


Abstract
Geometric and topological properties of protein structures, including surface pockets, interior cavities and cross channels, are of fundamental importance for proteins to carry out their functions. Computed Atlas of Surface Topography of proteins (CASTp) is a web server that provides online services for locating, delineating and measuring these geometric and topological properties of protein structures. It has been widely used since its inception in 2003. In this article, we present the latest version of the web server, CASTp 3.0. CASTp 3.0 continues to provide reliable and comprehensive identifications and quantifications of protein topography. In addition, it now provides: (i) imprints of the negative volumes of pockets, cavities and channels, (ii) topographic features of biological assemblies in the Protein Data Bank, (iii) improved visualization of protein structures and pockets, and (iv) more intuitive structural and annotated information, including information of secondary structure, functional sites, variant sites and other annotations of protein residues. The CASTp 3.0 web server is freely accessible at http://sts.bioe.uic.edu/castp/.
INTRODUCTION
Protein structures are complex and are sculpted with numerous surface pockets, internal cavities and cross channels. These topographic features provide structural basis and micro-environments for proteins to carry out their functions such as ligand binding, DNA interaction and enzymatic activity. Identification and quantification of these topographic features of proteins are therefore of fundamental importance for understanding the structure–function relationship of proteins (1), in engineering proteins for desired properties (2) and in developing therapeutics against protein targets (3).
MATERIALS AND METHODS
The Computed Atlas of Surface Topography of proteins (CASTp) server uses the alpha shape method (4) developed in computational geometry to identify topographic features, to measure area and volume and to compute imprint (5–8). The alpha shape method has also been applied in other studies of cavities and channels in protein structures (9,10). The secondary structures are calculated using DSSP (11). Residue annotations of proteins are obtained from UniProt database (12) and mapped to PDB structures with residue-level information from the SIFTS database (13). The biological assemblies are extracted from the .mmicf files of the PDB database. Only the assemblies with biological significance and designated by the authors of the PDB structures (http://mmcif.wwpdb.org) are processed and listed on the CASTp server.
THE CASTp SERVER
The CASTp server aims to provide comprehensive and detailed quantitative characterization of topographic features of proteins (14,15). Since its release 15 years ago, the CASTp server has ∼45 000 visits and fulfills ∼33 000 calculation requests annually. It has been proven to be a useful tool for a wide range of studies, including investigations of signaling receptors (1), discoveries of cancer therapeutics (16), understanding of mechanism of drug actions (17), studies of immune disorder diseases (18), analysis of protein–nanoparticle interactions (19), inference of protein functions (20) and development of high-throughput computational tools (21,22).
To provide additional useful information and to deliver improved user experience, we introduce here an updated server called CASTp 3.0. All important features of the previous versions of the server are retained, including detecting and characterizing cavities, pockets and channels of protein structures (Figure 1). In addition, we have substantially extended its functions by providing pre-computed topographic features of biological assemblies in the PDB database, as well as imprints of negative volumes of these topographic features. Furthermore, the user interface has been redesigned, making it more intuitive and informative.
Figure 1.
The surface of the ATP binding pocket of CDK2 (PDB ID: 2xmy), computed by CASTp 3.0.
New features of CASTp 3.0
Pre-computed results for biological assemblies
The atomic coordinates of a PDB ID in the Protein Data Bank describe an asymmetric unit, which is the minimum structure that can produce the unit cell of the crystal through duplication and crystal symmetry operations. For many PDB IDs, the asymmetric unit differs from the biological assembly, which is the unit that has either been shown or is thought to be the functional form of the protein. This difference can be significant, and can make the computation results of a PDB entry biologically irrelevant. For example, PDB ID: 2iwv of the outer membrane porin OmpG contains four units as deposited. Its direct computation leads to the detection of a giant artificial pocket (Figure 2A), which obscures the actual functional channel (Figure 2B). To uncover topographic features that are biologically most relevant, the new CASTp server pre-computes topographic features for the biological assemblies of PDB IDs. Users now are able to navigate effortlessly between results of the asymmetric unit and results of biological assemblies of a PDB ID.
Figure 2.

The case when the asymmetric unit differs from the biological assembly. (A) Asymmetric unit in the PDB ID: 2iwv and the artificial giant pocket. The giant pocket is formed by the four channels of the four biological units and the space among them with unclear biological relevance. (B) Biological structure of 2iwv (biological assembly 1) and its channel detected by the CASTp server.
Imprints of negative volumes of topographic features
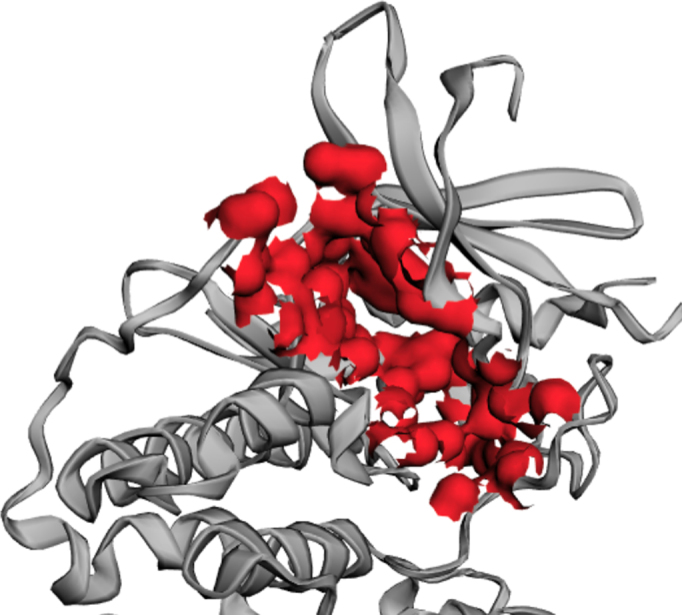
In the previous versions of CASTp servers, geometric and topological features, such as pockets, cavities and channels, were shown only through the representation of surface atoms participating in their formation (e.g. the atom surface in Figure 1 and the stick model shown in Figure 3). The new CASTp server has added imprints of the negative volumes (e.g. the red bulbs in Figures 2 and 3) as the default visualization option. The negative volume is the space encompassed by the atoms that form these geometric and topological features, which can give users direct and intuitive understanding of these important structural features of proteins (5,6).
Figure 3.

The main user interface of the CASTp server. (A) The pocket panel. (B) The sequence and annotation panels.
Improved user interface
The visualization techniques employed in previous versions of CASTp servers over 12-year ago are out-of-date and are incompatible with the modern browsers. The new CASTp server now uses 3Dmol.js (23) for structural visualization, which allows users to view, to interact with protein structures, and to examine computation results in modern web browsers such as Chrome and Firefox. Users can choose the representation style of the atoms that form each topographic feature. The imprint of these features can also be shown with user-selected colors (Figure 3A).
An intuitive sequence panel is also presented to users with secondary structures color-coded, where residues in user-selected pockets are highlighted. Residues annotated from UniProt (12) are also labeled. Both information on topographic feature-forming atoms and UniProt annotations are conveniently displayed when the cursor hovers over the relevant residues. The annotations are also summarized in the annotation panel (Figure 3B). In addition, both the sequence panel and the annotation panel are linked to the structure viewer, so users can conveniently click on one residue on the sequence map to have the structure viewer zoomed into that specific residue of interest.
Furthermore, a floating structure viewer has been added (Figure 3B). When a user inspects information in the sequence panel or the annotation panel, the structure viewer will automatically follow the web page scrolling of the user, which saves the user from unnecessary and unproductive efforts in scrolling up and down.
INPUT AND OUTPUT
Input
The CASTp server takes protein structures in the PDB format and a probe radius as input for topographic computation. Through the intuitive interface, users can either search for pre-computed results using a four-letter PDB ID, or submit their own protein structures to request customized computation. For pre-computed results, a default probe radius of 1.4 Å is used, which is the standard value for computing solvent accessible surface area. For customized computation request, users can specify any probe radius desired.
Output
The CASTp server identifies all surface pockets, interior cavities and cross channels in a protein structure and provides detailed delineation of all atoms participating in their formation. It also measures their exact volumes and areas, as well as sizes of the mouth openings if exist. These metrics are calculated analytically, using both the solvent accessible surface model (Richards’ surface) (24) and the molecular surface model (Connolly’s surface) (25). In addition, the CASTp server also provides imprints of topographic features. These results can be directly downloaded from CASTp server, which can be visualized using either the UCSF Chimera (26) or our PyMOL plugin, CASTpyMOL.
Case study
Figure 3 illustrates the adenosine triphosphate (ATP) binding pocket on the cyclin-dependent kinase 2 (CDK2, PDB ID: 2xmy) that is automatically identified by the CASTp server. CDK2 is an important enzyme that regulates cell cycle with other CDKs. Many functionally important residues are located in this pocket, including three residues Lys33, Asp86 and Asp145 in the ATP binding site, one residue Asp127 in the catalytic site, two residues Asn132 and Asp145 in Mg binding sites and one residue Lys89 in the binding site that interacts with CDK7, another important cell-cycle-controlling enzyme. Due to the importance of this pocket, it has been widely studied as a target site for antitumor agents (16).
In the CASTp server, users can choose different representations of pockets. For example, the pocket can be displayed as surface (Figure 1). In Figure 3, the imprint of the pocket is shown in red, and the atoms forming the pocket are shown as sticks. Volume and area measures, and the list of all atoms participating in the pocket formation are also given (Figure 3A). Users can view the pocket from the sequence panel. Important annotations can also be found easily in either the sequence panel or the annotation panel (Figure 3B).
Additional examples illustrating how CASTp computations have been used can be found in the literature (20–21,27–32).
DISCUSSION
This paper describes significant improvement of the CASTp server, including: (i) the addition of imprints of negative volumes as a visualization option; (ii) the inclusion of pre-computed results for biological assemblies, rather than reporting results for the asymmetric unit alone; (iii) an improved user interface, which contains an updated and adaptive structure viewer, a panel for sequence information and a more intuitive panel for UniProt annotations. We believe these updates with additional information and improved user experience will continue to facilitate studies of protein structures and functions.
FUNDING
NIH [R01CA204962]. Funding for open access charge: NIH [R01CA204962]; UIC Research Open Access Article Publishing (ROAAP) Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Toh S., Holbrook-Smith D., Stogios P.J., Onopriyenko O., Lumba S., Tsuchiya Y., Savchenko A., McCourt P.. Structure-function analysis identifies highly sensitive strigolactone receptors in Striga. Science. 2015; 350:203–207. [DOI] [PubMed] [Google Scholar]
- 2. Reetz M.T. Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology. 2016; Weinheim: Wiley-VCH. [Google Scholar]
- 3. Brouwer J.M., Lan P., Cowan A.D., Bernardini J.P., Birkinshaw R.W., van Delft M.F., Sleebs B.E., Robin A.Y., Wardak A., Tan I.K. et al. . Conversion of Bim-BH3 from activator to inhibitor of Bak through structure-based design. Mol. Cell. 2017; 68:659–672. [DOI] [PubMed] [Google Scholar]
- 4. Edelsbrunner H., Mücke E.P.. Three-dimensional alpha shapes. ACM Trans. Graph. 1994; 13:43–72. [Google Scholar]
- 5. Ebalunode J.O., Ouyang Z., Liang J., Zheng W.. Novel approach to structure-based pharmacophore search using computational geometry and shape matching techniques. J. Chem. Inf. Model. 2008; 48:889–901. [DOI] [PubMed] [Google Scholar]
- 6. Tian W., Liang J.. On quantification of geometry and topology of protein pockets and channels for assessing mutation effects. Proceedings - IEEE International Conference on Biomedical and Health Informatics. 2018; BHI; 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang J., Edelsbrunner H., Woodward C.. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998; 7:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edelsbrunner H., Facellob M., Liangc J.. On the definition and the construction of pockets in macromolecules. Discrete Appl. Math. 1998; 88:83–102. [Google Scholar]
- 9. Mach P., Koehl P.. Geometric measures of large biomolecules: surface, volume, and pockets. J. Comput. Chem. 2011; 32:3023–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masood T.B., Sandhya S., Chandra N., Natarajan V.. CHEXVIS: a tool for molecular channel extraction and visualization. BMC Bioinformatics. 2015; 16:119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabsch W., Sander C.. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983; 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 12. Consortium T.U. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Velankar S., Dana J.M., Jacobsen J., Van Ginkel G., Gane P.J., Luo J., Oldfield T.J., O’Donovan C., Martin M.J., Kleywegt G.J.. SIFTS: Structure Integration with Function, Taxonomy and Sequences resource. Nucleic Acids Res. 2013; 41:D483–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Binkowski T.A., Naghibzadeh S., Liang J.. CASTp: computed atlas of surface topography of proteins. Nucleic Acids Res. 2003; 31:3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J.. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006; 34:W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang S., Griffiths G., Midgley C.A., Barnett A.L., Cooper M., Grabarek J., Ingram L., Jackson W., Kontopidis G., McClue S.J. et al. . Discovery and characterization of 2-anilino-4- (thiazol-5-yl)pyrimidine transcriptional CDK inhibitors as anticancer agents. Chem. Biol. 2010; 17:1111–1121. [DOI] [PubMed] [Google Scholar]
- 17. Kadam R.U., Wilson I.A.. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shahine A., Van Rhijn I., Cheng T.-Y., Iwany S., Gras S., Moody D.B., Rossjohn J.. A molecular basis of human T cell receptor autoreactivity toward self-phospholipids. Sci. Immunol. 2017; 2:eaao1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ranjan S., Dasgupta N., Chinnappan S., Ramalingam C., Kumar A.. A novel approach to evaluate titanium dioxide nanoparticle-protein interaction through docking: an insight into mechanism of action. Proc. Natl. Acad. Sci. India SectB Biol. Sci. 2017; 87:937–943. [Google Scholar]
- 20. Tseng Y.Y., Liang J.. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: a Bayesian Monte Carlo approach. Mol. Biol. Evol. 2006; 23:421–436. [DOI] [PubMed] [Google Scholar]
- 21. Binkowski T.A., Joachimiak A., Liang J.. Protein surface analysis for function annotation in high-throughput structural genomics pipeline. Protein Sci. 2005; 14:2972–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Melaccio F., del Carmen Marín M., Valentini A., Montisci F., Rinaldi S., Cherubini M., Yang X., Kato Y., Stenrup M., Orozco-Gonzalez Y. et al. . Toward automatic rhodopsin modeling as a tool for high-throughput computational photobiology. J. Chem. Theory Comput. 2016; 12:6020–6034. [DOI] [PubMed] [Google Scholar]
- 23. Rego N., Koes D.. 3D mol. js: molecular visualization with WebGL. Bioinformatics. 2015; 31:1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee B., Richards F.M.. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971; 55:379–400. [DOI] [PubMed] [Google Scholar]
- 25. Connolly M. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983; 221:709–713. [DOI] [PubMed] [Google Scholar]
- 26. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 27. Aryal P., Dvir H., Choe S., Slesinger P.A.. A discrete alcohol pocket involved in GIRK channel activation. Nat. Neurosci. 2009; 12:988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao J., Dundas J., Kachalo S., Ouyang Z., Liang J.. Accuracy of functional surfaces on comparatively modeled protein structures. J. Struct. Funct. Genomics. 2011; 12:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jimenez-Morales D., Liang J., Eisenberg B.. Ionizable side chains at catalytic active sites of enzymes. Eur. Biophys. J. 2012; 41:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glukhova A., Hinkovska-Galcheva V., Kelly R., Abe A., Shayman J.A., Tesmer J.J.. Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 2015; 6:6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smoum R., Baraghithy S., Chourasia M., Breuer A., Mussai N., Attar-Namdar M., Kogan N.M., Raphael B., Bolognini D., Cascio M.G. et al. . CB2 cannabinoid receptor agonist enantiomers HU-433 and HU-308: An inverse relationship between binding affinity and biological potency. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:8774–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh A.N., Baruah M.M., Sharma N.. Structure based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against Prostate Cancer. Sci. Rep. 2017; 7:1955. [DOI] [PMC free article] [PubMed] [Google Scholar]