


Abstract
A server implementation of the UNRES package (http://www.unres.pl) for coarse-grained simulations of protein structures with the physics-based UNRES model, coined a name UNRES server, is presented. In contrast to most of the protein coarse-grained models, owing to its physics-based origin, the UNRES force field can be used in simulations, including those aimed at protein-structure prediction, without ancillary information from structural databases; however, the implementation includes the possibility of using restraints. Local energy minimization, canonical molecular dynamics simulations, replica exchange and multiplexed replica exchange molecular dynamics simulations can be run with the current UNRES server; the latter are suitable for protein-structure prediction. The user-supplied input includes protein sequence and, optionally, restraints from secondary-structure prediction or small x-ray scattering data, and simulation type and parameters which are selected or typed in. Oligomeric proteins, as well as those containing D-amino-acid residues and disulfide links can be treated. The output is displayed graphically (minimized structures, trajectories, final models, analysis of trajectory/ensembles); however, all output files can be downloaded by the user. The UNRES server can be freely accessed at http://unres-server.chem.ug.edu.pl.
INTRODUCTION
The biological functions of proteins depend on their structure and dynamics, therefore, research on these subjects is central in molecular biology and medicinal chemistry, including drug design. However, experimental techniques (x-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and, since very recently, cryoelectron microscopy), provide structures of only a small fraction of protein sequences discovered yearly (1) and only fragmentary information is available by experimental techniques regarding protein dynamics. Computer modeling, aided by experimental and database information is, therefore, routinely used for the prediction of unknown protein structures and simulation of protein dynamics. For protein-structure prediction comparative modeling has proved to be the most successful method nowadays (1); however, it fails when a protein represents a completely new fold. Protein dynamics is usually simulated by using all-atom molecular dynamics (MD). However, despite the continuous development of computer technology, including the construction of machines dedicated to run MD simulations (2), the discrepancy between the simulation and biological time scale restricts the applicability of this technique to solve concrete biological problems. Coarse-grained models of proteins, in which an amino-acid residue is represented by a few extended atoms and, consequently, the simulation time- and size-scale is extended by orders of magnitude are, therefore, a subject of intense development (3). Generally, these models can be divided into those that use the knowledge- (derived from database statistics) and physics-based (derived by translating the all-atom energy function to that corresponding to a reduced model) potentials.
The coarse-grained UNRES model developed in our laboratory (4,5) is a highly-reduced physics-based model of proteins, in which only two interaction sites per residue (united side chains and united peptide groups) are kept. The α-carbon (Cα) atoms are present in the model to assist in the definition of chain geometry. The effective energy function has been defined as a potential of mean force of polypeptide chains in water, which has been subsequently expanded into Kubo cluster cumulant functions, identified with the respective energy terms (6). MD (7,8) and its replica-exchange (REMD) (9) and multiplexed replica exchange (MREMD) (10) extensions have been implemented in UNRES (11). In contrast to most of the protein coarse-grained models, owing to its physics-based origin, the UNRES force field can be used in simulations, including those aimed at protein-structure prediction, without ancillary information from structural databases (12,13); however, the implementation includes the possibility of using restraints (14). UNRES has been used with success in protein-structure prediction (12–14), studying protein-folding kinetics and free-energy landscapes as well as to solve biological problems (4,5,15).
MATERIALS AND METHODS
Data processing
Oligomeric proteins (16), proteins that include D-amino-acid residues (17) and those with disulfide links (18) can be treated with UNRES. For general information on UNRES the reader is referred to the pertinent book chapter (4) and review article (5), while the theory behind the model is described in detail in our recent work (6).
Although the UNRES package is available for download (at http://www.unres.pl) since several years ago, a user needs to install it on its system and run in batch mode. This feature leaves out a large number of potential users who prefer to submit jobs using a web-based interface. Moreover, a parallel compute server is required for most of the functions of the package to run. Therefore, we have recently created a server based on the UNRES package, coined a name ‘UNRES server’, which enables a user to submit jobs using a web-based interface. The present article is devoted to the description of this server.
A scheme of the UNRES server, outlining its function and data pipelines are shown in Figure 1. As shown, three types of calculations are available: (i) single local energy minimization (the MIN button), (ii) single-trajectory canonical MD (the MD button) and (iii) multiplexed replica-exchange molecular dynamics (the MREMD button). The mode of calculations is selected by the user. For the MREMD type of calculation, a production simulation is followed by running weighted histogram analysis method (19) to enable us to compute the probabilities of conformations, thermodynamic quantities and ensemble averages at any temperature (20), and cluster analysis of the ensemble at the desired temperature (usually 10–20 K below that of the heat-capacity peak) to construct the final models. The number of clusters is fixed to five and the clustering is carried out by using Ward’s minimum-variance method (21). The final models are selected as the conformations closest to the average structures from the respective clusters (20). This procedure copies our physics-based protein-structure prediction procedure that has been tested in the CASP exercises (13). The final models are converted into all-atom representation by using the PULCHRA (22) and SCWRL (23) methods and subjected to a short energy minimization with the AMBER force field with the Generalized-Born method to treat hydration (24).
Figure 1.
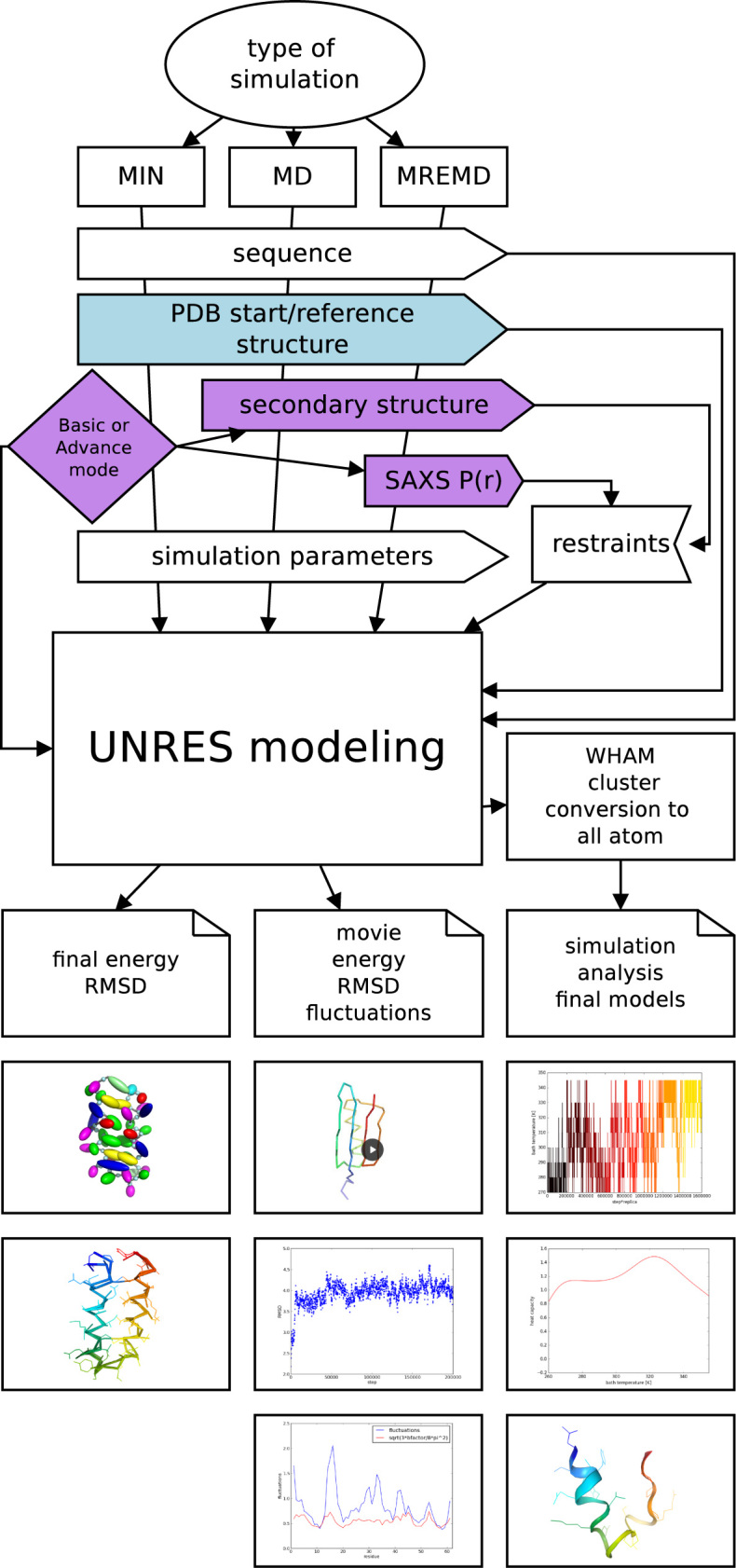
UNRES server operation flow.
Each calculation can be run in the basic or advanced mode; in the basic mode default parameters which were proved to work in most situations are applied, while in the advanced mode the user can select the parameters of the calculations. In particular, one of the two variants of UNRES force field can be selected, a ‘canonical’ one obtained by extensive search of parameter space (25) and subsequently supplemented with additional torsional potentials accounting for the coupling between backbone-local and sidechain-local conformational states (26), which improves the quality of local structure, hereafter referred to as FF2, and the UNRES force field recently calibrated with seven proteins of different structural classes, by using the maximum-likelihood approach developed in our laboratory, hereafter referred to as OPT-WTFSA-2 (27). Both variants of UNRES have been validated in our earlier work (12,13,25–27).
Input data
Amino-acid sequence is required for all types of simulations; it can be input by the user as one-letter code or read from a PDB file (only the first model is taken), which can be uploaded by the user or downloaded from the PDB database given the PDB code. The reference/initial structure, if the user chooses so, is also read from a PDB file. In the current implementation, no missing residues in a structure read from a PDB file are allowed, an error message is displayed if this occurs. For oligomeric proteins, different chains are automatically recognized while reading a PDB file; it is also possible to request calculations for a selected chain only. For example, by supplying the string 5G3Q the user specifies that a calculation should be run for all chains present in the PDB file, while 5G3Q:B means that chain B has been selected. User-supplied sequence of an oligomeric protein must contain the ’XX’ chain separator; for example the string AAGGAAXXAAGGAA means that the sequence is a dimer consisting of two AAGGAA chains. D-residues are recognized in PDB files; in the user-supplied sequence they are marked by lowercase letters. For example, the sequence AAaAA means that the chain contains D-alanine in the third position. The positions of the disulfide links are read from the SSBOND records of the PDB file.
For the energy-minimization type, the sequence is always read from a PDB file, along with the starting structure. Energy minimization is carried out with the quasi-Newton Secant Unconstrained Minimization Solver (SUMSL) algorithm (28). In the advanced mode, the user can select the number of minimization steps and the number of maximum function evaluations, and decide whether to use an initial Monte Carlo search of the local geometry of side-chain centroids to remove overlaps.
For the MD type of calculations, a single canonical MD trajectory can be run. The basic mode includes selection of the starting structure (extended chain, randomly generated structure, or the structure read from a PDB file), number of MD steps, temperature and the seed to initialize the random-number generator. Initial energy minimization is turned on if the starting structure is read from a PDB file or randomly generated. Calculations are run with the Langevin thermostat, by using an adaptive multiple-time-step (A-MTS) quasi-symplectic algorithm developed in our laboratory (29), which is similar to the RESPA algorithm (30). In the advanced mode, the thermostat (Langevin or Berendsen) can be selected and secondary-structure restraints can be input [in the PSIPRED (31) format]. The ‘molecular time unit’ (mtu) used in UNRES MD amounts to 48.9 fs (7). However, it should be noted that, because of averaging over the secondary degrees of freedom, the time scale of UNRES MD is extended by 1000–10 000 times compared to the all-atom time scale (8). Simulation parameters can be changed in the advanced mode.
The MREMD type of calculations implies running multiple trajectories in a parallel run. In the basic input mode, an REMD calculation (no multiplexing) is carried out with eight replicas run at temperatures from 270 K to 345 K. In the advanced mode, the user can choose the number of replica temperatures and the values of the temperatures, as well as multiplexing of each replica and replica-exchange frequency. In addition to this, the distance distribution from small x-ray scattering (SAXS) measurements can be input to run SAXS-restrained simulations (32). MD parameters can be selected in the advanced mode, as for MD-type runs. At last, the user can also select the temperature at which to cluster the final conformational ensemble.
Output
For each simulation type, the output is displayed graphically and can be accessed up to 2 weeks from the time of job completion provided that the user has saved the web address of the job. Moreover, the UNRES input file(s) and all output files can also be downloaded from the server; see http://www.unres.pl/docs for the description of input/output files.
For the respective types of calculations, the output is the following:
Minimization: UNRES representation of minimized structure, minimized structure superposed on the starting (reference) structure, Cα-root-mean square deviation (Cα-rmsd) from the starting structure, percentage of native contacts.
Canonical MD: Temperature distribution, plots of potential energy, radius of gyration, Cα-rmsd, fraction of native contacts, fluctuations versus time, movie of the trajectory.
MREMD: Plots of heat capacity and ensemble-average Cα-rmsd versus temperature, plots of walk in temperature space, Cα-rmsd from the reference structure (if present) versus energy, Cα-rmsd versus time, representative conformations of the five families obtained after clustering (cf. section Data processing). These structures, in all-atom form, obtained after the conversion of the UNRES structures, can be downloaded by clicking on the respective button. For each model, its probability (fraction in the ensemble), average cluster Cα-rmsd, as well the Cα-rmsd, TMscore and GDT_TS of the respective model are displayed.
RESULTS AND DISCUSSION
Examples included in server tutorial
The tutorial includes examples of running server jobs for all calculation types in the basic and in the advanced mode. Each calculation corresponding to a basic-mode example can be run within minutes, while those corresponding to advanced mode require up to a couple of hours. By pressing the ‘Load example data’ button all input data are loaded and parameters are set. The results of pre-run computations are displayed by selecting the ‘Tutorial’ item from the top navigation bar.
The following basic-mode examples include (i) minimization of the energy of the experimental structure of the N-terminal portion of the B-domain of staphylococcal protein A (PDB code: 1BDD); (ii) canonical MD simulation of the IGG binding domain of streptococcal protein G (PDB code: 1IGD), starting from the experimental structure; a fluctuation plot in residue index is displayed and compared with that of the B-factor; (iii) REMD simulation of the tryptophan cage mini-protein (PDB code: 1L2Y), starting from the extended chain; this is a full-blown structure-prediction run.
Advanced-mode examples include: (i) minimization of the P8MTCP1 disulfide-bonded α-helical hairpin miniprotein (PDB code: 1EI0); (ii) canonical MD run of the tryptophan-cage mini-protein (PDB code: 1L2Y) starting from the extended structure; (iii) MREMD run of CASP12 target T0882 also discussed in section ‘Selected test cases’; (iv) REMD run of the Bacteriocin CbnXY miniprotein (PDB code: 5UJG) starting from extended chain with simulated SAXS data in the form of the Gaussian-smoothed distance distribution calculated from the experimental structure showing that very good agreement between the distance distribution calculated from the models and the input distribution is achieved; (v) the central portion of Factor H, modules 10–15 starting from the NMR structure (PDB code: 2KMS structure) with including the experimental distance distribution from SAXS [the SASDA25 entry of the SASBDB database (33)]. Examples (iii) and (v) are discussed in more detail in section ‘Selected test cases’.
Selected test cases
CASP12 target T0882
The calculation has been run with secondary-structure restraints from PSIPRED (34) obtained at the CASP12 time when the experimental 5G3Q structure was not included in the PDB database. The FF2 variant of the UNRES force field (25,26) was used. Figure 2 displays the overlap of UNRES server model 1 and the experimental 5G3Q:B structure. The Cα-RMSD of that model from the experimental structure is 3.6 Å and the GDT_TS is 58.8%.
Figure 2.

Overlap of the experimental 5G3Q:B structure (gray) of CASP12 target T0882 with UNRES server Model 1, rainbow-colored from the N-terminus (blue) to the C-terminus (red).
Example of including experimental SAXS restraints in simulations
This example pertains to the central portion of Factor H, which has been solved by NMR (35) (PDB code: 2KMS). This is a relatively small two-domain β-sheet protein. The distance distribution calculated from the experimental structure does not fully overlap with that from SAXS; in particular, the plot calculated from the 2KMS structure decays to 0 quicker than that from SAXS. By running REMD with UNRES and with the SAXS distance-distribution and secondary-structure restraints, much better agreement, in particular in the long-distance part is obtained (Figure 3). The reason for this better agreement is that, in the UNRES-calculated structure, the domains are at a larger angle than those in the experimental structure (Figure 3).
Figure 3.

Comparison of the distance distribution from SAXS measurements of the central portion of Factor H, modules 10–15 with those calculated from the experimental 2KMS structure and from the five UNRES server models. The 2KMS experimental structure (top, gray) and UNRES model 1 [bottom, rainbow-colored from the the N- (blue) to the C-terminus (red)] are superposed on the graph.
SERVER ARCHITECTURE
The UNRES server is equipped with the web interface written using Django Python Web framework. At the time of submission the server checks the correctness of data provided by the user and reports possible errors. For accepted jobs, the server prepares UNRES input files and submits jobs to the queue on our local cluster. A total of 42 nodes of the cluster are assigned to run server jobs. Upon pressing the ‘Refresh’ button or every 30 s, the server checks if the job is finished and reports the percentage of accomplishment. At last, the results are post-processed (which includes making a movie of an MD trajectory, constructing the plots, etc.) and displayed graphically.
Third-party software employed in the server are (i) pymol (36), (ii) convvpdb.pl from the MMTSB Tool Set (37), (iii) AMBER tools (24), (iv) PULCHRA (22), (v) SCWRL (23), NGL Viewer (38), which is employed as an interactive molecular viewer on the web page.
DATA AVAILABILITY
The UNRES server is available at http://unres-server.chem.ug.edu.pl. This website is free and open to all users and there is no login requirement. However, there is optional registration; registered users can access old jobs from their homepage in the server website without having to copy the link corresponding to a particular job. The source code of the server is available from group GIT repository at mmka.chem.univ.gda.pl/repo/django_unres.
ACKNOWLEDGEMENTS
Computational resources were provided by (i) the Informatics Center of the Metropolitan Academic Network (CI TASK) in Gdańsk, (ii) the Interdisciplinary Center of Mathematical and Computer Modeling (ICM), University of Warsaw (grant GA-71-23), (iii) the Polish Grid Infrastructure (PL-GRID), (iv) our cluster at the Faculty of Chemistry, University of Gdańsk.
FUNDING
National Science Center of Poland (Narodowe Centrum Nauki) (NCN) [UMO-2017/25/B/ST4/01026, UMO-2017/26/M/ST4/00044]. Funding for open access charge: NCN [UMO-2017/25/B/ST4/01026, UMO-2017/26/M/ST4/00044].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tramontano A. The Ten Most Wanted Solutions in Protein Bioinformatics. 2005; Boca Raton: Taylor & Francis. [Google Scholar]
- 2. Shaw D.E., Maragakis P., Lindorff-Larsen K., Piana S., Dror R.O., Eastwood M.P., Bank J.A., Jumper J.M., Salmon J.K., Shan Y. et al. . Atomic-level characterization of the structural dynamics of proteins. Science. 2010; 330:341–346. [DOI] [PubMed] [Google Scholar]
- 3. Kmiecik S., Gront D., Kolinski M., Wieteska L., Dawid A.E., Kolinski A.. Coarse-grained protein models and their applications. Chem. Rev. 2016; 116:7898–7936. [DOI] [PubMed] [Google Scholar]
- 4. Liwo A., Czaplewski C., Ołdziej S., Rojas A.V., Kaźmierkiewicz R., Makowski M., Murarka R.K., Scheraga H.A.. Voth G. Simulation of protein structure and dynamics with the coarse-grained UNRES force field. Coarse-graining of Condensed Phase and Biomolecular Systems. 2008; Boca Raton: CRC Press; 1391–1411. [Google Scholar]
- 5. Liwo A., Baranowski M., Czaplewski C., Gołaś E., He Y., Jagieła D., Krupa P., Maciejczyk M., Makowski M., Mozolewska M.A. et al. . A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014; 20:2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sieradzan A.K., Makowski M., Augustynowicz A., Liwo A.. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. I. Backbone potentials of coarse-grained polypeptide chains. J. Chem. Phys. 2017; 146:124106. [DOI] [PubMed] [Google Scholar]
- 7. Khalili M., Liwo A., Rakowski F., Grochowski P., Scheraga H.A.. Molecular dynamics with the united-residue model of polypeptide chains. I. Lagrange equations of motion and tests of numerical stability in the microcanonical mode. J. Phys. Chem. B. 2005; 109:13785–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khalili M., Liwo A., Jagielska A., Scheraga H.A.. Molecular dynamics with the united-residue model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model α-helical systems. J. Phys. Chem. B. 2005; 109:13798–13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansmann U.H.E., Okamoto Y.. Comparative study of multicanonical and simulated annealing algorithms in the protein folding problem. Physica A. 1994; 212:415–437. [Google Scholar]
- 10. Rhee Y.M., Pande V.S.. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 2003; 84:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Czaplewski C., Kalinowski S., Liwo A., Scheraga H.A.. Application of multiplexing replica exchange molecular dynamics method to the UNRES force field: tests with α and α + β proteins. J. Chem. Theory Comput. 2009; 5:627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He Y., Mozolewska M.A., Krupa P., Sieradzan A.K., Wirecki T.K., Liwo A., Kachlishvili K., Rackovsky S., Jagiela D., Slusarz R. et al. . Lessons from application of the UNRES force field to predictions of structures of CASP10 targets. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14936–14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krupa P., Mozolewska M., Wiśniewska M., Yin Y., He Y., Sieradzan A., Ganzynkowicz R., Lipska A., Karczyńska A., Ślusarz M. et al. . Performance of protein-structure predictions with the physics-based UNRES force field in CASP11. Bioinformatics. 2016; 32:3270–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mozolewska M., Krupa P., Zaborowski B., Liwo A., Lee J., Joo K., Czaplewski C.. Use of restraints from consensus fragments of multiple server models to enhance protein-structure prediction capability of the UNRES force field. J. Chem. Inf. Model. 2016; 56:2263–2279. [DOI] [PubMed] [Google Scholar]
- 15. Maisuradze G.G., Senet P., Czaplewski C., Liwo A., Scheraga H.A.. Investigation of protein folding by coarse-grained molecular dynamics with the UNRES force field. J. Phys. Chem. A. 2010; 114:4471–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rojas A.V., Liwo A., Scheraga H.A.. Molecular dynamics with the united-residue force field: ab initio folding simulations of multichain proteins. J. Phys. Chem. B. 2007; 111:293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sieradzan A., Hansmann U.H.E., Scheraga H.A., Liwo A.. Extension of UNRES force field to treat polypeptide chains with d-amino acid residues. J. Chem. Theory Comput. 2012; 8:4746–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chinchio M., Czaplewski C., Liwo A., Ołdziej S., Scheraga H.A.. Dynamic formation and breaking of disulfide bonds in molecular dynamics simulations with the UNRES force field. J. Chem. Theory Comput. 2007; 3:1236–1248. [DOI] [PubMed] [Google Scholar]
- 19. Kumar S., Bouzida D., Swendsen R.H., Kollman P.A., Rosenberg J.M.. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992; 13:1011–1021. [Google Scholar]
- 20. Liwo A., Khalili M., Czaplewski C., Kalinowski S., Ołdziej S., Wachucik K., Scheraga H.A.. Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J. Phys. Chem. B. 2007; 111:260–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ward J. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963; 58:236–244. [Google Scholar]
- 22. Rotkiewicz P., Skolnick J.. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008; 29:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Q., Canutescu A.A., Dunbrack R.L.. SCWRL and MolIDE: computer programs for side-chain conformation prediction and homology modeling. Nat. Protoc. 2008; 3:1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Case D.A., Darden T.A., Cheatham T.E., Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M. et al. . AMBER 12. 2012; San Francisco: University of California. [Google Scholar]
- 25. He Y., Xiao Y., Liwo A., Scheraga H.A.. Exploring the parameter space of the coarse-grained UNRES force field by random search: selecting a transferable medium-resolution force field. J. Comput. Chem. 2009; 30:2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sieradzan A.K., Krupa P., Scheraga H.A., Liwo A., Czaplewski C.. Physics-based potentials for the coupling between backbone- and side-chain-local conformational states in the united residue (UNRES) force field for protein simulations. J. Chem. Theory Comput. 2015; 11:817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krupa P., Hałabis A., Żmudzińska W., Ołdziej S., Scheraga H.A., Liwo A.. Maximum likelihood calibration of the UNRES force field for simulation of protein structure and dynamics. J. Chem. Inf. Model. 2017; 57:2364–2377. [DOI] [PubMed] [Google Scholar]
- 28. Gay D.M. Algorithm 611. Subroutines for unconstrained minimization using a model/trust-region approach. ACM Trans. Math. Softw. 1983; 9:503–524. [Google Scholar]
- 29. Rakowski F., Grochowski P., Lesyng B., Liwo A., Scheraga H.A.. Implementation of a symplectic multiple-time-step molecular dynamics algorithm, based on the united-residue mesoscopic potential energy function. J. Chem. Phys. 2006; 125:204107. [DOI] [PubMed] [Google Scholar]
- 30. Tuckerman M., Berne B.J., Martyna G.J.. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992; 97:1990–2001. [Google Scholar]
- 31. McGuffin L., Bryson K., Jones D.T.. The PSIPRED protein structure prediction server. Bioinformatics. 2000; 16:404–405. [DOI] [PubMed] [Google Scholar]
- 32. Karczynska A.S., Mozolewska M.A., Krupa P., Giełdoń A., Liwo A., Czaplewski C.. Prediction of protein structure with the coarse-grained UNRES force field assisted by small X-ray scattering data and knowledge-based information. Proteins. 2017; 86:228–239. [DOI] [PubMed] [Google Scholar]
- 33. Valentini E., Kikhney A.G., Previtali G., Jeffries C.M., Svergun D.I.. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015; 43:D357–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999; 292:195–202. [DOI] [PubMed] [Google Scholar]
- 35. Schmidt C.Q., Herbert A.P., Mertens H.D.T., Guariento M., Soares D.C., Uhrin D., Rowe A.J., Svergun D.I., Barlow P.N.. The central portion of Factor H (modules 10-15) is compact and contains a structurally deviant CCP module. J. Mol. Biol. 2010; 395:105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schrödinger LLC The PyMOL molecular graphics system, version 1.3r1. 2010; https://pymol.org/2/support.html.
- 37. Feig M., Karanicolas J., Brooks C.L. III. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004; 22:377–395. [DOI] [PubMed] [Google Scholar]
- 38. Rose A.S., Hildebrand P.W.. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The UNRES server is available at http://unres-server.chem.ug.edu.pl. This website is free and open to all users and there is no login requirement. However, there is optional registration; registered users can access old jobs from their homepage in the server website without having to copy the link corresponding to a particular job. The source code of the server is available from group GIT repository at mmka.chem.univ.gda.pl/repo/django_unres.