 contains six types of miRNA sets: family, cluster, tissue specificity, disease, function and transcription factor (TF). Except the tissue specificity sets which had been derived from a comprehensive miRNA transcriptome atlas (16) when building previous TAM, the transcription factor sets were newly added, while other four types of miRNA sets were significantly updated in TAM 2.0 (Figure 1A). The miRNA-family sets were obtained from the updated miRBase annotations (http://www.mirbase.org/; version 21, queried in June 2017) (19). The miRNA cluster was defined as miRNAs grouped within 50 kb on the chromosome (according to the genomic coordinates provided by miRBase), following previous publication (20). The newly added TF-miRNA sets incorporated the TF-miRNA regulation pairs from the TransmiR (21) and mirTrans (22) databases. The disease and function miRNA sets were compiled through manual literature reading. We searched the PubMed with the keyword ‘Human microRNA’ OR ‘Human miRNA’, and limited the publication date from January 2014 to October 2017, as the publications before 2014 have been largely covered by previous TAM and HMDD database. We noted that many publications did not specify the mature miRNA (with -3p or -5p suffix) or duplicated miRNA genes (with -1 or -2 suffix). For better compatibility and coverage of the publication data, all miRNA set assignments were limited to the miRNA gene (pre-miRNA) level and there was no discrimination between mature miRNAs from the same pre-miRNA. Mature miRNAs were collapsed into the corresponding miRNA genes. And for duplicated miRNA genes, the disease or function term was assigned to all of them if the exact miRNA gene name was not specified in the publication. The disease and functional terms were summarized from the overrepresented keywords in the abstracts of the curated publications. During data curation process of TAM 2.0, we also managed to: (i) align the nomenclatures of disease and functional terms to the ICD-10-CM (https://www.cdc.gov/nchs/icd/icd10cm.htm), Disease Ontology (http://disease-ontology.org), MeSH (https://www.nlm.nih.gov/mesh), OMIM (http://omim.org), Human Phenotype Ontology (http://human-phenotype-ontology.github.io) and Gene Ontology (http://www.geneontology.org) approved synonyms, respectively, if applicable (see also Supplementary Table S1); (ii) merge or remove 98 ambiguous and redundant miRNA sets; (iii) discard miRNA genes unmappable to the miRBase release (version 21). Moreover, for miRNA-disease associations, we also classified miRNAs that were up-regulated in disease condition, or exhibited disease promoting function according to the phenotype from gene permutation assays. as the up-miRNA set of the disease. The down-miRNA set of the disease was curated in the same way. Such curation strategy is essential for our new comparison function in TAM 2.0. Finally, we discarded all miRNA-disease/function sets smaller than two miRNAs because these miRNA sets were often of worse quality.
Figure 1.

The overview of TAM 2.0 web server. (A) The comparison of miRNA set numbers in different categories, among the miRNA set enrichment analysis tools. (B) The workflow of TAM 2.0. Depends on the input, TAM 2.0 provides three major functionalities: (i) miRNA set query; (ii) overrepresentation analysis and 3) comparison between de-regulated miRNAs. All of these functionalities rely on the significantly updated curated reference miRNA sets of TAM 2.0.
Statistical analysis
To test if the input miRNAs are over- or under-represented in one miRNA set, the overrepresentation analysis is performed for each set. More specifically, a hypergeometric P-value was calculated based on the method identical to the previous version of TAM (16), i.e.:
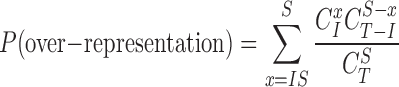 |
(1) |
 |
(2) |
Where I, S and IS represent the number of input miRNAs (covered by TAM 2.0), the number of miRNA in the tested miRNA set, and the number of miRNAs present in the intersection between the input and the tested miRNA set, respectively. The T is the total number of miRNAs covered by TAM 2.0.
For users who provide both up-regulated miRNAs and down-regulated miRNAs, TAM 2.0 could also compare these miRNAs with de-regulated miRNAs in other disease conditions, by using the previously established correlation-based method (23,24). This comparison is also partly inspired by the Connectivity Map project, where the correlation between transcriptome changes is utilized for drug repurposing (25). First, the lists of de-regulated miRNAs were transformed into a vector of association scores. The vector length was fixed to the total number of de-regulated miRNAs among all diseases, Tc. And in this vector, the association score di,j between miRNA j and condition i reads
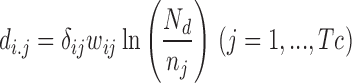 |
(3) |
In the above equation, the raw association δi,j has three possible values, +1 for up-regulated (or disease promoting) miRNAs, –1 for down-regulated (or disease suppressing) miRNAs, and 0 for miRNAs that are not de-regulated. The wi,j is the number of references supporting the association (for input miRNA lists, wi,j equals to 1). The last term corrects the specificity of association, where nj is the total number of diseases associated with miRNA j and Nd is the total number of diseases to be compared. Together, di,j encodes the weighted signed association score between miRNA j and condition i. Then the association score vectors of the input l and disease s could be compared by using cosine correlation as:
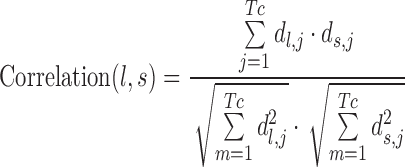 |
(4) |
Server construction
The web server was established in the ‘Linux + Apache + MySQL + PHP’ framework. The data visualization (bar plots, bubble plots and heatmaps) was implemented by using open source G2 package (https://github.com/antvis/g2/).
RESULTS
Overview of TAM 2.0 server
Currently, the reference miRNA set annotations in TAM 2.0 are constituted by 151 miRNA-family sets, 211 miRNA cluster sets, 547 miRNA-disease and 158 miRNA-function sets, 166 miRNA-TF sets and 6 tissue specificity sets. As depicted in Figure 1A, the number of miRNA sets were significantly increased in TAM 2.0, compared with previous TAM versions. And the coverage of the curated miRNA-function sets and miRNA-disease sets was also improved compared with those used by miSEA and miEAA (whereas miEAA used massive target gene-deduced miRNA-function sets instead of manually curated miRNA-function sets). The increased amount of miRNA sets in TAM 2.0 was not simply resulted from aggregation of uncommon miRNA sets. Instead, the average number of miRNAs per set was increased from ∼10.9 in TAM to ∼13.9 in TAM 2.0, indicating that more comprehensive miRNA associations are recorded in TAM 2.0. Finally, for more than half (445 out of 547) of miRNA-disease sets, we could discriminate up-miRNAs and down-miRNAs, which supports the condition comparison function of TAM 2.0 described later.
The workflow of TAM 2.0 is shown in Figure 1B. Depends on the input, TAM 2.0 provided three major functions. First, if users input a miRNA name or a keyword of miRNA set, a miRNA query function is provided in the ‘Query’ page of the server (Figure 2A). After first round of query, users can perform a filtration of the query results by clicking the toggle switches of miRNA set categories, or by inputting additional keywords. Second, if users submit a list of miRNAs, an overrepresentation analysis is performed via the ‘Analysis’ page. The tabular result is firstly provided (Figure 2B). For each associated miRNA set, users can click the [details] link to view the PubMed reference list supporting the association and the cross-refence of the miRNA set term to other public databases like ICD-10-CM and Gene Ontology. Users can also click the ‘Result Visualization’ button to the top of tabular result for generating custom graphical results. The bar plots and bubble plots (Figure 2C and D), together with a heat-map showing the detailed miRNA set associations (Supplementary Figure S1), will be provided. Third, if users have both up-regulated miRNAs and down-regulated miRNAs, a comparison with de-regulated miRNAs in other diseases is enabled in the ‘Comparison’ page. And the comparison results include a bi-color bar plot illustrating the correlations between de-regulated miRNAs (Figure 2E) and a heatmap summarizing the up- and down-miRNAs in other diseases (Supplementary Figure S2). All of the above analysis could typically be finished within 30 seconds unless the input miRNA list is large (>300 miRNAs).
Figure 2.

The sample results of TAM 2.0 web server. (A) The sample miRNA set query results. (B) The sample tabular view of the overrepresentation analysis results. (C) The sample bar plot illustrating the top 10 significant miRNA-function associations of the input sepsis miRNA biomarkers. (D) The sample bubble plot illustrating the top 10 significant miRNA-disease associations of the input sepsis miRNA biomarkers. (E) The sample bar plot illustrating the correlations between de-regulated miRNAs after metformin intervention and those in other disease conditions, only the top 10 diseases are shown.
Example for using the overrepresentation analysis
We exemplified the results of overrepresentation (enrichment) analysis of TAM 2.0 with the known miRNA biomarkers of sepsis, a severe systemic inflammatory response syndrome that causes massive death worldwide. Nine miRNA biomarkers for sepsis were recently summarized by Dumache et al. (26). A sample screenshot of tabular results is shown in Figure 2B. The full list of miRNA-set associations can be downloaded by clicking the ‘Text files of results’ button on the server. In this tabular view, user can move mouse over one miRNA set term to view and save the related miRNAs. User can also click the [details] link asides each term to view the cross-reference of this term and references supporting the miRNA-term associations. The significant disease and functional association of the sepsis miRNA biomarkers are shown in Supplementary Table S2. The ‘Sepsis’ disease term (FDR = 4.63E–10) and closely related ‘Inflammation’ (FDR = 3.91E–6) and ‘Immune System’ function terms (FDR = 5.88E–6) are of the top significance. And some immune- or inflammation-related disease terms like ‘Multiple Sclerosis’, ‘Psoriasis’, ‘Systemic Lupus Erythematosus’, ‘Autoimmune Diseases [unspecific]’, ‘Immune Thrombocytopenic Purpura’, ‘Chronic Hepatitis B’, ‘Osteoarthritis’, ‘Human Immunodeficiency Virus Infection’, ‘Tuberculosis, Pulmonary’ and ‘Rheumatoid Arthritis’ are also overrepresented. We also noted the significant associations with multiple cancer-related disease terms and the ‘Onco-miRNAs’ function term, which would imply plausible relationship between inflammation and cancer. Indeed, some miRNAs like has-mir-223 could link the mechanisms between inflammation and cancer (27), and this miRNA is also a known sepsis biomarker (26). Besides the tabular view, graphical visualization of the result is also available. By clicking ‘Result Visualization’ button (Figure 2B), the bar plot and the bubble plot could be generated with customized parameters (Figure 2C and D). In addition, a heatmap depicting the holistic view of miRNA-disease/function associations is also provided (Supplementary Figure S1). Finally, TAM 2.0 also permits analyzing up-/down-miRNAs separately. Supplementary Table S3 lists the results when up-/down-miRNA analysis function is enabled. Interestingly, the sepsis miRNA biomarkers are enriched for up-miRNAs in ‘Sepsis’ and ‘Rheumatoid Arthritis’, whereas they are enriched for down-miRNAs in ‘Systemic Lupus Erythematosus’. This result indicates that these miRNAs may play distinct roles in different immune-related diseases, which could not be observed without the up-/down-miRNA analysis newly available in TAM 2.0.
We also compared the TAM 2.0’s analysis results with the results from the most up-to-date published miRNA set enrichment tool miEAA (18). Similar to TAM 2.0, miEAA would accept a simple list of miRNA genes (precursors), whereas some other server required more specialized inputs (17). The overrepresentation analysis results of miEAA are shown in Supplementary Table S4. Indeed, miEAA also captured some important associations with immune- or inflammation-related disease terms such as ‘Sepsis’, ‘Psoriasis’, ‘HIV Infections’ ‘Multiple Sclerosis’ and ‘Hepatitis B’. Nevertheless, the overall disease associations of miEAA are less comprehensive than TAM 2.0, and there is no functional association provided by miEAA at the miRNA gene (precursor) level. Give the two tools adopted identical hypergeometric test method for overrepresentation analysis, this distinction should be mostly resulted from the difference in the coverage of curated miRNA sets. Moreover, we also tried to use the mature miRNA names (transferred by miEAA) as the input to analyze the function and pathway associations of the input miRNAs, because these functional annotations are only available at the mature miRNA level in miEAA (18). Unfortunately, however, no additional functional associations of the sepsis biomarkers were obtained. We noted that the function and pathway annotations used by miEAA were largely derived from miRNA target genes, which would be of insufficient accuracy to produce prominent results (9). Finally, miEAA did not enable the analysis of up-/down-miRNAs separately, and this function, as what we will show in the next sub-section, could provide additional interesting hypothesis in biomedical applications.
Example for using the miRNA set comparison analysis
In TAM 2.0, the correlations between the input de-regulated miRNAs and the de-regulated miRNAs in other disease conditions could also be assessed. Intuitively, a positive correlation with disease miRNAs would indicate potential deleterious changes of miRNA expression, while a negative correlation with disease miRNAs would suggest beneficial or protective effects. This novel function for miRNA set analysis would be useful for several biomedical applications like drug repositioning and discovery of novel gene-disease association, just like the Connectivity Map that is widely used for comparing expression changes of coding genes (25). Here as an example, we obtained 23 up-regulated and 25 down-regulated miRNAs (human homologs) after metformin intervention of non-alcoholic steatohepatitis mouse model from the previous study (28). We found that the de-regulated miRNAs after metformin intervention exhibit negative correlations with several types of cancers (Figure 2E), implying the anti-cancer potential of metformin. For example, the comparison analysis demonstrates noticeable negative correlations with pleural mesothelioma and gastric adenocarcinoma. Indeed, experimental evidence for metformin's anti-mesothelioma and anti-gastric carcinoma effects is accumulating in recent years (29,30), which suggests the repurposing potential of metformin as an anti-cancer drug.
DISCUSSION
Here we present the updated online miRNA set enrichment tool TAM 2.0. The improvement of TAM 2.0 should largely attribute to the efforts in manual data curation. With the miRNA sets in more details (e.g. stratifying by up- and down-miRNAs) and higher quality, a more comprehensive analysis of the disease- and function-associations of miRNAs has become possible. As illustrated above, the user would benefit from higher coverage of the curated miRNA sets during hypothesis generation and result interpretation. The correlation analysis of de-regulated miRNAs, which is based on the manually curated disease-promoting (or disease-activated) miRNAs and disease-suppressing (or disease-inhibited) miRNAs, could also provide additional biomedical implication that is not readily available for simple overrepresentation analysis (e.g., implication for drug repurposing). Finally, TAM 2.0 is also equipped with miRNA set query and graphical visualization functions which make the server more user-friendly than previous TAM.
Nevertheless, it should be also noted that TAM 2.0 and other miRNA set enrichment tools like miSEA and miEAA are not mutually exclusive but functionally complementary. For example, miSEA and miEAA provide GSEA-like statistics which is not available for TAM 2.0. If the users wish to take the expression values (or ranks thereof) into consideration, they would refer to these web servers instead. If the users focus on the correlation between de-regulated miRNAs in different conditions, TAM 2.0 should be a better choice. Besides, there are several more advanced enrichment analysis methods that have been demonstrated helpful for gene set analyses, e.g. gene set clustering (6), comparative enrichment analysis (8) and network-based analysis (31). Although the validity and usefulness of these methods in miRNA set analysis remain unchecked, it should be the future direction to further improve the functionality of the tool. Finally, manually curated data from publications are prone to be biased by the experiment designs, e.g. the onco-miRNAs and tumor suppressor miRNAs overwhelmed others in current TAM 2.0 dataset, and the experimental data are of various quality and may include artefacts. For now, TAM 2.0 provides options to mask cancer-related and non-standard miRNA terms when performing analyses. Nevertheless, more high-throughput functional screens of miRNAs are continuously in demand to provide less biased evaluation of miRNA-function associations.
Finally, there are several clear caveats or limitations of the current TAM 2.0. First, we only performed manual curation on the publications from January 2014 to October 2017, as the publication before 2013 was assumed to be covered by TAM 1.0 and the HMDD database behinds it. However, there are indeed some important publications that had been overlooked by the HMDD database, which turns out to limit the comprehensiveness of our miRNA sets. Currently, we have a project with the collaboration from the HMDD team to re-curate such historic publication data. This project is assumed to be done within this year and the updated miRNA set will become available with the next major update of TAM 2.0. Second, although the PubMed IDs are supplied for our manually curated data, no evidence code is provided to evaluate the confidence of the associations. We note that the HMDD team is also managing to update their evidence code of publication data, so that we can also incorporate their new evidence classification when it becomes accessible. Third, because many publications actually described the function of miRNA genes (pre-miRNAs) but did not specify the mature miRNAs, the TAM 2.0 actually works on human miRNA genes (pre-miRNAs) rather than mature miRNAs to ensure the data coverage and compatibility. Nevertheless, this is indeed a compromise between precision and coverage of the miRNA sets. One solution for this issue is to compile new miRNA sets from the public transcriptome data (where the mature miRNAs are specified) and provide a new function module for analyzing or comparing the transcriptome-derived mature miRNA sets. In all, we hope the coverage and quality of TAM 2.0 data will be further improved after several rounds of updates in the near future.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yan Huang and Zhou Huang for assistance in data curation. We also thank the anonymous referees for their constructive comments on server implementation and manuscript improvement.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81672113 to J.L., 81670462 to Q.C.]; Fundamental Reseaarch Funds for Central Universies of China [BMU2017YJ004 to Y.Z.]; Natural Science Foundation of Hebei Province [C2018202083 to J.L.]. Funding for open access charge: National Natural Science Foundation of China [81672113].
Conflict of interest statement. None declared.
REFERENCES
- 1. Alvarez-Garcia I., Miska E.A.. MicroRNA functions in animal development and human disease. Development. 2005; 132:4653–4662. [DOI] [PubMed] [Google Scholar]
- 2. Hata A., Lieberman J.. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015; 8:re3. [DOI] [PubMed] [Google Scholar]
- 3. Chen G., Wang J., Cui Q.. Could circulating miRNAs contribute to cancer therapy. Trends Mol. Med. 2013; 19:71–73. [DOI] [PubMed] [Google Scholar]
- 4. D’Alessandra Y., Devanna P., Limana F., Straino S., Di Carlo A., Brambilla P.G., Rubino M., Carena M.C., Spazzafumo L., De Simone M. et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010; 31:2765–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keller A., Meese E.. Can circulating miRNAs live up to the promise of being minimal invasive biomarkers in clinical settings. Wiley Interdiscipl. Rev. RNA. 2016; 7:148–156. [DOI] [PubMed] [Google Scholar]
- 6. Dennis G. Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A.. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003; 4:P3. [PubMed] [Google Scholar]
- 7. Ma W., Huang C., Zhou Y., Li J., Cui Q.. MicroPattern: a web-based tool for microbe set enrichment analysis and disease similarity calculation based on a list of microbes. Scientific Rep. 2017; 7:40200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J.. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016; 44:W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Godard P., van Eyll J.. Pathway analysis from lists of microRNAs: common pitfalls and alternative strategy. Nucleic Acids Res. 2015; 43:3490–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dweep H., Gretz N.. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods. 2015; 12:697. [DOI] [PubMed] [Google Scholar]
- 11. Dweep H., Sticht C., Pandey P., Gretz N.. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011; 44:839–847. [DOI] [PubMed] [Google Scholar]
- 12. Vlachos I.S., Zagganas K., Paraskevopoulou M.D., Georgakilas G., Karagkouni D., Vergoulis T., Dalamagas T., Hatzigeorgiou A.G.. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015; 43:W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bleazard T., Lamb J.A., Griffiths-Jones S.. Bias in microRNA functional enrichment analysis. Bioinformatics. 2015; 31:1592–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q.. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y.. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu M., Shi B., Wang J., Cao Q., Cui Q.. TAM: a method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010; 11:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corapcioglu M.E., Ogul H.. miSEA: microRNA set enrichment analysis. Biosystems. 2015; 134:37–42. [DOI] [PubMed] [Google Scholar]
- 18. Backes C., Khaleeq Q.T., Meese E., Keller A.. miEAA: microRNA enrichment analysis and annotation. Nucleic Acids Res. 2016; 44:W110–W116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baskerville S., Bartel D.P.. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005; 11:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J., Lu M., Qiu C., Cui Q.. TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res. 2010; 38:D119–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hua X., Tang R., Xu X., Wang Z., Xu Q., Chen L., Wingender E., Li J., Zhang C., Wang J.. mirTrans: a resource of transcriptional regulation on microRNAs for human cell lines. Nucleic Acids Res. 2018; 46:D168–D174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma W., Zhang L., Zeng P., Huang C., Li J., Geng B., Yang J., Kong W., Zhou X., Cui Q.. An analysis of human microbe-disease associations. Brief. Bioinformatics. 2017; 18:85–97. [DOI] [PubMed] [Google Scholar]
- 24. Zhou X., Menche J., Barabasi A.L., Sharma A.. Human symptoms-disease network. Nat. Commun. 2014; 5:4212. [DOI] [PubMed] [Google Scholar]
- 25. Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006; 313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 26. Dumache R., Rogobete A.F., Bedreag O.H., Sarandan M., Cradigati A.C., Papurica M., Dumbuleu C.M., Nartita R., Sandesc D.. Use of miRNAs as biomarkers in sepsis. Anal. Cell Pathol. (Amst). 2015; 2015:186716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haneklaus M., Gerlic M., O’Neill L.A., Masters S.L.. miR-223: infection, inflammation and cancer. J. Internal Med. 2013; 274:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katsura A., Morishita A., Iwama H., Tani J., Sakamoto T., Tatsuta M., Toyota Y., Fujita K., Kato K., Maeda E. et al. MicroRNA profiles following metformin treatment in a mouse model of non-alcoholic steatohepatitis. Int. J. Mol. Med. 2015; 35:877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shimazu K., Tada Y., Morinaga T., Shingyoji M., Sekine I., Shimada H., Hiroshima K., Namiki T., Tatsumi K., Tagawa M.. Metformin produces growth inhibitory effects in combination with nutlin-3a on malignant mesothelioma through a cross-talk between mTOR and p53 pathways. BMC Cancer. 2017; 17:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li P., Zhang C., Gao P., Chen X., Ma B., Yu D., Song Y., Wang Z.. Metformin use and its effect on gastric cancer in patients with type 2 diabetes: a systematic review of observational studies. Oncol. Lett. 2018; 15:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J., Vasaikar S., Shi Z., Greer M., Zhang B.. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017; 45:W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.