

ABSTRACT
Duchenne muscular dystrophy (DMD) is an X-linked muscle-wasting disorder caused by mutations in the dystrophin gene, with an incidence of 1 in 3500 in new male births. Mdx mice are widely used as an animal model for DMD. However, these mice do not faithfully recapitulate DMD patients in many aspects, rendering the preclinical findings in this model questionable. Although larger animal models of DMD, such as dogs and pigs, have been generated, usage of these animals is expensive and only limited to several facilities in the world. Here, we report the generation of a rabbit model of DMD by co-injection of Cas9 mRNA and sgRNA targeting exon 51 into rabbit zygotes. The DMD knockout (KO) rabbits exhibit the typical phenotypes of DMD, including severely impaired physical activity, elevated serum creatine kinase levels, and progressive muscle necrosis and fibrosis. Moreover, clear pathology was also observed in the diaphragm and heart at 5 months of age, similar to DMD patients. Echocardiography recording showed that the DMD KO rabbits had chamber dilation with decreased ejection fraction and fraction shortening. In conclusion, this novel rabbit DMD model generated with the CRISPR/Cas9 system mimics the histopathological and functional defects in DMD patients, and could be valuable for preclinical studies.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: CRISPR, Cardiomyopathy, Dystrophin, Gene editing, Muscular dystrophy, Rabbit
Summary: The DMD KO rabbit engineered by CRISPR genome editing faithfully recapitulates the DMD pathologies, and could be a valuable tool for basic and translational studies to combat this disease.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a fatal X-linked recessive disease characterized by progressive muscle weakening and wasting. DMD patients usually lose the ability to walk between 10 and 12 years of age and die of respiratory and/or cardiac failure by the age of 20-30 years (Spurney, 2011). DMD affects ∼1 in 3500 male births according to newborn screening (Mendell and Lloyd-Puryear, 2013; Mendell et al., 2012).
DMD is caused by mutations in the DMD gene, which encodes dystrophin protein (Bonilla et al., 1988; Hoffman et al., 1987). The DMD gene consists of 79 exons on the X chromosome (Hoffman et al., 1987), and mutations occur in any of these exons with the common hotspots in exons 3-7 and exons 45-55. The mutations include various forms: large deletion (68%), large duplication (11%), point mutations (11%) and small insertion/deletion (7%) (Bladen et al., 2015). These mutations cause frameshift and/or premature stop codon formation in the DMD gene, thus disrupting the expression of dystrophin and leading to the development of DMD (Beggs et al., 1991). Currently, there is no effective therapeutic treatment available for DMD.
At present, several animal models of DMD presenting the dystrophic phenotype are available, including mouse (Bulfield et al., 1984; Chapman et al., 1989; Sicinski et al., 1989), dog (Baltzer et al., 2007; Cooper et al., 1988; Jones et al., 2004; Sharp et al., 1992; Smith et al., 2011) and pig (Klymiuk et al., 2013). Many of the pathogenesis and preclinical studies were initially carried out in Mdx (Dmd) mice, a widely used animal model of DMD. However, the Mdx mice do not recapitulate human DMD patients in many aspects. For example, the phenotype of Mdx mice is much milder compared with that of DMD patients, with almost a normal life span (Chamberlain et al., 2007), although they exhibit muscular dystrophy (Bulfield et al., 1984; Chapman et al., 1989; Sicinski et al., 1989). This could be part of the reason for the poor translation of the findings achieved with these animals. Golden retriever muscular dystrophy (GRMD) dogs (Baltzer et al., 2007; Cooper et al., 1988; Jones et al., 2004; Sharp et al., 1992; Smith et al., 2011), on the other hand, show severe muscular dystrophy pathology, lethal respiratory distress and cardiomyopathy, resembling DMD patients. Dystrophin-deficient pigs were generated by deleting exon 52 in pig fibroblasts followed by nuclear transfer (Klymiuk et al., 2013). These pigs exhibit many features of muscular dystrophy including elevated serum creatine kinase, impaired mobility and progressive dystrophic changes in skeletal muscle (Klymiuk et al., 2013). Interestingly, pathological alterations were not observed in the heart of these pigs; however, some DMD pigs also die shortly after birth (Klymiuk et al., 2013; Yu et al., 2016). Although GRMD dogs and DMD pigs resemble human DMD patients better than Mdx mice do, the cost associated with the usage of these large animals poses a big hurdle for many laboratories.
Therefore, there is a need to develop DMD models in other species, which must recapitulate human DMD patients with reasonable costs for maintenance in ordinary laboratories. Of note, the rabbit shares more similarities with humans in terms of physiology, anatomy and genetics than does the mouse (Wang et al., 2014), and has been extensively used as an appropriate model for cardiovascular and metabolic disease research (Bősze et al., 2003). Lower maintenance cost and shorter gestational duration makes the rabbit a superior model compared with dogs or pigs.
In this study, we established a novel DMD rabbit model by cytoplasm microinjection of Cas9 mRNA and single guide RNA (sgRNA). These clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 knockout (KO) rabbits showed many features of muscular dystrophy including elevated serum creatine kinase (CK), muscle necrosis and regeneration, impaired mobility, and increased fibrosis in the heart and bladder. This novel DMD rabbit model could be a valuable resource for DMD research and preclinical studies.
RESULTS
Generation of DMD KO rabbits using CRISPR/Cas9
In order to disrupt the open reading frame (ORF) of DMD in rabbits, we designed a pair of sgRNAs targeting exon 51, which is commonly mutated in human DMD patients (Fig. 1A,B). To test the efficiency of CRISPR/Cas9-mediated gene targeting of DMD in zygotes, the mixed Cas9 mRNA and sgRNAs were microinjected into the zygotes, and cultured until the blastocyst stage. As shown in Table 1, 79.6% of injected embryos (n=123) developed into the blastocyst stage, among which 78.0% carried mutations in DMD at the target sites. There were no significant differences in the developmental rate between the noninjected embryos and microinjected embryos (P>0.05). These results demonstrated that the dual sgRNA-directed CRISPR/Cas9 system is efficient for generation of mutations in the DMD gene in rabbit zygotes.
Fig. 1.

Generation of DMD KO rabbits using CRISPR/Cas9. (A) The common mutations in exon 51 in human DMD patients. (B) Schematic diagram of two sgRNA target sites located in exon 51 of the rabbit DMD locus. DMD exons are indicated by purple boxes; target sites of the two sgRNA sequences (sgRNA1 and sgRNA2) are highlighted in red; protospacer-adjacent motif (PAM) sequences are highlighted in green. (C,D) Mutation detection by the T7E1 cleavage assay in pups 1-16 (C) and 17-33 (D). M, the DNA ladder (DL2000).
Table 1.
Summary of embryo microinjections of Cas9 mRNA/sgRNA in zygotes

In order to generate DMD KO rabbits, a total of 128 injected zygotes were transferred into the oviducts of four surrogate rabbits. All surrogates were pregnant to term and produced 33 live pups (Table 2). The genomic DNA from each pup was isolated, and mutations were determined by T7 endonuclease I (T7E1) assay and Sanger sequencing of the PCR products near the target sites. As shown in Fig. 1C and Fig. S1, 26 of the 33 (78.8%) newborn pups carried a DMD mutation, and 22 of them (84.6%) carried the biallelic DMD mutations. In order to examine the off-target effects in these DMD KO rabbits, the PCR products of the top ten potential off-target sites were subjected to Sanger sequencing and T7E1 cleavage assay. No off-target mutations were detected at these potential sites in the DMD KO rabbits (Fig. S2).
Table 2.
Generation of the DMD KO rabbits using the CRISPR/Cas9 system

Disruption of dystrophin expression in the DMD KO rabbits
To examine whether the mutations in the rabbits induced by CRISPR/Cas9 disrupt the expression of the DMD gene in the KO rabbits, we performed quantitative reverse transcription PCR (RT-PCR) analysis of DMD expression in the gastrocnemius muscle of KO and wild-type (WT) rabbits using two sets of primers. As shown in Fig. S3, the expression of DMD was significantly reduced in the skeletal muscle of DMD KO rabbits compared with WT controls. To further examine the expression of dystrophin protein in the muscle of these rabbits, we performed immunofluorescence staining. Dystrophin was detected at the sarcolemma of skeletal muscle fibers of WT rabbits, and its expression was completely disrupted in the KO rabbits (Fig. 2A). It has been shown previously that dystrophin forms a large dystrophin-glycoprotein complex (DGC) on the muscle membrane, and dystrophin deficiency would compromise the integrity of the entire DGC. This is also true for DMD KO rabbit skeletal muscle in which the glycosylated form of α-dystroglycan (Fig. 2B) and α-sarcoglycan (Fig. 2C) were greatly reduced. Therefore, our data suggest that engineered mutations in exon 51 lead to disruption of dystrophin and its associated complex.
Fig. 2.
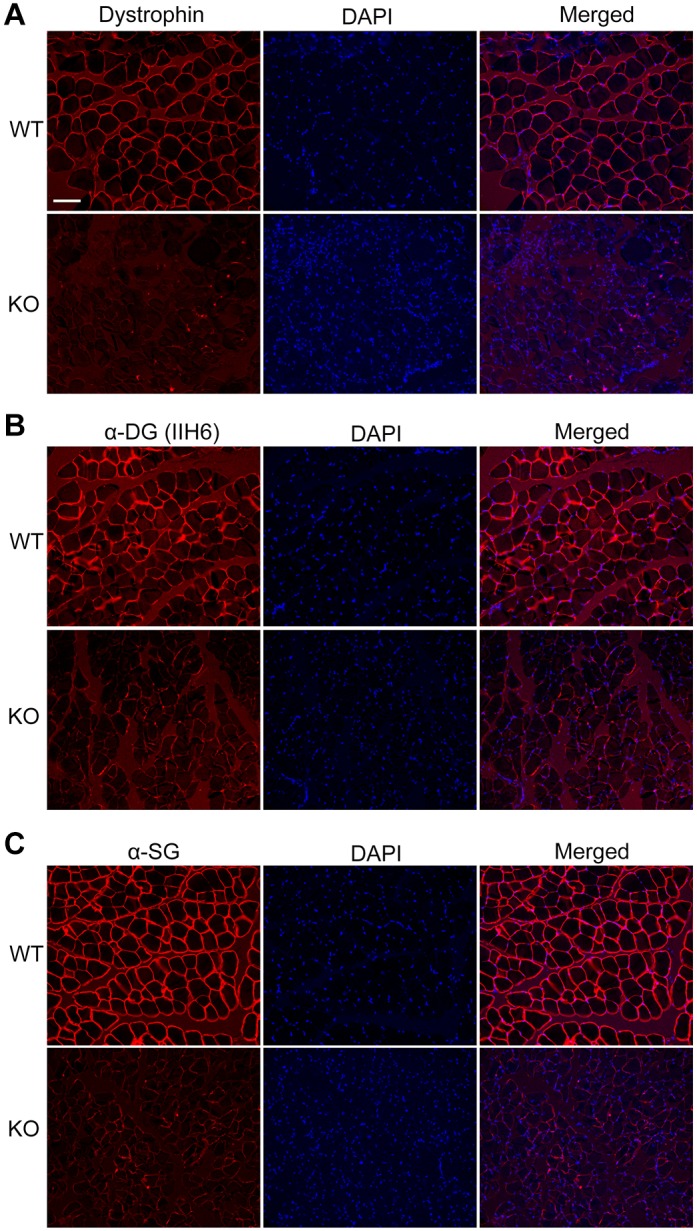
Disruption of dystrophin expression in the skeletal muscle of DMD KO rabbits. (A-C) Immunofluorescence staining of muscle sections from WT and DMD KO rabbits with mouse monoclonal antibodies against dystrophin (A), glycosylated α-dystroglycan (B) and α-sarcoglycan (C). Nuclei were stained by DAPI. Scale bar: 100 µm.
Muscular dystrophy presentation in DMD KO rabbits
The DMD KO rabbits were smaller in size compared with their WT littermates, and exhibited obvious forelimb paralysis (Fig. 3A). The DMD KO rabbits and control littermates were weighed weekly, and the data showed that both the female and male KO rabbits were significantly lighter than their control littermates (Fig. 3B,C). Approximately 20% of the DMD KO rabbits died within the first 2 weeks after birth, and close to half (42.6%) died by 20 weeks of age (Fig. 3D).
Fig. 3.

Impaired physical activity in DMD KO rabbits. (A) Photograph of a 12-week-old DMD KO rabbit and its WT littermate showing significant forelimb paralysis in the KO rabbit. (B,C) Body mass comparison of DMD KO and WT female (B) and male (C) rabbits from birth to 24 weeks of age (n=6). (D) Kaplan–Meier survival curves for the DMD KO and WT rabbits (n=22). (E) The walking steps during a 1-h recording period for the DMD KO and WT rabbits (n=6). (F) N-acetyl-L-cysteine measurements of serum CK levels (n=6). (G) Photographs of a DMD KO rabbit and WT control rabbit performing a stair-climb challenge. *P<0.05; **P<0.01; ***P<0.001.
DMD patients show progressive decline in ambulation, which can be evaluated by a 6-min walk test (6MWT) (McDonald et al., 2010). The 6MWT is also the most accepted primary clinical endpoint in ambulatory DMD trials and has also been used to assess dogs with heart disease (Boddy et al., 2004). To examine the physical activity of the rabbits and to design a simple 6MWT-like assay for preclinical trials using these rabbits, we mounted a human-used activity-monitoring device (Millet Sports Bracelet Wearable Device) on the right hind leg of rabbits and counted the walking steps within 1 h. The KO rabbits showed significantly decreased mobility compared with the healthy ones (Fig. 3E). This 1-h wearable device-assisted walking test (1HWDAWT) provides an extremely simple and inexpensive way to evaluate the physical activity of the freely moving rabbits.
Muscle injury is a hallmark of muscular dystrophy, which releases muscle proteins such as CK into the circulation. We observed that serum CK was significantly elevated in the KO rabbits from 8 to 20 weeks of age compared with their age-matched littermate controls (Fig. 3F). In addition, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were elevated in the DMD KO rabbits (data not shown), which could also be caused by muscle breakdown. When the rabbits were placed in front of a step, WT rabbits easily climbed up the step whereas the KO rabbits failed with obvious attempts (Fig. 3G; Movies 1 and 2).
To further examine the histopathology of the DMD KO rabbits, we performed Hematoxylin and Eosin (H&E) and Masson's trichrome staining of the gastrocnemius muscle sections from the rabbits at 16 and 20 weeks of age. As shown in Fig. 4, the DMD KO rabbits displayed typical muscular dystrophy signs, as evidenced by increased fiber size variation, centrally nucleated fibers, fibrosis and fatty replacement (Fig. 4A,B). The average fiber area was significantly decreased, owing to the cycles of degeneration and regeneration (Fig. 4C,E), and was accompanied by an increased percentage of muscle fibers with central nuclei (Fig. 4D,F). The fiber size distribution (Fig. 4G) showed that there was a significant increase in the smaller fibers, consistent with an increased regeneration in dystrophic muscle. The fibrotic area was significantly increased in the DMD KO rabbits compared with WT rabbits (Fig. 4H). These pathological changes in skeletal muscle resemble the typical features of human DMD. Similar pathological alterations were also observed in other muscles, including the tibialis anterior (Fig. S4) and quadriceps (Fig. S5) muscles. The diaphragm muscles, which are severely affected in DMD patients, mice and dogs (Moser, 1984; Valentine et al., 1988), were also found to have extensive muscle degeneration and fibrosis (Fig. S6).
Fig. 4.

Muscular dystrophy presentation in DMD-KO rabbits. (A,B) Analysis of H&E- and Masson's trichrome-stained sections of gastrocnemius from 5-month-old (A) and 12-month-old (B) WT and DMD KO rabbits. DMD KO rabbits displayed myopathy with excessive fiber size variation (red arrows), fiber fracture (green arrows), fibrosis (black arrows) and central nucleated fibers (blue rectangle). (C,E) Quantification of mean gastrocnemius muscle fiber area in WT and DMD KO rabbits at 5 (C) and 12 (E) months of age. (D,F) Quantification of centrally nucleated fiber (CNF) percentage in WT and DMD KO rabbits at 5 (D) and 12 (F) months of age. (G) Size distribution of WT and DMD KO gastrocnemius muscle at 5 months of age. (H) Quantification of relative fibrotic area in WT and DMD KO rabbits at 5 months of age. Scale bars: 50 µm. ***P<0.001; n=5.
Cardiomyopathy in the DMD KO rabbits
Approximately 95% of DMD patients develop cardiomyopathy by 20 years of age, and 20% of these patients die from cardiac complications (Muntoni, 2003; Shirokova and Niggli, 2013). The Mdx mice, a widely used animal model for DMD, do not show obvious cardiac pathology until a year later (Pastoret and Sebille, 1995), making it problematic for studying the DMD-associated cardiomyopathy. To examine whether disruption of DMD in rabbits causes any pathology in the heart, we performed histological and functional assessments of these rabbits. At 16 weeks of age, the hearts from the DMD KO rabbits were grossly similar in size to those from the WT controls (Fig. 5A). We measured cardiac function by echocardiography recording at 4 months of age. The left ventricular ejection fraction (EF) and fraction shortening (FS) of the DMD KO rabbits were significantly decreased compared with those of the control rabbits (Fig. 5B,C). Consistent with the detected cardiac dysfunction, the DMD KO rabbits also exhibited significant loss of cardiomyocytes and increased interstitial fibrosis, as shown by H&E and Masson's trichrome staining (Fig. 5D,E). These results suggest that the DMD KO rabbits developed cardiomyopathy.
Fig. 5.

Cardiomyopathy in the DMD KO rabbits. (A) The hearts from a DMD KO rabbit and WT control at 20 weeks of age. (B,C) The left ventricular ejection fraction (EF, B) and fractional shortening (FS, C) were decreased in DMD KO rabbits. (D,E) H&E-stained and Masson's trichrome-stained sections of cardiac muscle from WT and DMD KO rabbits at 5 (D) and 12 (E) months of age showed significant fibrosis (black arrows), mononuclear inflammatory cell infiltration (red arrow) and adipose tissue (blue arrow). Scale bars: 50 µm. **P<0.01; n=5.
DISCUSSION
In this study, we generated a novel rabbit model for DMD via the zygote injection of Cas9 mRNA and a pair of sgRNAs targeting exon 51 of the DMD gene, and demonstrated that the DMD KO rabbits exhibited almost all hallmarks of the disease observed in DMD patients, including muscular dystrophy, cardiomyopathy and high incidence of premature death. To the best of our knowledge, this is the first report of a DMD rabbit model that develops both muscular dystrophy and cardiomyopathy.
Approximately 74% of the live rabbit pups carried mutations at the target sites of the DMD gene and ∼85% of these targeted animals carried biallelic mutations. Many of these mutations are small deletions or insertions, disrupting the reading frame of the DMD gene. Sanger sequencing and T7E1 assay did not detect significant off-target activities at the top predicted off-target sites. These results are consistent with previous reports that cytoplasmic injection of sgRNA-directed CRISPR/Cas9 mRNA can be used as an efficient approach to generate targeted KO animals, including rabbits, with high fidelity (Honda et al., 2015; Yuan et al., 2016).
Previous studies have shown the DMD mutation hotspots are located in the regions of exons 3-7 and exons 45-55 (Badalian and Malygina, 1995; Koenig et al., 1989). We thus chose exon 51 as the target site. Small deletions/insertions (with the number of nucleotides deleted or inserted indivisible by three) in this exon would cause frameshift and thus disrupt the dystrophin expression. Indeed, we observed that the DMD transcript was greatly reduced in the skeletal muscle of DMD KO rabbits.
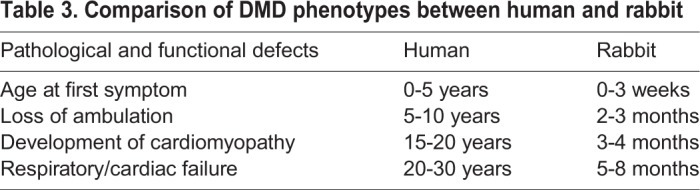
Using serum CK as an indicator of muscle injury, we found that it was significantly elevated in the DMD KO rabbits as early as 8 weeks of age. In comparison with WT littermates, DMD KO rabbits exhibited biochemical and pathological phenotypes characteristic of human DMD, including elevated serum CK levels, muscle degeneration and regeneration, interstitial fibrosis, fatty replacement and mononuclear inflammatory cell infiltration. Various skeletal muscle groups, including gastrocnemius, tibialis anterior, quadriceps and diaphragm, are affected in DMD KO rabbits. These observations are similar to those in other animal models of DMD, including mice (Sicinski et al., 1989; Spurney et al., 2009; Tanabe et al., 1986), pigs (Klymiuk et al., 2013; Yu et al., 2016) and dogs (Baltzer et al., 2007; Cooper et al., 1988; Jones et al., 2004; Sharp et al., 1992; Smith et al., 2011). We could not measure the contractility of skeletal muscle from the DMD KO rabbits owing to the unavailability of the force measurement setup. It was also not feasible to perform grip strength measurement on these animals owing to their large body size. However, functional defects of skeletal muscle in the DMD KO rabbits were obviously detected by the reduced physical activity and impaired ability to climb up the step, very similar to DMD pigs (Klymiuk et al., 2013) and boys with DMD in their early life (Zhu et al., 2013) (Table 3).
Table 3.
Comparison of DMD phenotypes between human and rabbit
Different from Mdx mice, the DMD KO rabbits showed clear cardiomyopathy at 4 months of age, as evidenced by reduced left ventricular ejection fraction (EF) and fractional shortening (FS) (Fig. 5B,C). Therefore, the DMD KO rabbit can be used in preclinical studies to evaluate the therapeutic effects of the tested medicine or treatment on muscular dystrophy and cardiomyopathy. The relatively lower maintenance cost and shorter gestational duration compared with dog and pig models make the DMD KO rabbit a particularly attractive model for preclinical studies. We showed that human-used activity-monitoring devices, such as the Millet Sports Bracelet Wearable Device used in this study, can be easily mounted on freely moving rabbits to monitor their physical activities. Moreover, echocardiography recording can be performed to assess their cardiac function.
To the best of our knowledge, this is the first report of a DMD rabbit model with close resemblance to its human counterpart. This new model could facilitate basic research to understand the pathogenesis of DMD, and translational studies to develop novel therapeutic strategies for this devastating disease.
MATERIALS AND METHODS
Animals and ethics statement
The New Zealand rabbits used in this study were maintained at the Laboratory Animal Center of Jilin University. All experiments involving rabbits in this study were performed in accordance with the guide of the Animal Care and Use Committee of Jilin University.
CRISPR/Cas9 sgRNA preparation, embryo microinjection and embryo transfer
The CRISPR/Cas9 sgRNA was designed and assembled as previously described (El Refaey et al., 2017; Xu et al., 2015b; Xu et al., 2016). The annealed sgRNA oligonucleotides were cloned into the BbsI sites of pUC57-T7-sgRNA cloning vector (Addgene ID 51306) as described (Shen et al., 2013). The vector of pUC57-T7-sgRNA was PCR amplified using T7 primers (Forward, 5′-GAAATTAATACGACTCACTATA-3′; Reverse, 5′-AAAAAAAGCACCGACTCGGTGCCAC-3′), and the PCR products were transcribed in vitro with a MAXIscript T7 Kit (Ambion; Applied Biosystems, CA, USA) and purified using a miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
The 3xFLAG-NLS-SpCas9-NLS vector (Addgene ID 48137) (containing Cas9 from Streptococcus pyogenes), was linearized with NotI and transcribed in vitro using a mMessage mMachine SP6 Kit (Ambion) and RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions.
The microinjection procedure and embryo transfer were performed essentially the same as previously described (Sui et al., 2016). Briefly, female New Zealand White rabbits at the age of 6-8 months were superovulated with 50 IU follicle stimulating hormone (FSH) at intervals of 12 h six times, mated with male rabbits, and injected with 100 IU human chorionic gonadotropin (HCG). The female rabbits were then euthanized and the oviducts were flushed with 5 ml Dulbecco's phosphate-buffered saline (DPBS)-bovine serum albumin (BSA) for zygote collection. Rabbit embryos were collected at the pronuclear stage. A mixture of in vitro-transcribed Cas9 mRNA (200 ng/µl) and sgRNA (50 ng/µl) was microinjected into the cytoplasm of zygotes. The injected embryos were transferred into Earle's balanced salt solution (EBSS) medium and cultured at 38.5°C in 5% CO2 for 20-30 min. Approximately 30-50 injected zygotes were then transferred into the oviducts of recipient rabbits.
Mutation detection in pups by PCR and sequencing
The genomic DNA from DMD KO and WT rabbits was extracted from a small piece of ear tissue using a TIANamp Genomic DNA Kit (Tiangen, Beijing, China) according to the manufacturer's instructions. The sgRNA target sites were amplified by PCR using primers (Forward, 5′-TAGTTTGGCTCAGATTGTAG-3′; Reverse, 5′-AGAATAGACAAAGCAGTGTG-3′). The PCR products were gel purified and cloned into pGM-T vector (Tiangen). A minimum of 14 positive clones were sequenced and analyzed using DNAman.
T7E1 cleavage assay
The T7E1 cleavage assay was performed as described previously (Xu et al., 2015b, 2013). Briefly, the PCR products as described above were purified, denatured and then re-annealed in NEBuffer 2 (NEB) using a thermocycler. Hybridized PCR products were digested with T7 endonuclease 1 (M0302L, NEB) for 30 min at 37°C and subjected to 2% agarose gel electrophoresis.
Off-target analysis
The top off-target sites were predicted using the online CRISPR Design tool developed by the Zhang group at Massachusett's Institute of Technology (http://crispr.mit.edu/). The PCR products for these potential off-target sites using the primers listed in Table S1 were subjected to T7E1 assay and Sanger sequencing.
RNA extraction, RT-PCR and quantitative RT-PCR
Total RNA was isolated from the gastrocnemius muscle of WT and DMD KO rabbits using TRNzol-A+ reagent (Tiangen), and treated with DNase I (Fermentas Inc., MD, USA). The first-strand cDNA was synthesized using the cDNA first strand synthesis kit (Tiangen). The cDNA was used for regular RT-PCR and quantitative RT-PCR (qRT-PCR) analyses to examine the expression of DMD. The primers were used for regular RT-PCR (Forward, 5′-GTCAACTATCTACTGCAAGAGC-3′; Reverse, 5′-CTGTACTTCATCCCACTGATTC-3′) and qRT-PCR (Forward, 5′-CCGAACTAGAAATGCCATCTT-3′; Reverse, 5′-CACAATCACTTGCTGCGATTT-3′). qRT-PCR was performed using a BioEasy SYBR Green I Real Time PCR Kit (Bioer Technology, Hangzhou, China), and the 2−ΔΔCT formula was used to analyze gene expression, Gapdh was used as a reference gene. All experiments were repeated three times for each gene. Data were expressed as the mean±s.e.m.
Body weight and survival curve
The body weight of age- and sex-matched WT and DMD KO rabbits were measured biweekly. All data were expressed as the mean±s.e.m., and a minimum of three individual animals of each genotype were used in all experiments.
Serum biochemistry analysis
The blood samples were collected into heparinized tubes from the ear vein, and sera were prepared by precipitation and centrifugation. Serum CK, ALT and AST levels were measured using a CK test kit (N-acetyl-L-cysteine method), ALT test kit (continuous monitoring method) and AST test kit (continuous monitoring method), respectively (Ningbo Ruiyuan Biotechnology Co., China).
Activity measurement
A Millet Sports Bracelet Wearable Device was used to record movement steps within a 1-h period for DMD KO and WT rabbits. The rabbits wearing the device on their right hind leg were placed in an ∼30 m2 room to allow free movement.
Echocardiography
Echocardiography recording was performed as described previously (Han et al., 2007; Xu et al., 2015a). Briefly, two-dimensional and M-mode transthoracic echocardiography were performed as previously described on WT and DMD KO rabbits (n≥3 per group) by an SIUI all digital color doppler ultrasound diagnostic system (Apogee 300, ShanTou, China). Rabbits were studied in right lateral recumbency from parasternal long and short axis views. The rabbits were held in the right position by restraining their limbs with people. A linear array probe and center frequency of 10.0 MHz were used. Cardiac dimensions [the interventricular septal thickness at end-diastole (IVSd), left ventricular end diastolic diameter (LVDd) and left ventricular systolic diameter (LVDs)] were determined and the percentage of FS and left ventricular EF calculated.
Histology analysis
Various tissues, including gastrocnemius, tibialis anterior, quadriceps, diaphragm and heart, were collected from DMD KO and WT rabbits (euthanized at 5 and 12 months of age). The tissues were fixed in 4% paraformaldehyde at 4°C, dehydrated in increasing concentrations of ethanol (70% for 6 h, 80% for 1 h, 96% for 1 h and 100% for 3 h), cleared in xylene and embedded in paraffin for histological examination. The 5-μm sections were cut for H&E (Han et al., 2007; Xu et al., 2015a) and Masson's trichrome staining as previously described. The stained sections were imaged with a Nikon TS100 microscope.
Immunofluorescence staining
Paraffin-embedded skeletal muscle tissues were deparaffinized and antigen retrieval was performed using a pressure cooker. After blocking with 10% goat serum in PBS for 1 h at room temperature, the tissue sections were incubated with primary antibodies diluted in PBS with 1% BSA at 4°C overnight. Primary antibodies against dystrophin (MANDYS1 clone 3B7, 1:20, Developmental Studies Hybridoma Bank), α-dystroglycan (sc-53987, 1:50, Santa Cruz Biotechnology), α-sarcoglycan (NCL-L-a-SARC, 1:100, Leica Biosystems) and caveolin 3 (610420, 1:500, BD Biosciences) were used. The slides were then extensively washed with PBS and incubated with secondary antibodies (Alexa Fluor 594-conjugated goat anti-mouse IgG, 1:500, Invitrogen) for 1 h at room temperature. Finally, the glass slides were mounted using VECTASHIELD Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Then the slides were imaged with a Nikon Ti-E inverted fluorescence microscope equipped with an Andor Zyla sCMOS camera and a Nikon Super Fluor 20×/0.75 NA objective lens. Images were recorded using the NISElements Advanced Research software package (Nikon) and processed using Photoshop CS5 (Adobe).
Morphometric analysis of myofibers
The H&E-stained cross-sections of gastrocnemius, tibial anterior and quadriceps muscles from the DMD KO and WT rabbits at 5 months of age were analyzed for fiber size, central nucleation and fibrosis. A minimum of three different regions were counted per section. The fiber size, percentage of central nucleated fibers and fibrotic area were calculated using ImageProPlus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Statistics
Data are expressed as mean±s.e.m. Statistical differences were determined by unpaired Student's t-test for two group comparisons, and one-way ANOVA with Bonferroni's post-tests for multiple group comparisons, using Prism 7.0 (GraphPad). P≤0.05 was considered significant.
Supplementary Material
Acknowledgements
We thank Peiran Hu for providing technical assistance at the Embryo Engineering Center.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: Z.L., R.H.; Methodology: L.X., Y.G., R.H.; Formal analysis: T.S.; Resources: Z.L., R.H.; Data curation: T.S., Y.S.L., D.L., T.L.; Writing - original draft: Z.L.; Writing - review & editing: R.H.; Supervision: Z.L., R.H.; Project administration: L.L., Z.L., R.H.; Funding acquisition: L.L., Z.L., R.H.
Funding
This work was supported by the National Key Research and Development Program of China Stem Cell and Translational Research (2017YFA0105101 to Z.L.), the National Heart, Lung, and Blood Institute (R01HL116546 to R.H.), and National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR064241 to R.H.).
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.032201.supplemental
References
- Badalian L. O. and Malygina N. A. (1995). [Molecular genetic research on Duchenne-Becker myodystrophy. The correlation of the severity of the disease with the type of deletion]. Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova 95, 90-92. [PubMed] [Google Scholar]
- Baltzer W. I., Calise D. V., Leving J. M., Shelton G. D., Edwards J. F. and Steiner J. M. (2007). Dystrophin-deficient muscular dystrophy in a weimaraner. J. Am. Anim. Hosp. Assoc. 43, 227-232. 10.5326/0430227 [DOI] [PubMed] [Google Scholar]
- Beggs A. H., Hoffman E. P., Snyder J. R., Arahata K., Specht L., Shapiro F., Angelini C., Sugita H. and Kunkel L. M. (1991). Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am. J. Hum. Genet. 49, 54-67. [PMC free article] [PubMed] [Google Scholar]
- Bladen C. L., Salgado D., Monges S., Foncuberta M. E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A. J., Chamova T. et al. (2015). The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36, 395-402. 10.1002/humu.22758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy K. N., Roche B. M., Schwartz D. S., Nakayama T. and Hamlin R. L. (2004). Evaluation of the six-minute walk test in dogs. Am. J. Vet. Res. 65, 311-313. 10.2460/ajvr.2004.65.311 [DOI] [PubMed] [Google Scholar]
- Bonilla E., Samitt C. E., Miranda A. F., Hays A. P., Salviati G., DiMauro S., Kunkel L. M., Hoffman E. P. and Rowland L. P. (1988). Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell 54, 447-452. 10.1016/0092-8674(88)90065-7 [DOI] [PubMed] [Google Scholar]
- Bősze Z., Hiripi L., Carnwath J. W. and Niemann H. (2003). The transgenic rabbit as model for human diseases and as a source of biologically active recombinant proteins. Transgenic Res. 12, 541-553. 10.1023/A:1025816809372 [DOI] [PubMed] [Google Scholar]
- Bulfield G., Siller W. G., Wight P. A. and Moore K. J. (1984). X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 81, 1189-1192. 10.1073/pnas.81.4.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain J. S., Metzger J., Reyes M., Townsend D. W. and Faulkner J. A. (2007). Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 21, 2195-2204. 10.1096/fj.06-7353com [DOI] [PubMed] [Google Scholar]
- Chapman V. M., Miller D. R., Armstrong D. and Caskey C. T. (1989). Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 86, 1292-1296. 10.1073/pnas.86.4.1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper B. J., Winand N. J., Stedman H., Valentine B. A., Hoffman E. P., Kunkel L. M., Scott M.-O., Fischbeck K. H., Kornegay J. N., Avery R. J. et al. (1988). The homolog of the Duchenne locus is defective in X-linked muscular-dystrophy of dogs. Nature 334, 154-156. 10.1038/334154a0 [DOI] [PubMed] [Google Scholar]
- El Refaey M., Xu L., Gao Y., Canan B. D., Adesanya T. A., Warner S. C., Akagi K., Symer D. E., Mohler P. J., Ma J. et al. (2017). In vivo genome editing restores dystrophin expression and cardiac function in dystrophic mice. Circ. Res. 121, 923-929. 10.1161/CIRCRESAHA.117.310996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R., Bansal D., Miyake K., Muniz V. P., Weiss R. M., McNeil P. L. and Campbell K. P. (2007). Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 117, 1805-1813. 10.1172/JCI30848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman E. P., Brown R. H. Jr. and Kunkel L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919-928. 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- Honda A., Hirose M., Sankai T., Yasmin L., Yuzawa K., Honsho K., Izu H., Iguchi A., Ikawa M. and Ogura A. (2015). Single-step generation of rabbits carrying a targeted allele of the tyrosinase gene using CRISPR/Cas9. Exp. Anim. 64, 31-37. 10.1538/expanim.14-0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. R., Brennan S., Mooney C. T., Callanan J. J., McAllister H., Guo L. T., Martin P. T., Engvall E. and Shelton G. D. (2004). Muscular dystrophy with truncated dystrophin in a family of Japanese Spitz dogs. J. Neurol. Sci. 217, 143-149. 10.1016/j.jns.2003.09.002 [DOI] [PubMed] [Google Scholar]
- Klymiuk N., Blutke A., Graf A., Krause S., Burkhardt K., Wuensch A., Krebs S., Kessler B., Zakhartchenko V., Kurome M. et al. (2013). Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum. Mol. Genet. 22, 4368-4382. 10.1093/hmg/ddt287 [DOI] [PubMed] [Google Scholar]
- Koenig M., Beggs A. H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Muller C. R., Lindlof M., Kaariainen H. et al. (1989). The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 45, 498-506. [PMC free article] [PubMed] [Google Scholar]
- McDonald C. M., Henricson E. K., Han J. J., Abresch R. T., Nicorici A., Elfring G. L., Atkinson L., Reha A., Hirawat S. and Miller L. L. (2010). The 6-min walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve 41, 500-510. 10.1002/mus.21544 [DOI] [PubMed] [Google Scholar]
- Mendell J. R. and Lloyd-Puryear M. (2013). Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 48, 21-26. 10.1002/mus.23810 [DOI] [PubMed] [Google Scholar]
- Mendell J. R., Shilling C., Leslie N. D., Flanigan K. M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D. M., Duval B., Aoyagi A. et al. (2012). Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 71, 304-313. 10.1002/ana.23528 [DOI] [PubMed] [Google Scholar]
- Moser H. (1984). Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum. Genet. 66, 17-40. 10.1007/BF00275183 [DOI] [PubMed] [Google Scholar]
- Muntoni F. (2003). Cardiomyopathy in muscular dystrophies. Curr. Opin. Neurol. 16, 577-583. 10.1097/00019052-200310000-00003 [DOI] [PubMed] [Google Scholar]
- Pastoret C. and Sebille A. (1995). mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 129, 97-105. 10.1016/0022-510X(94)00276-T [DOI] [PubMed] [Google Scholar]
- Sharp N. J. H., Kornegay J. N., Van Camp S. D., Herbstreith M. H., Secore S. L., Kettle S., Hung W.-Y., Constantinou C. D., Dykstra M. J., Roses A. D. et al. (1992). An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics 13, 115-121. 10.1016/0888-7543(92)90210-J [DOI] [PubMed] [Google Scholar]
- Shen B., Zhang J., Wu H., Wang J., Ma K., Li Z., Zhang X., Zhang P. and Huang X. (2013). Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 23, 720-723. 10.1038/cr.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokova N. and Niggli E. (2013). Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies. J. Mol. Cell. Cardiol. 58, 217-224. 10.1016/j.yjmcc.2012.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicinski P., Geng Y., Ryder-Cook A. S., Barnard E. A., Darlison M. G. and Barnard P. J. (1989). The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578-1580. 10.1126/science.2662404 [DOI] [PubMed] [Google Scholar]
- Smith B. F., Yue Y. P., Woods P. R., Kornegay J. N., Shin J.-H., Williams R. R. and Duan D. S. (2011). An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed. Lab. Investig. 91, 216-231. 10.1038/labinvest.2010.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney C. F. (2011). Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve 44, 8-19. 10.1002/mus.22097 [DOI] [PubMed] [Google Scholar]
- Spurney C. F., Gordish-Dressman H., Guerron A. D., Sali A., Pandey G. S., Rawat R., Van Der Meulen J. H., Cha H.-J., Pistilli E. E., Partridge T. A. et al. (2009). Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve 39, 591-602. 10.1002/mus.21211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui T., Yuan L., Liu H., Chen M., Deng J., Wang Y., Li Z. and Lai L. (2016). CRISPR/Cas9-mediated mutation of PHEX in rabbit recapitulates human X-linked hypophosphatemia (XLH). Hum. Mol. Genet. 25, 2661-2671. [DOI] [PubMed] [Google Scholar]
- Tanabe Y., Esaki K. and Nomura T. (1986). Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 69, 91-95. 10.1007/BF00687043 [DOI] [PubMed] [Google Scholar]
- Valentine B. A., Cooper B. J., de Lahunta A., O'Quinn R. and Blue J. T. (1988). Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J. Neurol. Sci. 88, 69-81. 10.1016/0022-510X(88)90206-7 [DOI] [PubMed] [Google Scholar]
- Wang Y., Fan N., Song J., Zhong J., Guo X., Tian W., Zhang Q., Cui F., Li L., Newsome P. N. et al. (2014). Generation of knockout rabbits using transcription activator-like effector nucleases. Cell Regen 3, 3 10.1186/2045-9769-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Zhao P., Mariano A. and Han R. (2013). targeted myostatin gene editing in multiple mammalian species directed by a single pair of tale nucleases. Mol. Ther. Nucleic. Acids 2, e112 10.1038/mtna.2013.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., El Refaey M., Xu L., Zhao L., Gao Y., Floyd K., Karaze T., Janssen P. M. L. and Han R. (2015a). Genetic disruption of Ano5 in mice does not recapitulate human ANO5-deficient muscular dystrophy. Skelet Muscle 5, 43 10.1186/s13395-015-0069-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Park K. H., Zhao L., Xu J., El Refaey M., Gao Y., Zhu H., Ma J. and Han R. (2015b). CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther. 24, 564-569. 10.1038/mt.2015.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Zhao L., Gao Y., Xu J. and Han R. (2016). Empower multiplex cell and tissue-specific CRISPR-mediated gene manipulation with self-cleaving ribozymes and tRNA. Nucleic Acids Res. 45, e28 10.1093/nar/gkw1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. H., Zhao H., Qing Y. B., Pan W. R., Jia B. Y., Zhao H. Y., Huang X. X. and Wei H. J. (2016). Porcine zygote injection with Cas9/sgRNA results in DMD-modified pig with muscle dystrophy. Int. J. Mol. Sci. 17, 1668 10.3390/ijms17101668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L., Sui T., Chen M., Deng J., Huang Y., Zeng J., Lv Q., Song Y., Li Z. and Lai L. (2016). CRISPR/Cas9-mediated GJA8 knockout in rabbits recapitulates human congenital cataracts. Sci. Rep. 6, 22024 10.1038/srep22024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.-F., Liu H.-H., Zhou T. and Tian L. (2013). Novel mutation in exon 56 of the dystrophin gene in a child with Duchenne muscular dystrophy. Int. J. Mol. Med. 32, 1166-1170. 10.3892/ijmm.2013.1498 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.