

ABSTRACT
Protein ubiquitylation is a dynamic post-translational modification that can be reversed by deubiquitylating enzymes (DUBs). It is unclear how the small number (∼100) of DUBs present in mammalian cells regulate the thousands of different ubiquitylation events. Here, we analysed annotated transcripts of human DUBs and found ∼300 ribosome-associated transcripts annotated as protein coding, which thus increases the total number of DUBs. By using USP35, a poorly studied DUB, as a case study, we provide evidence that alternative isoforms contribute to the functional expansion of DUBs. We show that there are two different USP35 isoforms that localise to different intracellular compartments and have distinct functions. Our results reveal that isoform 1 is an anti-apoptotic factor that inhibits staurosporine- and TNF-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10)-induced apoptosis. In contrast, USP35 isoform 2 is an integral membrane protein of the endoplasmic reticulum (ER) that is also present at lipid droplets. Manipulations of isoform 2 levels cause rapid ER stress, likely through deregulation of lipid homeostasis, and lead to cell death. Our work highlights how alternative isoforms provide functional expansion of DUBs and sets directions for future research.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Apoptosis, Deubiquitinase, Endoplasmic reticulum, Lipid droplets, Ubiquitin signalling
Highlighted Article: An analysis of USP35 shows that alternative isoforms can substantially contribute to expanding the functionality of deubiquitylating enzymes, hence allowing them to regulate multiple intracellular ubiquitylation events.
INTRODUCTION
Covalent attachment of ubiquitin to target proteins, or ubiquitylation, regulates many critical intracellular processes such as signal transduction, protein turnover and trafficking. Ubiquitylation is achieved through a sequential action of E1 activating, E2 conjugating and E3 ligating enzymes, and can be reversed by deubiquitylating enzymes or deubiquitinases (DUBs) (Hershko and Ciechanover, 1998). Ubiquitylation usually occurs on lysine residues of a substrate protein. Importantly, one of the seven internal lysine residues or the N-terminal methionine of ubiquitin can serve as an attachment site for another ubiquitin, thus allowing for the formation of ubiquitin chains (polyubiquitin) whose linkage type determines the biological outcome of ubiquitylation (Yau and Rape, 2016).
The importance of protein ubiquitylation and deubiquitylation is underscored by the fact that deregulation of these processes has been linked to the pathogenesis of many human diseases, such as cancer, and neurodegenerative and metabolic disorders (Heideker and Wertz, 2015; Popovic et al., 2014). In many cases, such detrimental outcomes stem from deregulation of key signalling pathways that are normally controlled by the ubiquitin system. For example, several DUBs have been implicated in the regulation of cell death either through controlling the stability of various pro- and anti-apoptotic factors or by regulating the formation of essential signalling complexes (Engel et al., 2016; Jeong et al., 2017; Lee et al., 2015; Mahul-Mellier et al., 2012; Mei et al., 2011; Schwickart et al., 2010; Weber et al., 2016).
Considering their profound roles in regulating cell fate and function, we still know surprisingly little about the precise functions and substrates of many DUBs. Furthermore, it is intriguing that a relatively small number of DUBs (∼100 are predicted to exist based on the human genome sequence) can regulate at least the ∼20,000 ubiquitin-modified sites that can be currently detected at steady-state conditions in mammalian cells (Clague et al., 2015; Kim et al., 2011; Udeshi et al., 2013). Although we are still far from understanding how this is achieved, several potential modes of expanding the functionality of DUBs have been proposed. These include alternative subcellular localisation of DUBs, regulation by interacting proteins and post-translational modifications. An unappreciated mechanism is the expansion of DUB functionality via alternative splicing, which provides a way to increase the total number of DUBs that can perform distinct functions (Leznicki and Kulathu, 2017). However, with a few exceptions, investigation into the presence and specific functions of alternative DUBs isoforms have been almost completely neglected.
In order to understand to what extent alternative isoforms contribute to the functional expansion of DUBs, we analysed a recent dataset of ribosome-associated transcripts from HEK293 cells (Floor and Doudna, 2016). Strikingly, we found ∼300 DUB transcripts annotated as protein coding that were associated with translating ribosomes in human cells. To address whether DUB isoforms perform non-redundant functions, we chose a poorly studied DUB, USP35, as a case study. Here, we characterised USP35 using biochemical, structural and cell biological approaches. Intriguingly, we detect at least two different USP35 isoforms at the endogenous level that localise to distinct intracellular compartments. In contrast to isoform 1, which is a soluble protein, isoform 2 is a novel integral membrane protein that localises to the ER and lipid droplets. Remarkably, isoform 2 expression induces rapid ER stress most likely because of perturbed lipid homeostasis and leads to cell death. Isoform 1 has a distinct function and acts as an anti-apoptotic factor that inhibits TNF-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10)- and staurosporine-induced apoptosis. Our results highlight how products of a single DUB-encoding gene can have contrasting functions.
RESULTS
Alternative isoforms expand the number of DUBs
A number of transcripts associate with translating ribosomes in HEK293 cells, suggesting widespread production of alternative isoforms (Floor and Doudna, 2016). We analysed the results of this high-throughput study to get insights into the extent to which the functionality of DUBs could be expanded by the presence of DUB isoforms. Strikingly, we find that in HEK293 cells there are ∼500 ribosome-associated DUB transcripts, of which ∼300 are annotated as protein coding (Table S1). The number of protein-coding DUB transcripts is likely an underestimation because mRNAs coding for some of the known DUBs such as OTUD6A and USP26 were not detected in the above-mentioned study (Floor and Doudna, 2016). Furthermore, this estimation is based on HEK293 cells and given that isoforms are expressed in a tissue-specific manner (Buljan et al., 2012; Ellis et al., 2012; Yang et al., 2016), it is likely that the total number of DUB isoforms expressed is even greater. As many of the DUB transcripts lack annotated 5′ or 3′ ends, DUB-centred transcriptomics analyses will be required to define the precise boundaries of these isoforms. Importantly, isoform-specific peptides have been detected for at least some of these putative isoforms, confirming their existence in living cells (Wilhelm et al., 2014).
The DUB USP35 is active and shows promiscuous selectivity towards ubiquitin linkage types
To explore how alternative isoforms expand DUB function, we here studied USP35, a member of the ubiquitin-specific protease (USP) DUB family. Importantly, transcripts with annotated 5′ and 3′ ends coding for three different USP35 isoforms with an intact catalytic domain were shown to associate with translating ribosomes (Floor and Doudna, 2016; Fig. 1A). The different USP35 isoforms are: full-length (denoted isoform 1, USP35iso1), an isoform with an N-terminal truncation of 269 residues (denoted isoform 2, USP35iso2) and one lacking 432 residues at the N-terminus (denoted isoform 3, USP35iso3). Furthermore, high-throughput proteomic experiments have identified unique acetylated N-terminal peptides specific for USP35iso1 and USP35iso2 (Wilhelm et al., 2014). USP35 is a poorly studied DUB suggested to play a role in mitophagy (Wang et al., 2015) and as a tumour suppressor (Liu et al., 2015). The latter finding is, however, at odds with reports showing that USP35 is amplified in a number of cancers, particularly breast and ovarian (Chin et al., 2007; www.cbioportal.org).
Fig. 1.

USP35 is an active and promiscuous DUB. (A) Upper panel, organisation of USP35 transcripts encoding isoforms presented in this study. Orange and grey boxes represent exons and non-coding regions, respectively. Exon numbers are given. Lower panel, domain organisation of USP35 isoforms. The USP domain of USP35 is shown together with catalytic Cys450 and an unstructured insertion spanning residues 604–753. The HEAT repeat motif is predicted based on the crystal structure of the N-terminus of a related protein, USP38 (PDB: 4RXX). Protein regions used to generate anti-USP35 antibodies are indicated. Uniprot accession numbers of each isoform are shown on the left. (B) Purified recombinant full-length human USP35 (375 nM) was incubated with tetraubiquitins of defined linkage types (733 nM) at 37°C for the indicated times. Reactions were resolved by SDS-PAGE followed by silver staining of the gel. (C) Crystal structure of the fragment of human USP35 (residues 423–944) lacking the insertion region (residues 604–753) and complexed with ubiquitin is presented. The subdomains thumb, palm and fingers are shown in beige, light pink and yellow, respectively, whereas the non-covalently bound distal ubiquitin is in green. The dashed line has been used to show the disordered regions. The co-ordinated zinc can be seen as a black sphere in the finger subdomain. A potential binding site of proximal ubiquitin is indicated. Close up views show the catalytic centre and selected residues mediating USP35–ubiquitin interactions.
USP35 is characterised by the presence of a USP catalytic domain where cysteine 450 acts as the cysteine active site residue (Fig. 1A). Similar to other USP DUBs that have insertions within their catalytic domains, USP35 also has a long highly charged ∼150-residue loop insertion in its USP domain that spans residues 604–753 and is predicted to be unstructured (Ye et al., 2009). Based on the crystal structure of the N-terminal fragment of a closely related DUB, USP38 (PDB: 4RXX), it can be expected that USP35 also contains N-terminal HEAT repeats (Fig. 1A), a motif known to mediate protein–protein interactions.
In order to investigate whether USP35 is an active enzyme, we expressed and purified full-length human USP35iso1. When tested for enzymatic activity against tetraubiquitins of defined linkage types, USP35 showed promiscuous activity and cleaved several linkages of which Lys11-, Lys33-, Lys48- and Lys63-linked polyubiquitin were cleaved most efficiently (Fig. 1B). Such promiscuity is often found amongst members of the USP family of DUBs whose selectivity seems to be determined by a substrate rather than ubiquitin linkage type (Faesen et al., 2011).
To understand USP35 at the molecular level, we attempted to crystallise catalytically inactive (C450S mutant) USP35423–944, the fragment that spans the catalytic domain shared by all three isoforms, in complex with Lys48-linked diubiquitin. Since the full-length catalytic domain was refractory to crystallisation, we speculated that removing the disordered loop insertion within the catalytic domain would result in diffracting crystals. Indeed, we obtained crystals of the catalytic domain lacking this loop in complex with Lys48 diubiquitin that diffracted to 1.8 Å (Fig. 1C). The structure was solved by molecular replacement and refined to the statistics shown in Table S2. Importantly, a bacterially purified catalytic domain of USP35 lacking the loop insertion shows similar activity and ubiquitin linkage type preference to that of the wild-type catalytic domain (Fig. S1). Surprisingly, we could only detect a single ubiquitin in complex with USP35, which might result from the residual DUB activity of the C450S mutant used (data not shown). Hence, the structure represents USP35 in complex with the distal ubiquitin. The crystal structure of USP35 reveals a typical USP domain architecture that superposes well onto the structures of previously crystallised USP domains (Fig. 1C; Fig. S2). Clearly defined subdomains referred to as thumb, palm and fingers can be observed in the structure (Fig. 1C; Yin et al., 2015). The bulk of ubiquitin until Arg72 is clasped between the thumb, palm and fingers subdomains through a strong network of hydrophobic, ionic and hydrogen bonds (Fig. 1C; Fig. S3). The cation-π and aromatic–aromatic interactions between the fingers subdomain and the ubiquitin molecule stabilise the complex (Fig. S3). Clear electron density is visible for the catalytic triad of USP35, composed of Cys450, His862 and Asp895 (Fig. 1C; Fig. S3E).
USP35 shows isoform-specific subcellular localisation
Since the three USP35 isoforms all contain an intact catalytic domain (Fig. 1A), we wondered whether the differences in the N-terminal region regulate their intracellular localisation. To address this possibility, we analysed the subcellular localisation of C-terminally GFP-tagged USP35 isoforms (Fig. 2A). Strikingly, we observed that the intracellular distribution of the proteins analysed was isoform specific. While USP35iso1 and USP35iso3 display a rather diffuse staining, USP35iso2 shows a reticular pattern, suggestive of endoplasmic reticulum (ER) localisation. Importantly, since the C-terminus of all isoforms analysed is identical, the differences observed in the staining pattern do not result from protein tagging. To determine which subcellular compartment USP35iso2 resides in, we performed colocalisation experiments that showed a clear overlap in the staining pattern of USP35iso2 and the endogenous ER marker BAP31 (also known as BCAP31) (Ng et al., 1997) (Fig. 2B), confirming that USP35iso2 localises to the ER.
Fig. 2.
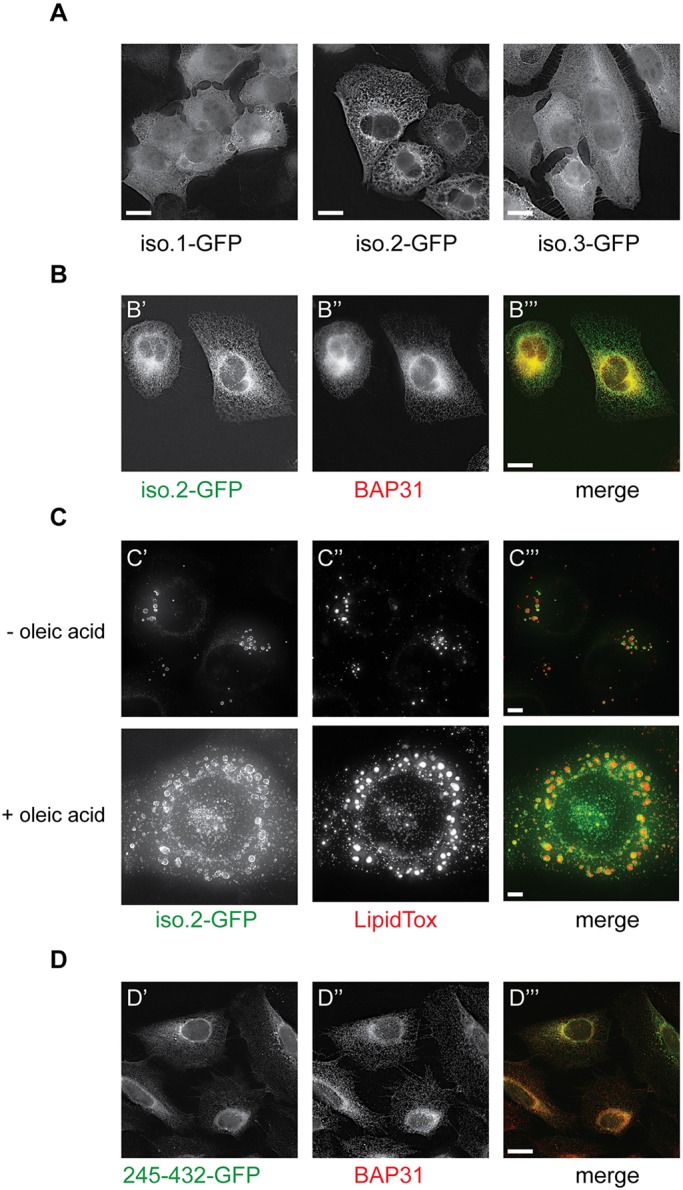
USP35 isoforms localise to distinct intracellular compartments. (A) U2OS FlpIn inducible cells expressing USP35 isoforms bearing a C-terminal GFP tag were fixed with paraformaldehyde (PFA) and stained with anti-GFP antibody. Protein localisation was examined by fluorescence microscopy. (B) As in A, but samples were simultaneously stained with anti-GFP and anti-BAP31 (an ER marker) antibodies. (C) U2OS FlpIn USP35iso2 cell line was left untreated or loaded with 0.3 mM oleic acid for 16 h at which point expression of isoform 2 was induced. Following an 8 h incubation, cells were fixed with PFA and stained with LipidTox Deep Red. LipidTox staining and intrinsic GFP fluorescence was analysed by microscopy. (D) Expression of USP35245-432 with a C-terminal GFP tag in stably transfected U2OS FlpIn cells was induced with tetracycline. Cells were fixed, stained with anti-GFP and anti-BAP31 antibodies, and samples analysed by immunofluorescence microscopy. Scale bars: 15 µm (A,B,D), 5 µm (C).
Interestingly, in addition to the reticular ER staining, USP35iso2 forms doughnut-like structures that could potentially correspond to protein aggregates, intracellular vesicles or lipid droplets (Fig. S4A). Interestingly, in a number of cells such doughnut-like staining of USP35iso2 was predominant, in particular when the detergent permeabilisation step during sample preparation was omitted. This suggests that these are lipid-based structures, such as lipid droplets. Lipid droplets are intracellular organelles that originate from the ER and serve as primary storage sites of neutral lipids, such as triglycerides and cholesterol esters (Welte, 2015). Given our finding that USP35iso2 localises to the ER and that the USP35iso2-positive doughnut-like structures are likely detergent sensitive, we investigated whether they correspond to lipid droplets. Indeed, USP35iso2 colocalises with the dye LipidTox, which stains lipid droplets (Fig. 2C, top panel), and this colocalisation is even more pronounced in cells treated with oleic acid (Fig. 2C, bottom panel), a condition that induces the formation and accumulation of lipid droplets (Eastman et al., 2009). These data indicate that, in addition to ER localisation, USP35iso2 is also present at lipid droplets making it, to the best of our knowledge, the first lipid droplet-localising DUB.
USP35 isoform 2 is an integral ER membrane protein
ER localisation of proteins results either from their integration into the lipid bilayer or peripheral binding to other proteins or lipids. To understand how ER targeting of USP35iso2 is achieved, we analysed its amino acid sequence. Two potential, albeit relatively hydrophilic, transmembrane domains (TMDs) predicted to span residues 317–337 and 797–815 (numbering based on isoform 1) were detected (Hessa et al., 2007). Helix 797–815 is located within the USP domain, suggesting that it does not mediate ER localisation. The potential TMD comprising residues 317–337 is more hydrophobic and also predicted by an alternative TMD prediction algorithm, TMpred (http://www.ch.embnet.org/software/TMPRED_form.html). Prediction algorithms do not indicate an N-terminal signal peptide in USP35iso2 amino acid sequence and hence the predicted transmembrane domain could also act as an ER targeting signal (Shao and Hegde, 2011). Indeed, a short USP35 fragment, USP35245–432, still colocalises with BAP31, indicating that the ER-targeting motif is located within this sequence, in agreement with the prediction for a putative TMD (Fig. 2D).
To experimentally address whether USP35iso2 is an integral membrane protein, we performed protease protection experiments using U2OS FlpIn cells expressing USP35 isoforms fused to GFP either at the N- or C-terminus. Cells were treated with digitonin, which selectively permeabilises the cholesterol-rich plasma membrane but does not permeabilise the cholesterol-poor ER membrane. This in turn allows for trypsin-mediated digestion of cytosolically exposed protein regions while leaving the ER luminal proteins intact owing to the inability of the protease to access the ER lumen. When calnexin (CNX, also known as CANX), a model ER membrane protein, was tested, addition of trypsin resulted in the cleavage of its cytosolic portion as assessed by the increase in its electrophoretic mobility (Fig. 3A). When the same analysis was carried out for GFP-tagged USP35 isoforms, an anti-GFP antibody-reactive protease-protected species was apparent only when N-terminally tagged USP35iso2 was expressed (Fig. 3A, lane 6, red dot). Importantly, the ∼37 kDa size of this fragment corresponds to the size of the GFP-tagged USP35270-360 fragment that would be generated by trypsin cleavage if residues 317–337 constitute the TMD (∼27 kDa GFP tag and ∼10 kDa mass of amino acids 270–360). No trypsin-resistant fragments were observed when isoform 1 or C-terminally tagged isoform 2 were used, indicating complete proteolysis of their GFP tag (Fig. 3A). The results obtained using the protease protection assay clearly show that isoform 2 is an integral membrane protein with its N-terminus in the ER lumen.
Fig. 3.

USP35 isoform 2 is a novel integral membrane DUB. (A) Expression of the indicated variants of USP35 in U2OS FlpIn cells was induced with tetracycline, cells were semi-permeabilised with digitonin and trypsin (tryp.) or buffer control was added. Samples were incubated for 1 h on ice, resolved by SDS-PAGE and immunoblotted (IB) with anti-GFP antibody. Immunoblotting for calnexin (CNX) was used to validate the assay. The red dot indicates the protease-resistant USP35 fragment; the arrow marks a protein species most likely corresponding to high amount of trypsin used in the assay. (B) U2OS FlpIn cells were transfected with USP35iso2 tagged at the N- or C-terminus with a peptide derived from bovine rhodopsin (OPG) that contains two N-glycosylation sites. Cells were lysed, and samples incubated with EndoH and resolved by SDS-PAGE. Results were visualised by immunoblotting with an anti-rhodopsin antibody. Numbers on the left indicate the number of N-glycans added to USP35iso2; the double arrowhead indicates an EndoH-sensitive truncated product of N-terminally tagged USP35iso2. (C) As in B, but an USP35 truncation spanning residues 270–432 was used. Numbers on the left indicate the number of N-glycans attached to the USP35 fragment. (D) As in B, but the indicated truncations of USP35 bearing an N-terminal OPG tag were used. Note that isoform 2 corresponds to the ‘270-end’ variant. Red dots represent N-glycosylated species. (E) Experimentally validated topology of USP35iso2. Amino acid sequence and conservation of the predicted transmembrane region is shown. ‘*’, fully conserved residues; ‘:’, conserved residues of strong groups; ‘.’, conserved residues of weak groups, as based on ClustalX. The sequences used were as follows: NP_065849.1 (H. sapiens), NP_001170883.1 (M. musculus), XP_015136357.1 (G. gallus), XP_002936594.2 (X. tropicalis) and NP_725167.1 (D. melanogaster). (F) HPAF-II cells were lysed in the absence of detergent, and post-nuclear supernatant (PNS), cytosolic and membrane fractions were isolated. Endogenous USP35 was identified by immunoblotting using the anti-USP35 antibody raised against the extreme C-terminus of the protein. Lysates of HPAF-II cells transfected with untagged USP35 isoforms 1 and 2 were used as markers for the electrophoretic migration of USP35 isoforms. CNX and tubulin were used as markers for the membrane and cytosolic fractions, respectively. Short and long exposures of the membrane are shown. (G) HPAF-II cells were lysed in RIPA buffer, and USP35 immunoprecipitated (IP) by using the indicated antibodies. Samples were resolved by SDS-PAGE and immunoblotted with the anti-USP35 antibody recognising the C-terminal region of the protein. Ectopically expressed untagged USP35 isoforms were used as markers for the electrophoretic mobility.
To further confirm our results, we used an assay that monitors N-glycosylation of ER-destined proteins (Borgese et al., 2001; Leznicki et al., 2010). Importantly, N-glycosylation takes place in the ER lumen and hence can be used as an independent read-out for the luminal localisation of a protein or a fragment of the protein. Therefore, we fused USP35iso2 to a short peptide derived from bovine rhodopsin (OPG) that contains two N-glycosylation sites (Borgese et al., 2001), and transiently expressed it in U2OS FlpIn cells. Consistent with the results of the protease protection assay, N-glycosylation of isoform 2 was only observed when the OPG tag was placed at its N-terminus, as assessed by the change in the electrophoretic mobility following endoglycosidase H (EndoH) treatment (Fig. 3B, lanes 1 and 2, OPG-iso2 panel). Consistent with amino acids 317–337 acting as the TMD, a short fragment of USP35, USP35270-432, is efficiently N-glycosylated when fused to the OPG tag (Fig. 3C). Interestingly, in this case, the orientation of the TMD is random as evidenced by the modification of both the N- and C-terminally tagged USP35270-432. This is most likely caused by the small size of the fragment used and suggests that the C-terminal portion (i.e. the catalytic domain) may play a role in dictating the topology.
The ER-targeting motif of USP35iso2 is also present in isoform 1; however, we found no evidence of its ER membrane integration. We speculated that this might be caused by the presence of the intact N-terminal HEAT repeats in isoform 1 that are truncated in isoform 2 (Fig. 1A). To investigate whether the length of the HEAT repeats affects ER integration of USP35, we generated truncated variants of USP35 bearing an N-terminal OPG tag and assessed their integration into the ER membrane using N-glycosylation as a read-out. Indeed, we observed that whereas USP35iso2 and USP35180-1018 are efficiently inserted into the ER lipid bilayer, increasing the length of the N-terminal HEAT repeats abolishes USP35 integration into the ER membrane (Fig. 3D). This argues for the role of HEAT repeats in limiting ER integration of USP35iso1. Taken together, we conclude that USP35iso1 is a soluble and predominantly cytosolic protein, while USP35iso2 is an integral membrane protein of the ER, with its N-terminus in the ER lumen, and C-terminus, together with the USP domain, facing the cytosol (Fig. 3E). Importantly, the sequence of the proposed transmembrane region is highly conserved in vertebrates (Fig. 3E).
It was previously reported (Buljan et al., 2012; Ellis et al., 2012; Yang et al., 2016) that the relative abundance of protein isoforms differs depending on the cell type and tissue analysed, and we wondered whether the same applied to USP35. We therefore raised antibodies that recognise the N-terminal (AbN), middle (Ab2) or C-terminal (AbC) portions of USP35 (Fig. 1A), and used AbC (which should recognise both isoforms 1 and 2) to screen a panel of different cell lines (Fig. S4B). In all cell lines tested, we could clearly detect a protein species that we identify as USP35iso1 based on its electrophoretic properties (Fig. S4B) and sensitivity to siRNA-mediated depletion (Fig. S4C).
Interestingly, in two cell lines, the pancreatic cancer cell line HPAF-II and lung cancer cell line NCI-H1435, we could also detect an additional USP35 protein species that could correspond to USP35iso2 as based on electrophoretic mobility (Fig. S4B). To verify whether this protein is indeed USP35iso2, we tested its localisation by performing subcellular fractionation of HPAF-II cells (Fig. 3F). To aid in the assignment of USP35 isoform bands, we used lysates of HPAF-II cells transfected with plasmids encoding untagged USP35iso1 and USP35iso2. Our results showed that USP35iso1 is predominantly cytosolic (Fig. 3F). Strikingly, we could also detect a protein species with electrophoretic migration identical to that of ectopically expressed untagged USP35iso2 (Fig. 3F, USP35, long exp.). Importantly, as expected for an integral membrane protein, this anti-USP35 antibody-reactive species was recovered exclusively in the membrane fraction (Fig. 3F, USP35, long exp.). To further confirm the existence of USP35iso2 at the endogenous level, we immunoprecipitated USP35 from HPAF-II cells (Fig. 3G). This approach allowed us to recover two USP35 species that migrate with similar electrophoretic mobility to isoforms 1 and 2 (Fig. 3G). Based on their electrophoretic properties, reactivity with three distinct anti-USP35 antibodies and results of the subcellular fractionation experiments, we infer that these two species correspond to USP35 isoforms 1 and 2.
USP35 isoform 2 induces ER stress and apoptosis
We next wanted to understand whether USP35 isoforms 1 and 2 perform isoform-specific functions. Intriguingly, when investigating the intracellular localisation of USP35, we noticed that cells expressing USP35iso2 in many cases showed altered BAP31 staining that resembled aggregate-like structures (Fig. 4A). To test whether this was due to a general aggregation of the ER membrane, we monitored the staining for signal peptide peptidase (SPP), another integral ER membrane protein. Localisation of SPP was unchanged, suggesting that the punctate staining of BAP31 was specific (Fig. 4A). Formation of BAP31 puncta reflects BAP31 cleavage by caspase-8 during apoptosis (Määttä et al., 2000), and expression of USP35iso2 results in BAP31 cleavage, as indicated by its increased electrophoretic mobility (Fig. 4B). Furthermore, BAP31 cleavage is accompanied by activation of caspase-8 and caspase-3 (Fig. 4B). Expression of USP35iso2 also causes ER stress, as is evident from the increased levels of the ER luminal chaperone BiP (also known as HSPA5) (Fig. 4B). Importantly, a control ER-membrane localising construct, GFP fused to the ER targeting sequence of cytochrome b5 (GFP–b5) (Bulbarelli et al., 2002), does not induce the same phenotype, indicating the specificity of USP35iso2 action (Fig. 4B, lanes 1 and 2). Consistent with the observed apoptotic events, overexpression of USP35iso2 blocks cell proliferation (Fig. 4C).
Fig. 4.

USP35 isoform 2 overexpression induces ER stress and apoptosis. (A) Expression of USP35iso2 tagged at the C-terminus with GFP was induced in U2OS FlpIn cells with tetracycline for 48 h. Cells were fixed with PFA, co-stained with anti-GFP, anti-BAP31 and anti-SPP (signal peptide peptidase) antibodies, and analysed by microscopy. Scale bar: 15 µm. (B) Expression of USP35iso2 or a control ER-localised construct GFP-b5 was initiated by adding tetracycline to U2OS FlpIn cell lines. After 48 h, cells were lysed, and samples resolved by SDS-PAGE and immunoblotted (IB) using antibodies against the indicated proteins. cl. BAP31, cleaved BAP31. p43, p41 and p18, cleaved products of pro-caspase 8. (C) As in B, but the cell lines were seeded into a 96-well plate and, at 48 h post-induction, an MTS proliferation assay was performed. The values on the y-axis represent the ratio of absorbance read for induced to non-induced cells. Error bars indicate s.e.m. (n=3 biological replicates). ****P<0.0001, ***P=0.0002 (one-way ANOVA). (D) USP35iso2 tagged at the C-terminus with GFP was expressed in the inducible U2OS FlpIn cell line for 24 h in the presence of 5 µM GSK2606414 or with DMSO (control). Cells were lysed, and samples resolved by SDS-PAGE and immunoblotted using antibodies against the indicated proteins. (E) As in B, but HeLa FlpIn cell lines induced to express GFP-b5 or USP35iso2 were used. (F) Expression of USP35iso2 bearing a C-terminal GFP tag was induced in U2OS FlpIn cell line. Levels of the GFP-tagged protein and upregulation of BiP were monitored at the indicated time points by immunoblotting cell lysates with antibodies against the indicated proteins. (G) As in B, but U2OS FlpIn cells stably transfected with GFP–USP19 were used as a control. ++ indicates a higher concentration of tetracycline used for GFP–USP19 expression.
ER stress initiates signalling events that result in improved folding capacity of the ER, for example, by upregulating the expression of luminal chaperones and blocking general protein translation (Tabas and Ron, 2011). Unresolved ER stress, however, triggers apoptosis through the action of protein kinase PERK (also known as EIF2AK3) and its target CHOP (also known as DDIT3), a transcription factor. CHOP activates transcription of several pro-apoptotic genes including death receptor 5 (DR5; also known as TNFRSF10B), a cell surface receptor for ligands, such as TNF-related apoptosis-inducing ligand (TRAIL), that stimulate the extrinsic apoptotic pathway (Lu et al., 2014). Importantly, ER stress-induced CHOP-mediated upregulation of DR5 has been proposed to activate caspase-8 leading to apoptosis (Lu et al., 2014). However, the precise function of caspase-8 in ER stress-induced cell death is debated, with some studies arguing against such a role for caspase-8 (Glab et al., 2017). In order to establish whether USP35iso2 expression also upregulates markers of ER stress-induced apoptosis, we monitored protein levels of CHOP and DR5 following USP35iso2 expression (Fig. 4D). Inducing USP35iso2 expression for 24 h resulted in significantly elevated levels of CHOP and DR5, and this effect was blocked by simultaneous treatment with GSK2606414, an inhibitor of PERK (Axten et al., 2012) (Fig. 4D). Importantly, in addition to the U2OS FlpIn cells used so far, we observe the same role of USP35iso2 in inducing ER stress and cell death in HeLa FlpIn cells (Fig. 4E), indicating that USP35iso2 function is conserved. Interestingly, while caspase-3 is clearly activated in HeLa FlpIn cells expressing USP35iso2, no processing of caspase-8 was detected, which is at odds with our findings using U2OS FlpIn cells. This is, however, consistent with conflicting reports about the role of caspase-8 in inducing cell death during conditions of ER stress (Glab et al., 2017; Lu et al., 2014). We then sought to understand the kinetics of USP35iso2-mediated ER stress. To this end, expression of C-terminally tagged USP35iso2 was induced, and ER stress monitored over time. Strikingly, BiP expression was already elevated after 10 h induction of USP35iso2, which is only 4 h after the protein is detectable by western blotting (Fig. 4F). To further assess the specificity of USP35iso2-induced ER stress and apoptosis, we created an U2OS FlpIn cell line expressing GFP-tagged USP19, the only reported integral membrane DUB of the ER. Importantly, expression of USP19 at levels comparable to USP35iso2 did not induce apoptosis or detectable ER stress (Fig. 4G). Collectively, our results suggest that ectopic USP35iso2 expression causes specific and rapid ER stress and apoptosis.
USP35iso2-mediated ER stress results from misregulated lipid homeostasis and is uncoupled from apoptosis induction
Given the lipid droplet localisation of USP35iso2, and links between the ER and lipid metabolism, we speculated that USP35iso2 induces ER stress and cell death by interfering with some aspects of lipid homeostasis. Because lipid droplets are the primary storage sites for cholesterol esters, we investigated whether USP35iso2-induced ER stress and apoptosis might result from aberrations of cholesterol metabolism. Hence, we followed the effect of USP35iso2 expression on ER stress in the presence of mevastatin and lovastatin, inhibitors of 3-hydroxy-3-methyl-CoA reductase (HMGCR), a rate-limiting enzyme of cholesterol biosynthesis. Strikingly, we observed that inhibiting HMGCR significantly reduces USP35iso2-induced ER stress, as measured by BiP upregulation (Fig. 5A,B). Importantly, inhibiting cholesterol biosynthesis had no effect on ER stress initiated by disrupting ER Ca2+ homeostasis (Fig. 5A). Taken together, these results imply that inhibiting cholesterol synthesis specifically interferes with the early steps of ER stress induced by USP35iso2 rather than downstream events general to the ER stress response. Surprisingly, inhibiting cholesterol synthesis prevents USP35iso2-induced ER stress but does not impede caspase-8 and caspase-3 activation, suggesting that ER stress and apoptosis induced by USP35iso2 could be unrelated events. To exclude the possibility that the inhibitory effect of the statins was an indirect consequence of depleting cholesterol, we expressed USP35iso2 in U2OS FlpIn cells grown in medium containing lipoprotein deficient serum (LPDS) and assessed ER stress by monitoring BiP levels (Fig. S5A). We found that while LPDS treatment efficiently lowered cellular cholesterol levels, as measured by upregulation of low density lipoprotein receptor (LDLR), it did not block USP35iso2-induced ER stress. Hence, we conclude that cholesterol per se is not required for the induction of ER stress by USP35iso2, but rather USP35iso2 expression interferes with lipid homeostasis possibly by affecting the cholesterol and/or isoprenoid biosynthetic route.
Fig. 5.

Inhibition of cholesterol biosynthesis blocks USP35iso2-induced ER stress. (A) U2OS FlpIn parental cells were pre-treated with 10 µM mevastatin or lovastatin for 2 h and incubated with 100 nM thapsigargin for an additional 18 h (lanes 2–4). In parallel, U2OS FlpIn cells were induced (ind.) to express USP35iso2 for 48 h in the presence of 10 µM statins. Samples were analysed as in Fig. 4B. Control samples represent cells without thapsigargin treatment or USP35iso2 induction. A long and short exposure is shown for the BiP blot. (B) Samples after statin treatment and USP35iso2 induction as shown in A (lanes 6–9) were analysed by quantitative western blotting, and the fold change of BiP induction relative to control plotted. Error bars indicate the s.e.m. with n=3 biological replicates. ****P<0.0001; NS, not significant (one-way ANOVA test). (C) As in Fig. 4B, but U2OS FlpIn cells expressing indicated variants of USP35iso2 in a tetracycline-inducible manner were used. (D) U2OS FlpIn cells expressing indicated variants of USP35iso2 for 24 h were subjected to subcellular fractionation followed by western blotting analysis. WT, wild-type. (E) U2OS FlpIn cells were induced to express indicated GFP-tagged variants of USP35iso2 or GFP-b5 for 24 h. Cells were lysed, and GFP-tagged proteins and their binding partners recovered and samples analysed by western blotting against the indicated proteins. (F) The indicated USP35 fragments (numbering based on isoform 1) were tagged at the C-terminus with GFP and at the N-terminus with the first 61 residues of SPP, which encompasses its first transmembrane region (TM1). USP35iso2 and GFP fused to the same N-terminal fragment of SPP served as controls. Cells were treated and samples processed as described for Fig. 4B.
To further explore the links between USP35iso2-mediated ER stress and apoptosis, we expressed various USP35iso2 truncations and assessed their capacity to induce ER stress and apoptosis. We observed that USP35iso2 lacking the insertion loop within the catalytic domain (Fig. 1A) was more efficient at inducing apoptosis than the wild-type protein, despite lower capacity to induce ER stress (Fig. 5C). Importantly, expression of USP35iso2 lacking the predicted ER luminal residues (amino acids 270–316) no longer triggered ER stress and apoptosis (Fig. 5C). Although it is unclear why deletion of the insertion loop from USP35iso2 increases its capacity to induce apoptosis yet lowers the extent of ER stress that it induces, this result again shows that apoptosis is not a simple consequence of ER stress caused by USP35iso2 expression. Hence, these observations suggest that the ER stress and apoptosis induced by USP35iso2 are two distinct phenomena.
To further investigate this possibility, we tested the effect of the PERK inhibitor GSK2606414 on cells expressing USP35iso2 (Fig. S5B). Again, we found that GSK2606414 almost completely blocked ER stress induced by USP35iso2 (Fig. S5B, compare lanes 1 and 4; see also Fig. 4D). Despite the loss of ER stress, the cells still exhibited apoptosis, albeit to a lesser extent than in control cells. Interestingly, a recent study showed that GSK2606414 is also a potent inhibitor of RIPK1-mediated apoptosis and necroptosis (Rojas-Rivera et al., 2017). Hence, an ‘off-target’ effect of GSK2606414 on apoptosis that is independent of ER stress is likely, and could explain the apparent discrepancy between the effect of GSK2606414 and statins on USP35iso2-induced apoptosis. Nonetheless, the fact that GSK2606414 failed to completely block USP35iso2-induced apoptosis led us to conclude that ER stress and apoptosis induced by USP35iso2 are unrelated events.
To investigate whether the ability of USP35iso2 to induce ER stress and apoptosis is linked to ER membrane/lipid droplet localisation, we performed subcellular fractionation and pull-down assays from cells expressing GFP-tagged wild-type USP35 and its variants lacking either the insertion loop or luminal residues. Indeed, we observed that variants that induce ER stress and apoptosis efficiently co-fractionate with cellular membranes (Fig. 5D) and interact with the lipid droplet-associated protein PLIN2 (Fig. 5E). Such shared requirements for membrane localisation and lipid droplet association are in agreement with previous reports showing that localisation to these two organelles can be mediated by the same hydrophobic domain (Bersuker et al., 2018; Zehmer et al., 2008). USP35iso2 lacking residues 270–316 only weakly associated with membranes (Fig. 5D), suggesting that these residues contribute to the ER membrane localisation of USP35iso2. These findings demonstrate the importance of membrane/lipid droplet localisation of USP35iso2 for the induction of ER stress and apoptosis. To further verify this conclusion, we fused GFP-tagged USP35 fragments to the first 61 residues of SPP, which encompass its first TMD, with a topology that ensures cytosolic localisation of the USP35 catalytic domain. Strikingly, we observed that such enforced ER membrane localisation of soluble fragments of USP35 spanning residues 338–1018 and 433–1018 (which corresponds to isoform 3) was sufficient to induce both ER stress and apoptosis (Fig. 5F). However, targeting GFP alone to the ER using the same tag does not produce the effects as observed with USP35. Hence, ER membrane anchoring of the USP35 catalytic domain is sufficient to induce ER stress and apoptosis.
USP35iso1 is an anti-apoptotic protein
In order to compare the functions of isoforms 1 and 2 of USP35, we tested the effect of their overexpression on the induction of ER stress and apoptosis. Consistent with our previous data (Fig. 4), increased levels of USP35iso2 led to the induction of apoptosis (Fig. 6A). In contrast, USP35iso1 did not induce the same phenotype (Fig. 6A), suggesting distinct functions for these two isoforms, in line with their distinct intracellular localisation.
Fig. 6.

USP35 isoform 1 is an anti-apoptotic protein. (A) Expression of the indicated isoforms of USP35 in stably transfected U2OS FlpIn cell lines was carried out for 48 h. Cells were lysed, and samples were resolved by SDS-PAGE and immunoblotted (IB) with antibodies against the indicated proteins. cl. BAP31, cleaved BAP31. (B) HEK293 FlpIn stably transfected cell lines were induced to express FLAG-tagged USP35iso1 wild-type (WT) protein, its catalytically inactive variant (C450A) or empty vector control. At 24 h post induction, cells were treated with 50 ng/ml TRAIL. Cells were lysed, samples resolved by SDS-PAGE and processing of caspase-8 analysed by immunoblotting. (C) HEK293 FlpIn parental cell line and two USP35 knockout clones (denoted 7-6 and 7-10) were treated with 50 ng/ml TRAIL and samples were analysed as for B. (D) As in C, but the cell lines were seeded into a 96-well plate, incubated with 50 ng/ml TRAIL for the indicated times and MTS proliferation assay was performed. The values on the y-axis represent the ratio of absorbance read for treated to untreated cells. Error bars indicate the s.e.m. for n=3 biological replicates. (E) HEK293 FlpIn parental cell line and the two USP35 knockout clones were treated with increasing concentrations of staurosporine for 24 h. Cells were lysed, and samples analysed by immunoblotting with an antibody against cleaved caspase-3. (F) HeLa cells were transfected with wild-type USP35iso1 or a variant with Asp743 replaced with an alanine residue, both tagged at the C-terminus with a GFP tag. At 24 h post transfection, 1 µM staurosporine was added and cells incubated for the indicated time. Cells were lysed, and samples were resolved by SDS-PAGE and analysed by immunoblotting against the indicated proteins.
The D. melanogaster orthologue of USP35 and USP38, DUBAI, has previously been shown to be an anti-apoptotic protein (Yang et al., 2014). To test whether USP35iso1 has the same function in mammalian cells, we monitored apoptosis in HEK293 cells overexpressing USP35iso1 following treatment with the protein TRAIL, an apoptotic stimulus, by monitoring cleavage of caspase-8, the main initiator caspase of the extrinsic apoptotic pathway. Compared to control cells, cells expressing increased levels of USP35iso1 exhibit delayed processing of caspase-8 during TRAIL-induced apoptosis (Fig. 6B). Importantly, this anti-apoptotic effect required the catalytic activity of USP35iso1 (Fig. 6B, lanes 9–16 and 17–24). Since overexpression has an anti-apoptotic effect, we posited that depletion of USP35 would result in an opposite effect (i.e. sensitise cells to apoptotic stimuli). To address this possibility, we deleted USP35 using CRISPR/Cas9-mediated gene editing. Indeed, we observed that USP35 knockout cells are substantially more sensitive to TRAIL-induced apoptosis as assessed by activation of caspase-8 (Fig. 6C). Consistent with such increased processing of caspase-8 upon USP35 depletion, USP35 knockout cells are significantly more sensitive to TRAIL treatment (Fig. 6D). Furthermore, we also observed increased sensitivity of USP35 knockout clones to staurosporine-induced apoptosis, as assessed by activation of caspase-3 (Fig. 6E). Our results reveal that, in contrast to USP35iso2, isoform 1 has an anti-apoptotic function.
A common feature of many anti-apoptotic proteins, such as inhibitors of apoptosis proteins (IAPs), is their proteolytic processing during apoptosis (Hao et al., 2004; Hörnle et al., 2011), which leads to their inactivation and allows for progression of cell death. We therefore wanted to investigate whether isoform 1 of USP35 is also a subject of such processing. To test this possibility, we induced apoptosis with staurosporine in HeLa cells, which express USP35iso1 at relatively high levels (Fig. S4B). Strikingly, endogenous USP35 was efficiently cleaved during staurosporine-induced cell death (Fig. S6A,B). The cleaved fragments could be recovered by immunoprecipitation using antibodies raised against the N- or C-terminal portion of USP35 with the N-terminal fragment being ∼85 kDa and the C-terminal one ∼30 kDa (Fig. S6B). This USP35 proteolysis could be blocked by zVAD-fmk, a pan-caspase inhibitor, suggesting that the processing is mediated by caspase(s) (Fig. S6A,B). Indeed, an in vitro caspase cleavage assay indicates that proteolysis of USP35 is mediated by the executioner caspases, caspase-3 and/or -6 (Fig. S6C). Mass spectrometric analyses identified Asp743 as the cleavage site, a finding consistent with the size of USP35 fragments observed in HeLa cells undergoing apoptosis (Fig. S6A,B). Indeed, mutation of the cleavage site Asp743 to alanine completely blocked USP35 proteolysis during staurosporine-induced apoptosis (Fig. 6F). In summary, our findings reveal that USP35iso1 is an anti-apoptotic protein and suggest a model where proteolytic cleavage by caspases at Asp743 within the USP35 catalytic domain inactivates the DUB, and thereby its anti-apoptotic function.
USP35 isoform-specific interactome
The fact that USP35iso1 is anti-apoptotic and USP35iso2 pro-apoptotic suggests that these two proteins might exert their effects by differentially regulating common interacting partner(s). To investigate this possibility, we identified the binding partners of both USP35 isoforms by using HEK293 FlpIn cell lines expressing USP35 isoforms C-terminally tagged with BirAR118G. This allows for the use of the BioID methodology capable of identifying interactions that are transient in nature or occur in organelles resistant to conventional immunoprecipitation techniques (Roux et al., 2012). In agreement with the distinct subcellular localisation of USP35iso1 and USP35iso2, we found that the GO terms associated with their interacting partners are differently enriched (Fig. 7A). Hence, USP35iso2 preferentially interacts with proteins linked to intracellular membranes, in particular the ER. In contrast, USP35iso1 interacts predominantly with cytosolic and centrosomal proteins. Importantly, USP35iso2 interacted with a number of enzymes linked to lipid metabolism (HMGCR, CYP51A1 and AGPAT4) and protein quality control (TRIM13, BAG6, UBE2J1, UBR3) (Fig. 7B; Table S3). Only ∼15% of the total binding partners were shared by the two USP35 isoforms confirming their distinct functions. Interestingly, among the common interacting proteins was BIRC6, a known IAP (Bartke et al., 2004; Hao et al., 2004), which could potentially be targeted by both USP35 isoforms. To test whether USP35 isoforms act on the same pathway regulating apoptosis, we tested whether USP35iso1 can inhibit USP35iso2-induced apoptosis. Cells expressing USP35iso2 in an inducible manner were transfected with USP35iso1 at the time of USP35iso2 induction, and upregulation of BiP and caspase-3 cleavage were monitored. This revealed that USP35iso1 cannot block USP35iso2-induced apoptosis (Fig. S6D), suggesting that the two isoforms regulate different aspects of apoptosis.
Fig. 7.

Isoform-specific interactome of USP35. (A) HEK293 FlpIn cells expressing USP35iso1 or USP35iso2 tagged at their C-terminus with BirAR118G were induced for 24 h. Biotinylated proteins were recovered on streptavidin–Sepharose, identified by mass spectrometry and subjected to SAINT analysis and DAVID GO analysis. Green bars show GO terms for the percentage of genes identified; red lines indicates the −log10 P value. (B) Isoform-specific and shared interacting partners are shown.
DISCUSSION
In the current study, we show that mammalian cells express at least two USP35 isoforms that localise to distinct subcellular compartments and have distinct functions. Isoform 1 is an anti-apoptotic protein that regulates progression of TRAIL- and staurosporine-induced apoptosis. At the same time, isoform 2 is a novel integral membrane protein that localises to the ER and lipid droplets; its overexpression perturbs lipid homeostasis and causes ER stress and apoptosis.
Interestingly, until now USP19 was the only known DUB that is an integral ER membrane protein (Hassink et al., 2009). USP35iso2, which we identified in this study, becomes the second integral ER membrane DUB. Intriguingly, as is observed for USP35, USP19 also exists in at least two forms, cytosolic and ER membrane-localised, that have non-redundant functions (Hassink et al., 2009; Lee et al., 2016). Moreover, USP35iso2 is, to the best of our knowledge, the first known lipid droplet-associated DUB. Lipid droplets perform important functions that extend beyond lipid storage and mobilisation. These include protein sequestration and degradation, as evidenced by the presence of ubiquitin-conjugating machinery at lipid droplets (Klemm et al., 2011; Spandl et al., 2011; Welte, 2015). Moreover, lipid droplet dynamics is also regulated by protein ubiquitylation (Eastman et al., 2009), and hence our discovery of a lipid droplet-associated DUB opens new avenues of research.
Surprisingly, USP35iso1 and USP35iso2 both contain the same ER membrane-targeting motif, yet we observe that only isoform 2 inserts into the ER lipid bilayer. Based on our data, we postulate that the N-terminal HEAT repeats present in isoform 1 but truncated in isoform 2 mediate either co-translational folding or recruitment of interacting factors that prevent ER delivery of isoform 1. Indeed, the inhibitory effect of co-translational folding on protein translocation into the ER is well documented (Conti et al., 2014; Denzer et al., 1995). Removal of the N-terminal 269 residues as seen in USP35iso2 would disrupt the HEAT repeat motif, which is composed of consecutive α-helices that form extensive contacts with each other. This, in turn, would allow for efficient ER targeting. Interestingly, addition of an N-terminal GFP tag to USP35iso2 does not block its integration into the ER membrane, indicating that specific interactions of the N-terminal portion of isoform 1 are required to prevent its ER delivery.
Strikingly, we observe that ectopic expression of USP35iso2 induces rapid ER stress and apoptosis in different cell lines. We show that ER stress initiated by USP35iso2 can be attenuated by inhibition of the rate-limiting enzyme of cholesterol biosynthesis, HMGCR, suggesting that it most likely results from perturbed lipid homeostasis. Imbalance in lipid metabolism can alter the composition of the ER membrane causing ‘lipid bilayer stress’ (Han et al., 2010; Pineau et al., 2009; Promlek et al., 2011; Thibault et al., 2012), and our data suggest that USP35iso2 might contribute to this effect. Importantly, we also show that anchoring soluble fragments of USP35 to the ER membrane is sufficient to induce both ER stress and apoptosis. The precise mechanistic details behind USP35iso2-mediated ER stress remain to be uncovered. However, growing incidence of metabolic disorders linked to cholesterol metabolism and ER stress, such as atherosclerosis (Hotamisligil, 2010), warrants future studies on USP35iso2.
Interestingly, our data indicate that the two phenotypes, ER stress and apoptosis, induced by USP35iso2 are unrelated. Intriguingly, our transcriptomics analyses indicate that total mRNAs coding for USP35iso2 is more abundant than mRNA encoding isoform 1 in cell lines and human tissues. However, at the protein level, USP35iso1 is the predominant USP35 species (Figs S4B and S7A,B). It is tempting to speculate that protein levels of USP35iso2 are kept low in order to avoid ER stress and apoptosis. However, the abundance of USP35iso2 mRNA would allow for its rapid translation in response to a yet unknown stimulus. Because USP35iso2 levels do not appear to increase following proteasome inhibition with MG132 (Fig. S4C), we think that USP35iso2 protein levels are controlled at the translation step rather than through regulated protein degradation. A similar relief of miRNA-inhibited protein translation during conditions of stress has been reported for the cationic amino acid transporter 1 (CAT-1) (Bhattacharyya et al., 2006), establishing a paradigm for such a mechanism. Interestingly, USP35 is amplified in a number of cancers (Chin et al., 2007; see Table S4 for USP35 copy number in selected cancer cell lines) and our analysis indicates that in most primary tumours, USP35iso2 is also preferentially transcribed (Fig. S7C). Identification of stimuli that induce USP35iso2 production at the protein level will be of key importance for deciphering its molecular function.
We show that, in contrast to USP35iso2, USP35iso1 is a novel anti-apoptotic protein. This is evident from both overexpression and knockout systems, and by assaying apoptosis in response to TRAIL and staurosporine treatments. Hence, USP35 is a hitherto unstudied DUB that regulates programmed cell death. This is consistent with the role of DUBAI, the D. melanogaster orthologue of both mammalian USP35 and the related DUB USP38 (Yang et al., 2014). DUBAI protects cells from apoptosis by stabilising DIAP1, a major IAP in fly. Although details of apoptotic pathways differ to some extent between fly and mammals, we provide evidence that the function of DUBAI as an anti-apoptotic factor is evolutionarily conserved, and in mammals is carried out by USP35iso1. Consistent with anti-apoptotic roles, USP35 is amplified in a number of cancers, in which preferential translation of USP35iso1 would block apoptosis and ensure cancer cell survival. Interestingly, we observe that both USP35iso1 and USP35iso2 interact with the mammalian IAP Birc6 (Bartke et al., 2004; Hao et al., 2004). It remains to be tested whether this interaction plays a role in the observed anti- and pro-apoptotic effects of USP35iso1 and USP35iso2, respectively.
Consistent with the distinct subcellular localisation, our proteomic analysis indicates that USP35 isoforms mostly interact with distinct sets of proteins. While USP35iso2 interacts predominantly with membrane-associated proteins, USP35iso1 preferentially binds cytoplasmic and centrosomal proteins. The latter finding suggests that USP35iso1 could participate, for example, in regulating cell division. Indeed, during the preparation of this manuscript, it was reported that USP35 stabilises Aurora B, a key kinase regulating mitotic progression, and USP35 depletion results in cytokinesis defects (Park et al., 2018). At the moment, it is unclear whether and how the anti-apoptotic effect of USP35iso1 is related to its function in stabilising Aurora B. Intriguingly, Birc6 has also been implicated in cytokinesis (Pohl and Jentsch, 2008) and future studies will establish whether the cytokinesis defects observed following USP35 depletion result solely from its effect on Aurora B or whether a USP35–Birc6 interaction also plays a role.
Current proteomic datasets indicate that at least ∼20,000 sites are modified with ubiquitin at steady state conditions in human cells (Kim et al., 2011; Udeshi et al., 2013), and it is therefore a massive endeavour for the relatively small number of ∼100 DUBs to regulate them. Our results provide an important paradigm showing that alternative protein isoforms are of key importance for the expansion of DUB functionality and stress the need to study DUB isoforms. However, dissecting isoform-specific cellular functions is challenging and will require development of novel approaches, such as the ability to selectively deplete only one isoform. We anticipate that studying functions of different isoforms in different experimental settings will reveal not only new DUB biology but also the true extent to which DUBs regulate cell physiology.
MATERIALS AND METHODS
Materials
Anti-USP35 antibodies were made to order by immunising sheep with glutathione-S-transferase (GST)-tagged recombinant fragments corresponding to USP35 residues 1–437 (AbN), 604–753 (Ab2) and 925–1018 (AbC). Anti-GST-reactive antibodies were subsequently depleted by passing the sera through a resin containing immobilised recombinant GST. These antibodies were used at 0.1 µg/ml for western blotting and 2–3 µg per 1 mg of cell lysate for immunoprecipitation. The following commercial antibodies were used for western blotting: rabbit anti-GFP (1:7500, Abcam, ab6556), rabbit anti-calnexin (1:1000, Cell Signaling Technology, 2679), mouse anti-α-tubulin (1:10,000, Cell Signaling Technology, 3873), mouse anti-caspase-8 (1:1000, Cell Signaling Technology, 9746), rabbit anti-caspase-8 (1:1000, Tocris, AF1650-SP), rabbit anti-cleaved caspase-3 (1:1000, Cell Signaling Technology, 9661), rabbit anti-BiP (1:1000, Cell Signaling Technology, 3177), rabbit anti-BAP31 (1:1000, ProteinTech, 11200-1-AP), mouse anti-CHOP (1:1000, Cell Signaling Technology, 2895P), rabbit anti-DR5 (1:1000, Cell Signaling Technology, 8074), and mouse anti-GAPDH (1:10,000, Abcam, ab8245) antibodies. Mouse monoclonal anti-OPG tag antibodies (used at 1:1000 dilution) were a kind gift of Prof. Stephen High (University of Manchester, Manchester, UK) and were described previously (Adamus et al., 1991). Antibodies used for immunofluorescence microscopy were as follows: chicken anti-GFP (1:2000, Abcam, ab13970), rat anti-BAP31 (1:2000, Abcam, ab15044), and rabbit anti-SPP (1:200, Bethyl Laboratories, A304-404A) antibodies.
Oleic acid–albumin complexes were purchased from Sigma-Aldrich (O3008), LipidTox Deep Red Neutral Lipid Stain was from Thermo Fisher Scientific (H34477), Endoglycosidase H was from New England Biolabs (P0703), the MTS assay kit was from Promega (G3582), human recombinant TRAIL was from Peprotech (310-04), and zVAD-fmk was from Enzo Life Sciences (ALX-260-020-M001). GSK2606414 (5107), mevastatin (1526) and lovastatin (1530) were obtained from Tocris.
Cell culture methods
HeLa FlpIn T-rex and HEK293 FlpIn T-rex cells were obtained from ThermoFisher Scientific, HPAF-II cells were from the ATCC, and U2OS FlpIn T-rex cells were as previously reported (Bago et al., 2014). All cell lines were regularly tested for contamination. U2OS FlpIn T-rex, HeLa FlpIn T-rex and HEK293 FlpIn T-rex cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. HPAF-II cells were grown in Eagle's minimum essential medium (EMEM) supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, 1 mM sodium pyruvate and non-essential amino acids. Stable cell lines were generated by co-transfecting cells with pOG44 and plasmids encoding indicated proteins, followed by selection in complete medium containing 100 µg/ml hygromycin and 7.4 µg/ml blasticidin. Protein expression in stable cell lines was induced by addition of tetracycline to a final concentration of 200 ng/ml (U2OS FlpIn T-rex cells) or 1 µg/ml (HEK293 FlpIn T-rex cells).
DNA transfection was carried out using GeneJuice (Merck Millipore) according to the manufacturer's instruction. For cell treatment with oleic acid, the medium was replaced with DMEM supplemented with 0.3 mM oleic acid–bovine serum albumin (BSA) complexes (Sigma-Aldrich), and cells were incubated for an additional 24 h. Cells were also treated with 20 µM MG132 for 7 h, and 50 ng/ml recombinant human TRAIL (Peprotech) or 1 µM staurosporine for the times indicated.
Mevastatin and lovastatin were used at 10 µM and added simultaneously with tetracycline-mediated induction of USP35iso2. Cells were then incubated for 48 h. Alternatively, parental cells were treated with statins for 2 h, then 100 nM thapsigargin was added and cells were further incubated for 18 h.
USP35 knockout clones were generated by targeting exon 7 shared by all isoforms tested (sense gRNA, 5′-gCATCCTTCAGGCCTTATTCA-3′, antisense gRNA, 5′-gTGACATAGCATGTGTTGCCC-3′). Cells transfected with plasmids encoding these gRNAs and Cas9 were sorted into 96-well plates, and clones derived from single cells were screened by western blotting. Candidate knockout clones used for subsequent experiments were verified by sequencing of the targeted locus.
Fluorescence microscopy
Cells were grown on coverslips in six-well plates and processed as described in figure legends. After washing with PBS, cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. PFA was quenched by three consecutive washes with PBS supplemented with 100 mM glycine pH 8.0 (5 min each) and cells permeabilised with 0.1% (w/v) Triton X-100 (TX-100) for 5 min at room temperature. TX-100 was washed off with PBS and samples blocked with 1% (w/v) BSA in PBS for 15 min at room temperature. Coverslips were stained with chicken anti-GFP, rat anti-BAP31 and rabbit anti-SPP antibodies for 1 h at room temperature. Incubation with secondary antibodies (Alexa Fluor conjugates, 1:750) was carried out for 30 min and coverslips were mounted using ProLong Gold reagent.
For lipid droplet staining, cells were fixed, PFA quenched as described above and coverslips stained with LipidTox Deep Red (Life Technologies, 1:400) for 45 min at room temperature.
Samples were imaged using DeltaVision fluorescence microscopes, and images processed by deconvolution with Softworx software.
Protein purification and in vitro assays
The pFastBac expression vectors encoding full-length human GST–USP35 and GST–USP35 C450A were used to generate recombinant baculoviruses using the Bac-to-Bac system (Invitrogen) following the manufacturer's protocol. These baculoviruses were used to infect Spodoptera frugiperda 21 cells (1.5×106/ml) at a multiplicity of infection of five and the infected cells were harvested 48 h post-infection. GST–USP35 and GST–USP35 C450A were purified on GSH-Sepharose and dialysed into 50 mM Tris-HCl pH 7.5, 0.1 mM EGTA, 150 mM NaCl, 270 mM sucrose, 0.03% Brij-35, 0.1% 2-mercaptoethanol, 1 mM benzamidine, 0.1 mM phenylmethylsulfonyl fluoride (PMSF).
The in vitro caspase cleavage assay was performed with the Caspases Set III human recombinant kit (#PK-RP577-K232, PromoKine), as per the manufacturer’s instruction. In brief, 2 U of active recombinant human caspases were incubated with 4 μg purified human USP35C450A in buffer C [50 mM Hepes, pH 7.2, 50 mM NaCl, 0.1% (w/v) CHAPS, 10 mM EDTA, 5% (w/v) glycerol and 10 mM DTT] for 2 h at 37°C.
DUB assays were carried out with 375 nM of USP35 and 733 nM of tetraUb of different linkage types (Met1, Lys6, Lys11, Lys29, Lys33, Lys48 and Lys63) in a 10 µl volume in buffer D (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM DTT) at 37°C for the times indicated. Reactions were quenched by adding LDS sample loading buffer.
Protein crystallisation
A construct of USP35 encoding amino acids Ser423–Pro944 with a deletion of Glu604–Cys753 (codon optimised for expression in Sf9cells) was generated by gene synthesis (GeneArt, Thermo Fisher Scientific) and expressed from a pDEST20 vector (Thermo Fisher Scientific) in Trichoplusia ni insect cells (High Five, Thermo Fisher Scientific) in Insect-XPRESS medium (Lonza) as an N-terminal GST fusion protein (cleavable by tobacco etch protease; TEV). Cells were collected after 48 h and homogenised with a Dounce tissue grinder in PBS pH 7.3, 10% glycerol and 5 mM DTT, in the presence of protease inhibitors (Complete, Roche Applied Science). After centrifugation at 11,000 g (Beckman JA-10), the supernatant was batch-bound to a glutathione resin (glutathione–Sepharose, GE Healthcare). The resin slurry was washed with PBS, 10% glycerol and 5 mM DTT, pH 7.3. The fusion protein was cleaved with TEV protease (produced in-house) at 4°C overnight to remove the GST tag. Resin was packed into a column while collecting protein in the flow-through. Protein eluting upon an additional wash step with buffer was combined with the flow-through. After a buffer exchange to 50 mM HEPES pH 7.3, 50 mM NaCl, 5 mM DTT and 5% glycerol, on a HiPrep Desalting column (GE Healthcare) the protein was applied to cation- and anion-exchange columns coupled in series [Mono S(1st)-Mono Q(2nd) (GE Healthcare)], pre-equilibrated with 50 mM HEPES pH 7.3, 50 mM NaCl, 5 mM DTT and 5% glycerol. After separating the columns, the recombinant protein was eluted from the Mono Q column with a gradient of 50 mM NaCl to 1 M NaCl in 50 mM HEPES pH 7.3, 5 mM DTT and 5% glycerol. The eluted protein fraction was further purified on a gel filtration column (HiLoad Superdex S200, GE Healthcare) pre-equilibrated with 50 mM HEPES pH 7.3, 150 mM NaCl, 5 mM DTT and 5% glycerol. The purified protein was concentrated to ∼13 mg/ml and stored at −80°C before further use.
The purified protein was incubated with a 2-fold molar excess of a Lys48-linked ubiquitin dimer for 30 min and crystallised by the vapour diffusion method. 1 µl of protein solution (10.5 mg/ml in 50 mM HEPES, 50 mM NaCl, 5% Glycerol, 5 mM DTT pH 7,3) was mixed with 1 µl of reservoir solution containing 100 mM HEPES pH 6.0 and 0.95 M ammonium sulfate at 20°C. Data were collected on an in-house Cu sealed tube (Rigaku MicroMax-003) and processed using AutoProc (Vonrhein et al., 2011). The structure was solved by sulfur single-wavelength anomalous dispersion (S-SAD) and automatically built using the AutoSol wizard of Phenix (Adams et al., 2010). The autobuilt model contained 378 of the 448 residues present in the asymmetric unit and was further refined using COOT (Emsley et al., 2010) and AutoBUSTER (Bricogne et al., 2016). See Table S2 for data collection and refinement statistics.
Determination of membrane protein topology
Potential transmembrane regions of USP351–1018 were predicted by using the ΔG predictor (Hessa et al., 2007) and TMpred (http://www.ch.embnet.org/software/TMPRED_form.html).
The protease protection assay was carried out using U2OS FlpIn cells induced with 200 ng/ml tetracycline for 48 h in order to express the indicated proteins. Cells from a 15-cm dish were trypsinised, spun down and resuspended in 4 ml ice-cold KHM buffer [20 mM HEPES-NaOH pH 7.5, 110 mM KOAc, 2 mM Mg(OAc)2]. Digitonin was added to a final concentration of 80 µg/ml and samples incubated on ice for 5 min, at which point they were diluted to 14 ml with KHM buffer and cells spun down. Cells were resuspended in 5 ml ice-cold HEPES buffer (90 mM HEPES-NaOH, pH 7.5, 50 mM KOAc), incubated for 10 min on ice and again isolated by centrifugation. Cells were resuspended in 1 ml ice-cold KHM buffer, counted and after pelleting resuspended in ice-cold KHM buffer. Proteolysis was carried out using equal number of cells expressing each construct in reactions supplemented with 0.5 mg/ml trypsin or 0.1 mM HCl used as a control. Reactions were performed for 1 h on ice, stopped by the addition of soya bean trypsin inhibitor (final concentration 1.25 mg/ml) and incubated for additional 10 min on ice. Sample buffer was added and samples denatured by heating at 70°C for 10 min.
In order to determine the topology using N-glycosylation as a read-out, U2OS FlpIn cells transfected with USP35 constructs tagged with a peptide derived from bovine rhodopsin (SRMNGTEGPNFYVPFSNKTVD) were lysed and denatured in SDS sample buffer at 70°C for 10 min. 500 U of Endoglycosidase H (EndoH) was added and samples incubated for 5 h at 37°C.
Subcellular fractionation
Cells were washed with PBS, harvested and cell pellets resuspended in hypotonic buffer [10 mM HEPES-NaOH pH 7.5, 1.5 mM Mg(OAc)2, 10 mM KOAc, 0.5 mM DTT] supplemented with 1 mM AEBSF and complete protease inhibitor cocktail. After 10 min incubation on ice, cells were lysed by passing them ten times through a 27G needle. Cell debris and nuclei were pelleted by centrifugation (1000 g, 10 min, 4°C) yielding a post-nuclear supernatant. Cytosolic and membrane fractions were separated by ultracentrifugation at 100,000 g, 15 min, 4°C.
Proliferation assay
U2OS FlpIn cell lines were seeded in full DMEM into a 96-well plate at 5000 cells/well (in total volume of 100 µl/well) and the next day protein expression was induced with 200 ng/ml tetracycline. Non-induced cells were analysed in parallel and each condition was replicated in three separate wells. After 48 h incubation, 20 µl of the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) reagent (Promega) was added and plates incubated for 90 min in a humidified incubator at 37°C. Absorbance was measured at 490 nm and the values obtained for medium only were used as controls and subtracted from the results obtained for the samples analysed. Results were processed with Microsoft Excel software and graphs prepared with Prism 7 software. Three biological replicates were carried out. The same assay was used to estimate the viability of HEK293 FlpIn parental and knockout clones in response to treatment with TRAIL but cells were grown in DMEM without Phenol Red.
Immunoprecipitation
HeLa cells were treated with 1 µM staurosporine for the times shown in the presence of 20 µM zVAD-fmk where indicated. Cell pellets were resuspended in buffer A [20 mM HEPES-NaOH pH 7.5, 110 mM KOAc, 2 mM Mg(OAc)2, 1 mM EGTA and 0.1% (w/v) NP-40] supplemented with 1 mM Na3VO4, 1 mM NaF, 25 mM iodoacetamide, 1 mM AEBSF and complete protease inhibitor cocktail. Samples were incubated for 30 min on ice followed by two rounds of snap freezing in liquid N2 and thawing at 30°C. Insoluble material was pelleted by centrifugation (16,000 g, 30 min, 4°C) and NaCl concentration adjusted to 150 mM. After pre-clearing with protein G, the agarose soluble fraction was subjected to immunoprecipitation with anti-USP35 antibodies or GFP trap resin (ChromoTek) for 2 h at 4°C. Beads were washed three times with buffer A supplemented with 150 mM NaCl, resuspended in SDS sample buffer and proteins denatured for 10 min at 70°C. For USP35 immunoprecipitation from HPAF-II cells, samples were processed as described above but cells were lysed in RIPA buffer [50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (w/v) NP-40, 0.5% (w/v) sodium deoxycholate and 0.1% (w/v) SDS] supplemented with 1 mM AEBSF, complete protease inhibitor cocktail and 0.1% benzonase.
BioID
The BioID experiment was carried out essentially as described by Coyaud et al. (2015) with slight modifications. Briefly, BirAR118G (BirA*) was fused to the C-terminus of human USP35iso1 and USP35iso2 in pcDNA5 FRT/TO, and stable HEK293 FlpIn cell lines were generated. Experiments were performed with four 150 cm2 dishes, and expression was induced at ∼60% confluency with 1 µg/ml tetracycline (Sigma) for 24 h. The medium was then replaced with a fresh medium supplemented with 50 µM biotin (Sigma), and cells were further incubated for 10 h. Cells were washed twice with PBS and scraped, and cell pellets were snap frozen in liquid nitrogen and stored at −80°C.
Cell pellets were lysed in 1 ml of modified RIPA lysis buffer [1% (v/v) NP-40, 50 mM Tris–HCl, pH 7.5, 0.5% (w/v) sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% (w/v) SDS and 1:500 protease inhibitor cocktail (Sigma), with 250 U benzonase nuclease (Sigma)] in an agitator for 1 h at 4°C. The lysate was centrifuged for 30 min at 20,817 g at 4°C, and clarified supernatant incubated with 30 µl prewashed Streptavidin–Sepharose beads (GE Healthcare, cat. no. 17-5113-01) for 3 h at 4°C. Beads were collected by centrifugation (400 g, 2 min), washed twice with RIPA buffer (1 ml per wash), twice with TAP lysis buffer [50 mM HEPES-KOH, pH 8.0, 2 mM EDTA, 10% (v/v) glycerol, 100 mM KCl and 0.1% (v/v) NP-40], and four times with 50 mM ammonium bicarbonate (ABC; pH 8.0). Beads were then resuspended in 200 µl ammonium bicarbonate buffer and treated with 1 µg of sequencing grade modified trypsin (Promega, V5113) overnight at 37°C with agitation. Following overnight incubation, an additional 1 μg of trypsin was added, samples were incubated for 2 h at 37°C with agitation and then reduced with 5 μl of 10 mM DTT and alkylated with 5 μl of 10 mM iodoacetamide (IAA). The supernatant containing the tryptic peptides was collected, de-salted using C18 micro-spin columns (The Nest Group, Inc.) and vacuum centrifuged. Peptides were re-suspended in 20 μl of 0.1% formic acid and 5 µl were used per mass spectrometry (MS) run.
In-gel digestion
Protein bands were excised from Coomassie Brilliant Blue-stained SDS-PAGE gel, washed twice with water and twice with 50% acetonitrile. Proteins were reduced and alkylated with 10 mM DTT and 55 mM iodoacetamide, respectively, in 100 mM ammonium bicarbonate and digested with 200 ng sequencing grade modified trypsin (Promega, V5113) for 14–16 h. Peptides were extracted twice with 50% acetonitrile and twice with 50% acetonitrile containing 2% formic acid, and combined supernatants were dried using a Speedvac (Thermo Savant).
Mass spectrometry
The resulting peptides were analysed by nano-liquid chromatography (nLC, Dionex ultimate 3000) coupled to mass spectrometry (MS, LTQ-Orbitrap Velos; Thermo Finnigan). Peptides were loaded on a pre-column (Acclaim pepmap, C18, 100 Å, 100 µm; Thermo) at a 5 μl/min flow rate and resolved on an analytical column (Acclaim PepMap C18, 2 µm, 100 Å, 75 µm×50 cm), with a 185 min gradient program with buffer A [2% acetonitrile, 3% DMSO, 0.1% (v/v) formic acid] and 1 to 45% buffer B [90% acetonitrile, 3% DMSO, 0.08% (v/v) formic acid]. Data was acquired in data-dependent mode, and a parent ion scan was performed in the Orbitrap, using a resolving power of 60,000 at m/z 400. Simultaneously, collision-induced dissociation (CID) was performed in an ion trap activation time of 10 ms, with the top 15 precursor ions. Protein identification was carried out by using the Sequest search algorithm against the Uniprot database by selecting semi-tryptic enzymatic digestion option.
Protein identification
The MS raw files were searched using MaxQuant (version 1.5.8.3) (Cox and Mann, 2008), with the integrated Andromeda search engine. For the identification of peptides and proteins, the UniProt human FASTA database (March 2015) was used with a false discovery rate (FDR)<0.01. Oxidised methionine (M) and acetylation (protein N-terminal) were set as variable modifications and carbamidomethyl (C) as a fixed modification. MaxQuant LFQ (label free quantification) was enabled with the stand ‘match between runs’. The bona fide USP35-interacting proteins were identified with spectral counting as input for SAINTexpress (Significance Analysis of INTeractome) (Teo et al., 2014) with a Bayesian FDR value of 2%. The resulting high confidence interacting proteins (HCIPs) were then subjected to DAVID enrichment analysis for cell compartment terms (Huang et al., 2009).
Bioinformatics analysis
To analyse isoform expression in primary tumours, the TCGA transcript expression levels have been downloaded from UCSC Xena (http://xena.ucsc.edu/) version 2016-04-12. RNA-Seq reads were processed with the RSEM tool based on Gencode v23 transcript annotation. TPM expression values have been extracted from the RSEM result files and transformed to log2 [transcripts per million reads (tpm)+0.001] values.
Expression of USP35 isoforms in selected cell lines was addressed by submitting the cell lines to Illumina Truseq RNA-Seq sequencing (poly-A selection) on an Illumina HiSeq instrument. Reads were processed with cufflinks version 2.0.2-foss-2015a with parameters -u --max-bundle-frags 10000000 --max-bundle-length 10000000 --no-effective-length-correction --compatible-hits-norm --max-frag-multihits 1. Ensembl v70 (hg19) gene annotation was used to determine the transcript expression levels. Fragments per kilobase of transcript per million mapped reads (FPKM) expression values were extracted from the cufflinks result files for all USP35 isoforms.
Accession code
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 5TXK.
Supplementary Material
Acknowledgements
We are extremely grateful for the excellent technical assistance that we received during the preparation of this manuscript. In particular, we would like to thank: Axel Knebel, James Hastie, Hilary McLauchlan and Samantha Raggett for the purification of USP35 proteins used for DUB assays and in vitro caspase cleavage reactions, Thomas Macartney for designing and preparing DNA constructs used to generate USP35 knock-out clones, Richard Ewan for help in preparing reagents during the initial stages of the project and Fiona Brown for purifying anti-USP35 antibodies. We are grateful to Stephen High (University of Manchester) for the gift of anti-rhodopsin antibody and help during the revision stage of this manuscript. We would like to thank all members of the Kulathu group for their help and comments received during the preparation of this manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: P.L., J.N., G. Bader, W.S., A.S., A.Z., G. Boehmelt, Y.K.; Methodology: P.L., J.N., G. Bader; Validation: P.L.; Formal analysis: P.L., J.N., G. Bader, W.S., A.S., S.A.A.R., M.C.B., N.L.B.-M.; Investigation: P.L., J.N., G. Bader, W.S., A.S., D.P., Y.K.; Resources: S.W.; Writing - original draft: P.L., W.S., G. Boehmelt, Y.K.; Writing - review & editing: P.L., Y.K.; Visualization: P.L., S.A.A.R.; Supervision: P.L., W.S., G. Boehmelt, Y.K.; Project administration: P.L., W.S., G. Boehmelt, Y.K.; Funding acquisition: G. Boehmelt, Y.K., P.L.
Funding
This work was supported by the Medical Research Council UK (MC_UU_12016/6 to Y.K.), European Research Council (677623 to Y.K.), the EMBO Young Investigator Programme (Y.K.), European Molecular Biology Organization (EMBO) Installation Grant (3057 to N.L.B.-M.), FCT Investigator Starting Grant from Fundação para a Ciência e a Tecnologia (Portugal) (IF/00595/2014 to N.L.B.-M.), Tenovus Scotland (T16/24 to P.L.) and the pharmaceutical companies supporting the Division of Signal Transduction Therapy. Y.K. is a Lister Institute Prize Fellow. Deposited in PMC for release after 6 months.
Data availability
The raw mass spectrometry data have been deposited in ProteomeXchange Consortium via the PRIDE repository (Vizcaíno et al., 2014) with ID: PXD009286. Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 5TXK.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.212753.supplemental
References
- Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W. et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213-221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamus G., Zam Z. S., Arendt A., Palczewski K., McDowell J. H. and Hargrave P. A. (1991). Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vision Res. 31, 17-31. 10.1016/0042-6989(91)90069-H [DOI] [PubMed] [Google Scholar]
- Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., Grant S. W., Li W. H. H., Heerding D. A., Minthorn E., Mencken T. et al. (2012). Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193-7207. 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- Bago R., Malik N., Munson M. J., Prescott A. R., Davies P., Sommer E., Shpiro N., Ward R., Cross D., Ganley I. G. et al. (2014). Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 463, 413-427. 10.1042/BJ20140889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke T., Pohl C., Pyrowolakis G. and Jentsch S. (2004). Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol. Cell 14, 801-811. 10.1016/j.molcel.2004.05.018 [DOI] [PubMed] [Google Scholar]
- Bersuker K., Peterson C. W. H., To M., Sahl S. J., Savikhin V., Grossman E. A., Nomura D. K. and Olzmann J. A. (2018). A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 44, 97-112.e7. 10.1016/j.devcel.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S. N., Habermacher R., Martine U., Closs E. I. and Filipowicz W. (2006). Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125, 1111-1124. 10.1016/j.cell.2006.04.031 [DOI] [PubMed] [Google Scholar]
- Borgese N., Gazzoni I., Barberi M., Colombo S. and Pedrazzini E. (2001). Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell 12, 2482-2496. 10.1091/mbc.12.8.2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C. et al. (2016). BUSTER Version 2.11.7. Cambridge, UK: Global Phasing Ltd. [Google Scholar]
- Bulbarelli A., Sprocati T., Barberi M., Pedrazzini E. and Borgese N. (2002). Trafficking of tail-anchored proteins: transport from the endoplasmic reticulum to the plasma membrane and sorting between surface domains in polarised epithelial cells. J. Cell Sci. 115, 1689-1702. [DOI] [PubMed] [Google Scholar]
- Buljan M., Chalancon G., Eustermann S., Wagner G. P., Fuxreiter M., Bateman A. and Babu M. M. (2012). Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871-883. 10.1016/j.molcel.2012.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin S. F., Teschendorff A. E., Marioni J. C., Wang Y., Barbosa-Morais N. L., Thorne N. P., Costa J. L., Pinder S. E., van de Wiel M. A., Green A. R. et al. (2007). High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 8, R215 10.1186/gb-2007-8-10-r215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague M. J., Heride C. and Urbé S. (2015). The demographics of the ubiquitin system. Trends Cell Biol. 25, 417-426. 10.1016/j.tcb.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Conti B. J., Elferich J., Yang Z., Shinde U. and Skach W. R. (2014). Cotranslational folding inhibits translocation from within the ribosome-Sec61 translocon complex. Nat. Struct. Mol. Biol. 21, 228-235. 10.1038/nsmb.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367-1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- Coyaud E., Mis M., Laurent E. M. N., Dunham W. H., Couzens A. L., Robitaille M., Gingras A.-C., Angers S. and Raught B. (2015). BioID-based identification of Skp Cullin F-box (SCF)β-TrCP1/2 E3 ligase substrates. Mol. Cell. Proteomics 14, 1781-1795. 10.1074/mcp.M114.045658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzer A. J., Nabholz C. E. and Spiess M. (1995). Transmembrane orientation of signal-anchor proteins is affected by the folding state but not the size of the N-terminal domain. EMBO J. 14, 6311-6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman S. W., Yassaee M. and Bieniasz P. D. (2009). A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J. Cell Biol. 184, 881-894. 10.1083/jcb.200808041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. D., Barrios-Rodiles M., Çolak R., Irimia M., Kim T. H., Calarco J. A., Wang X., Pan Q., O'Hanlon D., Kim P. M. et al. (2012). Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 46, 884-892. 10.1016/j.molcel.2012.05.037 [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. and Cowtan K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486-501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K., Rudelius M., Slawska J., Jacobs L., Ahangarian Abhari B., Altmann B., Kurutz J., Rathakrishnan A., Fernández-Sáiz V., Brunner A. et al. (2016). USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 8, 851-862. 10.15252/emmm.201506047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen A. C., Luna-Vargas M. P. A., Geurink P. P., Clerici M., Merkx R., van Dijk W. J., Hameed D. S., El Oualid F., Ovaa H. and Sixma T. K. (2011). The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem. Biol. 18, 1550-1561. 10.1016/j.chembiol.2011.10.017 [DOI] [PubMed] [Google Scholar]
- Floor S. N. and Doudna J. A. (2016). Tunable protein synthesis by transcript isoforms in human cells. Elife 5, e10921 10.7554/eLife.10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glab J. A., Doerflinger M., Nedeva C., Jose I., Mbogo G. W., Paton J. C., Paton A. W., Kueh A. J., Herold M. J., Huang D. C. S. et al. (2017). DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 24, 944-950. 10.1038/cdd.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Lone M. A., Schneiter R. and Chang A. (2010). Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc. Natl. Acad. Sci. USA 107, 5851-5856. 10.1073/pnas.0911617107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Sekine K., Kawabata A., Nakamura H., Ishioka T., Ohata H., Katayama R., Hashimoto C., Zhang X., Noda T. et al. (2004). Apollon ubiquitinates SMAC and caspase-9 and has an essential cytoprotection function. Nat. Cell Biol. 6, 849-860. 10.1038/ncb1159 [DOI] [PubMed] [Google Scholar]
- Hassink G. C., Zhao B., Sompallae R., Altun M., Gastaldello S., Zinin N. V., Masucci M. G. and Lindsten K. (2009). The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 10, 755-761. 10.1038/embor.2009.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heideker J. and Wertz I. E. (2015). DUBs, the regulation of cell identity and disease. Biochem. J. 465, 1-26. 10.1042/BJ20140496 [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425-479. 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I. M., White S. H. and von Heijne G. (2007). Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026-1030. 10.1038/nature06387 [DOI] [PubMed] [Google Scholar]
- Hörnle M., Peters N., Thayaparasingham B., Vörsmann H., Kashkar H. and Kulms D. (2011). Caspase-3 cleaves XIAP in a positive feedback loop to sensitize melanoma cells to TRAIL-induced apoptosis. Oncogene 30, 575-587. 10.1038/onc.2010.434 [DOI] [PubMed] [Google Scholar]
- Hotamisligil G. S. (2010). Endoplasmic reticulum stress and atherosclerosis. Nat Med. 16, 396-399. 10.1038/nm0410-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Jeong M., Lee E.-W., Seong D., Seo J., Kim J.-H., Grootjans S., Kim S.-Y., Vandenabeele P. and Song J. (2017). USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene 36, 458-470. 10.1038/onc.2016.215 [DOI] [PubMed] [Google Scholar]
- Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J. et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325-340. 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm E. J., Spooner E. and Ploegh H. L. (2011). Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 286, 37602-37614. 10.1074/jbc.M111.284794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.-W., Seong D., Seo J., Jeong M., Lee H.-K. and Song J. (2015). USP11-dependent selective cIAP2 deubiquitylation and stabilization determine sensitivity to Smac mimetics. Cell Death Differ. 22, 1463-1476. 10.1038/cdd.2014.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-G., Takahama S., Zhang G., Tomarev S. I. and Ye Y. (2016). Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 18, 765-776. 10.1038/ncb3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznicki P. and Kulathu Y. (2017). Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 130, 1997-2006. 10.1242/jcs.201855 [DOI] [PubMed] [Google Scholar]
- Leznicki P., Clancy A., Schwappach B. and High S. (2010). Bat3 promotes the membrane integration of tail-anchored proteins. J. Cell Sci. 123, 2170-2178. 10.1242/jcs.066738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Wang L., Chen W., Zhao S., Yin C., Lin Y., Jiang A. and Zhang P. (2015). USP35 activated by miR let-7a inhibits cell proliferation and NF-kappaB activation through stabilization of ABIN-2. Oncotarget 6, 27891-27906. 10.18632/oncotarget.4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Lawrence D. A., Marsters S., Acosta-Alvear D., Kimmig P., Mendez A. S., Paton A. W., Paton J. C., Walter P. and Ashkenazi A. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98-101. 10.1126/science.1254312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Määttä J., Hallikas O., Welti S., Hildén P., Schröder J. and Kuismanen E. (2000). Limited caspase cleavage of human BAP31. FEBS Lett. 484, 202-206. 10.1016/S0014-5793(00)02159-1 [DOI] [PubMed] [Google Scholar]
- Mahul-Mellier A.-L., Pazarentzos E., Datler C., Iwasawa R., AbuAli G., Lin B. and Grimm S. (2012). De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 19, 891-899. 10.1038/cdd.2011.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y., Hahn A. A., Hu S. and Yang X. (2011). The USP19 deubiquitinase regulates the stability of c-IAP1 and c-IAP2. J. Biol. Chem. 286, 35380-35387. 10.1074/jbc.M111.282020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F. W. H., Nguyen M., Kwan T., Branton P. E., Nicholson D. W., Cromlish J. A. and Shore G. C. (1997). p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 139, 327-338. 10.1083/jcb.139.2.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Kwon M.-S., Kim E. E. K., Lee H. and Song E. J. (2018). USP35 regulates mitotic progression by modulating the stability of Aurora B. Nat. Commun. 9, 688 10.1038/s41467-018-03107-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau L., Colas J., Dupont S., Beney L., Fleurat-Lessard P., Berjeaud J.-M., Bergès T. and Ferreira T. (2009). Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic 10, 673-690. 10.1111/j.1600-0854.2009.00903.x [DOI] [PubMed] [Google Scholar]
- Pohl C. and Jentsch S. (2008). Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell 132, 832-845. 10.1016/j.cell.2008.01.012 [DOI] [PubMed] [Google Scholar]
- Popovic D., Vucic D. and Dikic I. (2014). Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242-1253. 10.1038/nm.3739 [DOI] [PubMed] [Google Scholar]
- Promlek T., Ishiwata-Kimata Y., Shido M., Sakuramoto M., Kohno K. and Kimata Y. (2011). Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 22, 3520-3532. 10.1091/mbc.E11-04-0295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Rivera D., Delvaeye T., Roelandt R., Nerinckx W., Augustyns K., Vandenabeele P. and Bertrand M. J. M. (2017). When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 24, 1100-1110. 10.1038/cdd.2017.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K. J., Kim D. I., Raida M. and Burke B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801-810. 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J. et al. (2010). Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103-107. 10.1038/nature08646 [DOI] [PubMed] [Google Scholar]
- Shao S. and Hegde R. S. (2011). Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol. 27, 25-56. 10.1146/annurev-cellbio-092910-154125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandl J., Lohmann D., Kuerschner L., Moessinger C. and Thiele C. (2011). Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J. Biol. Chem. 286, 5599-5606. 10.1074/jbc.M110.190785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. and Ron D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184-190. 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G., Liu G., Zhang J., Nesvizhskii A. I., Gingras A.-C. and Choi H. (2014). SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37-43. 10.1016/j.jprot.2013.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault G., Shui G., Kim W., McAlister G. C., Ismail N., Gygi S. P., Wenk M. R. and Ng D. T. W. (2012). The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 48, 16-27. 10.1016/j.molcel.2012.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi N. D., Svinkina T., Mertins P., Kuhn E., Mani D. R., Qiao J. W. and Carr S. A. (2013). Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics 12, 825-831. 10.1074/mcp.O112.027094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N. et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223-226. 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T. and Bricogne G. (2011). Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293-302. 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Serricchio M., Jauregui M., Shanbhag R., Stoltz T., Di Paolo C. T., Kim P. K. and McQuibban G. A. (2015). Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11, 595-606. 10.1080/15548627.2015.1034408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A., Heinlein M., Dengjel J., Alber C., Singh P. K. and Häcker G. (2016). The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep. 17, 724-738. 10.15252/embr.201541392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte M. A. (2015). Expanding roles for lipid droplets. Curr. Biol. 25, R470-R481. 10.1016/j.cub.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M., Schlegl J., Hahne H., Gholami A. M., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H. et al. (2014). Mass-spectrometry-based draft of the human proteome. Nature 509, 582-587. 10.1038/nature13319 [DOI] [PubMed] [Google Scholar]
- Yang C.-S., Sinenko S. A., Thomenius M. J., Robeson A. C., Freel C. D., Horn S. R. and Kornbluth S. (2014). The deubiquitinating enzyme DUBAI stabilizes DIAP1 to suppress Drosophila apoptosis. Cell Death Differ. 21, 604-611. 10.1038/cdd.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Coulombe-Huntington J., Kang S., Sheynkman G. M., Hao T., Richardson A., Sun S., Yang F., Shen Y. A., Murray R. R. et al. (2016). Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164, 805-817. 10.1016/j.cell.2016.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R. and Rape M. (2016). The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579-586. 10.1038/ncb3358 [DOI] [PubMed] [Google Scholar]
- Ye Y., Scheel H., Hofmann K. and Komander D. (2009). Dissection of USP catalytic domains reveals five common insertion points. Mol. Biosyst. 5, 1797-1808. 10.1039/b907669g [DOI] [PubMed] [Google Scholar]
- Yin J., Schoeffler A. J., Wickliffe K., Newton K., Starovasnik M. A., Dueber E. C. and Harris S. F. (2015). Structural insights into WD-repeat 48 activation of ubiquitin-specific protease 46. Structure 23, 2043-2054. 10.1016/j.str.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Zehmer J. K., Bartz R., Liu P. and Anderson R. G. W. (2008). Identification of a novel N-terminal hydrophobic sequence that targets proteins to lipid droplets. J. Cell Sci. 121, 1852-1860. 10.1242/jcs.012013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.