

ABSTRACT
Actomyosin II contractility in epithelial cell plays an essential role in tension-dependent adhesion strengthening. One key unsettling question is how cellular contraction transmits force to the nascent cell–cell adhesion when there is no stable attachment between the nascent adhesion complex and actin filament. Here, we show that myosin-1c is localized to the lateral membrane of polarized epithelial cells and facilitates the coupling between actin and cell–cell adhesion. Knockdown of myosin-1c compromised the integrity of the lateral membrane, reduced the generation of tension at E-cadherin, decreased the strength of cell–cell cohesion in an epithelial cell monolayer and prevented force-dependent recruitment of junctional α-actinin. Application of exogenous force to cell–cell adhesions in a myosin-1c-knockdown cell monolayer fully rescued the localization defect of α-actinin, indicating that junction mechanoregulation remains intact in myosin-1c-depleted cells. Our study identifies a role of myosin-1c in force transmission at the lateral cell–cell interface and underscores a non-junctional contribution to tension-dependent junction regulation.
KEY WORDS: α-actinin, Synaptopodin, Myosin, Junction, Mechanobiology, Cell–cell adhesion
Highlighted Article: Myosin-1c is a regulator of intercellular tension that can promote tension generation from the extracellular domain of E-cadherin, propagating actomyosin-generated force to intercellular junctions.
INTRODUCTION
Epithelial cells are frequently subjected to chemical and mechanical insults, leading to a local loss of cells from the epithelium. To restore a continuous barrier, epithelial cells must quickly migrate into the open wound and reseal the monolayer (Begnaud et al., 2016; Park et al., 2017; Richardson et al., 2016; Humphreys, 2014; Crosby and Waters, 2010). However, covering the wounded area is not sufficient for the new epithelium to provide resilience at the tissue level. The regeneration process must create a mature junction with strong cell–cell adhesion and attachment to the actin cytoskeleton. Failure to strengthen cell–cell adhesion and re-establish a mature junction upon wound closure predisposes the epithelium to future injuries, resulting in chronic conditions (Humphreys, 2014; Powers et al., 2016; Gohy et al., 2016; Tschumperlin et al., 2000; Van Der Gracht et al., 2016). Understanding how a confluent epithelial monolayer strengthens cell–cell adhesion can provide insight into the final stages of epithelial regeneration and helps identify ways to facilitate the reformation of a strong epithelial protective layer (Sharples, 2007; Shaw and Martin, 2016; Schilders et al., 2016).
Junction formation starts with a nucleation process initiated by trans-interaction of the cadherin–catenin complex from apposing cells (Bajpai et al., 2008; Biswas et al., 2015), followed by a maturation process that requires mechanical input from actomyosin II contractility (Yao et al., 2014; Wu et al., 2014), stabilization of the cadherin–catenin complex (Adams et al., 1996; Mcneill et al., 1993; Weng and Wieschaus, 2016; Salomon et al., 2017), recruitment of additional junctional molecules (Yao et al., 2014; Kannan and Tang, 2015; Knudsen et al., 1995; Drenckhahn and Franz, 1986; Nieset et al., 1997; Le Duc et al., 2010; Ishiyama et al., 2013; Weiss et al., 1998; Kim et al., 2015; Yonemura et al., 2010) and attachment to actin filaments (Johnson and Craig, 1995; Vasioukhin et al., 2000; Yamada et al., 2004; Yonemura, 2011; Yonemura et al., 1995; Zhang et al., 2005). Previous studies have shown that tethering an actin filament to a minimal E-cadherin (also known as CDH1) complex containing the cytoplasmic tail of E-cadherin, β-catenin and an α-catenin (herein α-catenin refers to all family members) results in the engagement of the actin filament with the minimal complex (Buckley et al., 2014). Inside cells, additional actin-binding proteins, namely, vinculin and α-actinin-1 and α-actinin-4 hetero- and homo-dimers, are recruited to cell–cell adhesions under the regulation of actomyosin II contractility (Yao et al., 2014; Kannan and Tang, 2015; Ishiyama et al., 2013; Watabe-Uchida et al., 1998). Both α-catenin and α-actinin are essential for junctional recruitment of vinculin (Kannan and Tang, 2015; Yonemura et al., 2010). Both vinculin and α-actinin can interact with α-catenin and actin (Bois et al., 2005; Kelly et al., 2006; Mcgregor et al., 1994; Wachsstock et al., 1987; Rangarajan and Izard, 2012; Tang and Brieher, 2012) and with each other (Bois et al., 2005, 2006; Kelly et al., 2006; Mcgregor et al., 1994; Wachsstock et al., 1987). Thus, the formation of a tension-dependent tertiary complex consists of three actin-binding proteins, α-catenin, α-actinin and vinculin, could provide multiple actin linkages at the junction. In cells, the recruitment of α-actinin to the cell junction requires synaptopodin (Kannan and Tang, 2015), which also interacts with actin (Kannan and Tang, 2015; Kremerskothen et al., 2005). Therefore, the coupling between α-catenin–vinculin and α-actinin–synaptopodin, would create a quaternary junctional complex to support stable actin attachment, adhesion strengthening and junction maturation.
Although it is well-established that tension is required for junction assembly, it is unclear how force is propagated to the nascent cell–cell adhesion before the recruitment of α-actinin and vinculin. It is also unclear how force is transmitted from actin to the nascent E-cadherin–α-catenin complex to promote the initial loading of actin filament (Arif et al., 2011; Greenberg et al., 2012; Hokanson et al., 2006; Hokanson and Ostap, 2006; Lieto-Trivedi and Coluccio, 2008; Lu et al., 2015; Manceva et al., 2007; Pyrpassopoulos et al., 2012). Here, we provide evidence that a membrane-associated actin-binding motor, myosin-1c, plays a role in tension generation at E-cadherin and promotes tension-dependent recruitment of α-actinin to the cell junction.
RESULTS
Recruitment of α-actinin during junction maturation is associated with endogenous epithelial cell contraction
Previously, we have shown that α-actinin-4 accumulates at the cell junction during epithelial maturation over a period of several days, during which the cell–cell contact develops from a young to a mature junction with increased adhesion and barrier function (Kannan and Tang, 2015). In addition, we showed that exogenous application of cyclic intercellular force can induce α-actinin-4 recruitment to the cell junction, indicating a tension-dependent mechanism of regulation (Kannan and Tang, 2015). Here, using live-cell imaging of Venus–α-actinin, we found that endogenous cellular contraction is associated with the accumulation of Venus–α-actinin at the cell junction over time (Fig. 1A–C; Movie 1). In the maturing junction, repeated whole-cell contractions are followed by a ∼20% increase in Venus–α-actinin accumulation at the cell junction in less than 5 h (Fig. 1C). In a young epithelial monolayer (1-day confluent), whole-cell contraction is accompanied by a gradual increase in Venus–α-actinin at α-actinin-negative apical junctions (Fig. 1D,E; Movie 2). These findings imply that, prior to α-actinin-4 recruitment and the stable attachment of actin filament, a mechanism of force transmission is in place to propagate actomyosin II-generated force from the lateral plasma membrane to cell–cell adhesion to initiate tension-dependent junctional changes.
Fig. 1.

Recruitment of α-actinin during junction maturation is associated with endogenous epithelial cell contraction. (A) Time-lapse frames from Movie 1 showing the transient decrease in cell width as marked by Venus–α-actinin during a whole-cell contraction in a maturing cell monolayer (red and yellow asterisks). The yellow line marks the cell width. Data are representative of six sets of live-cell time-lapse movies. Scale bar: 10 µm. (B) Measurements of cell width of the contracting cell shown in A and Movie 1. The green bar marks the time sequence shown in A. The red asterisk corresponds to the contraction shown in A. Left and right insets show junctional Venus–α-actinin at t=0 min and t=120 min, respectively. Scale bar: 2 µm. (C) Quantification of the level (A.U., arbitrary units) of junctional Venus–α-actinin of the cell shown in A,B at t=0 and t=4.5 h. Bar graph shows the mean±s.e.m. of six separate junctional regions of the contracting cell. P<0.001 for α-actinin between t=0 and 4.5 h. Data are representative of six sets of live-cell time-lapse movies. (D) Time-lapse frames from Movie 2 showing whole-cell contraction along the lateral membrane (apical z=5 µm to basal z=0 µm) in a young monolayer with no original accumulation of α-actinin at the apical junction (yellow arrows). Pink arrows point to increasing Venus–α-actinin accumulation at the apical junction after contraction. Green arrows mark the lateral cell boundary of the contracting cell. Data are representative of 12 sets of live-cell time-lapse movies. Scale bar: 10 µm. (E) Measurements of the cell shown in D and Movie 2. The level of Venus–α-actinin at the apical junction (z=4 µm) gradually increases after the initial contraction (purple asterisk). (F) Cell width and intracellular Ca2+ concentration measured using the lipid-modifiable intramolecular FRET sensor Lyn-D3cpV (from Movie 3). Whole-cell contraction is preceded by a Ca2+ spike and characterized by the inward movement of the lateral plasma membrane. Orange solid line marks the time-lapse sequence shown in G. Three contraction events are shown (I, II, III), corresponding to the labeled images in G. The green dotted line represents the beginning of a Ca2+ spike; the orange dotted line represents the beginning of contraction. The Ca2+ spike is indicated by a change in the ratio of FRET to cpVenus. (G) Time-lapse frames from Movie 3 showing three Ca2+ spikes, corresponding to the three contraction events in F (marked by orange I, II, III). Lyn-D3cpV FRET (excitation of donor and emission of acceptor), cpVenus (excitation of acceptor and emission of acceptor contraction) and merge images of FRET and cpVenus are shown. Data are representative of six sets of live-cell time-lapse movies.
To directly observe plasma membrane movement during cellular contraction, we used the lipid-modifiable Ca2+-sensitive FRET-based biosensor Lyn-D3cpV to simultaneously label the plasma membrane and monitor intracellular Ca2+, a global regulator of cellular contraction (Fig. 1F,G; Movie 3). We found that whole-cell contraction in a young monolayer is characterized by a transient rise of intracellular Ca2+ followed by a decrease in cell width (Fig. 1F). These observations indicate that cellular contraction is associated with an inward movement of the lateral plasma membrane that can generate pulling force at the cell–cell adhesion interface.
Myosin-1c overlaps with F-actin and colocalizes with E-cadherin and α-catenin at discrete junctional puncta on the lateral plasma membrane
The inward movement of the lateral plasma membrane during whole-cell contraction would require the coupling of the lateral cell–cell adhesion interface to the actomyosin II cytoskeleton, a function that can be satisfied by a membrane-associated actin-binding protein. Myosin-1c is a membrane-tethered actin-binding motor that has previously been shown to regulate E-cadherin adhesion (Tokuo and Coluccio, 2013). Thus, myosin-1c could potentially play a role in force transmission at the cell–cell adhesion interface. To characterize the relationship between myosin-1c and junctional proteins in polarized MDCK cells, we performed immunofluorescence using optical-sectioning structured illumination microscopy (OS-SIM). In polarized epithelial cells, myosin-1c overlaps strongly with the tension-sensitive junctional protein α-actinin-4 (z=2.2 µm, R≈0.73) on the lateral linear junction (Fig. 2A,B, lozenge). In addition, myosin-1c colocalizes with both synaptopodin (R≈0.42–0.81) and α-actinin-4 (R≈0.53–0.82) at the junctional vertices (Fig. 2A,B, circle). Importantly, myosin-1c overlaps strongly with F-actin (z=1–4.2 µm, R≈0.79–0.83) along the z-axis of the lateral junction (Fig. 2C,D). Myosin-1c was found to localize at discrete regions overlapping with E-cadherin and α-catenin (Figs S1, S2). These observations indicate that myosin-1c is a major component on the lateral membrane and that it colocalizes with F-actin, α-actinin-4 and the E-cadherin complex, suggesting a possible role of myosin-1c in facilitating the coupling between actin and cell–cell adhesion.
Fig. 2.

Myosin-1c colocalizes with F-actin and a tension-sensitive α-actinin-4 protein on the lateral junction in polarized epithelial monolayer. (A) OS-SIM showing immunofluorescence of myosin-1c (M1c), synaptopodin (Syp, synpo), and α-actinin-4 (A4) at the apical (z=0 µm), the sub-apical (z=0.4 µm) and the lateral (z=2.2 µm) junctions. For each z-image, the top panel shows the x-z, y-z and x-y images, and the bottom panel shows a representative linear junction from the x-y image. Data are representative of ten sets of images from one experiment out of six independent experiments. Scale bars: 10 µm (orange); 2 µm (white). (B) Merge images of the representative linear junction shown in A. The Pearson's correlation coefficients between myoin-1c, synaptopodin or α-actinin-4 at the junctional vertices (white circle) and linear junctions (white lozenge) are represented by Rv and Rj, respectively. Correlations were calculated from 500–1500 pixels per protein per z-image in one set of data. Six sets of data were analyzed and a representative set of data is shown. Scale bar: 2 µm. (C) OS-SIM showing immunofluorescence of myosin-1c, actin (phalloidin, Phal), and myosin IIB (MIIB) at the sub-apical (z=1 µm) and the lateral (z=2.6 and 4.2 µm) junctions. For each z-image, the top panel shows the x-z, y-z and x-y images, and the bottom panel shows a representative linear junction from the x-y image. Data are representative of ten sets of images from one experiment out of three independent experiments. Scale bars: 10 µm (orange); 2 µm (white). (D) Merge images of the representative linear junction shown in C. The Pearson's correlation coefficient (R) between two proteins at the linear junction was calculated from 1000–2000 pixels per protein per z-image for one set of data. Data set is representative of six sets of data from one experiment out of three independent experiments. Scale bar: 2 µm.
Myosin-1c knockdown disrupts actin compaction on the lateral membrane and actin organization on the apical domain
To examine the role of myosin-1c in actin regulation at the junction, we depleted myosin-1c in MDCK cells by shRNA (Fig. 3A). We found that myosin-1c knockdown did not dramatically change the overall content of actin at the junction (Fig. 3B). However, the layer of actin on the cell boundary appeared to be thicker (Fig. 3C; Fig. S3). In wild-type cells, actin overlaps with β-catenin when myosin-1c is present (Fig. 3D, white arrowheads). However, in myosin-1c-knockdown cells, actin was often spatially separated from β-catenin (Fig. 3E, red arrowheads). Measurements of actin intensities tangential to the junction revealed a ∼1.8-fold increase (half-maximum distance change from ∼0.36 µm to ∼0.68 µm) in the thickness of cortical actin in the myosin-1c-knockdown cells (Fig. 3E) whereas the thickness of β-catenin was unchanged (Fig. 3D). These observations indicate that myosin-1c promotes actin compaction on the lateral membrane and the association of actin with the E-cadherin complex.
Fig. 3.

Myosin-1c knockdown disrupts actin compaction on the lateral cell-cell interface. (A) Constrained iterative deconvolution microscopy showing immunofluorescence of myosin-1c (myo1c), actin (phalloidin) and β-catenin (β-cat) at apical (z=0 µm), sub-apical (z=1 µm) and lateral (z=3, 5 and 5 µm) junctions. Images were generated from 0.2 µm wide-field optical sections. Wild-type and myosin-1c knockdown cell areas are marked by WT and KD, respectively. Data are representative of ten sets of images from one experiment out of three different experiments. Scale bar: 10 µm. (B) Junctional myosin-1c and actin (phalloidin) normalized to that of β-catenin in wild-type and myosin-1c-knockdown cells. Each point represents the measurement of one junctional area at z=3 µm. Data are representative of ten sets of images from one experiment out of three independent experiments. (C) Constrained iterative deconvolution microscopy showing immunofluorescence of myosin-1c, actin (phalloidin) and β-catenin at the lateral junction (z=2 µm). The top panel is a merge image of actin (phalloidin), β-catenin and myosin-1c. The bottom image shows the same area with actin (phalloidin) and β-catenin. Wild-type and myosin-1c-knockdown cells are marked by WT and KD, respectively. Scale bars: 2 µm. (D) Actin associates with β-catenin in the presence of myosin-1c. Immunofluorescence of actin (phalloidin), myosin-1c (myo1c) and β-catenin (β-cat) at the lateral junction (z=2 µm). White arrowheads mark regions with overlapping actin (phalloidin), myosin-1c and β-catenin. Red arrowheads mark regions with actin dissociated from β-catenin in the absence of myosin-1c. Line graphs show intensities of actin (phalloidin) and β-catenin across the lateral junctions (A.U., arbitrary units). The bar graph shows the mean±s.e.m. of the half-maximal width of β-catenin across 12 junctions in wild-type (WT) and myosin-1c knockdown (Myo1c KD) cells. Scale bar: 2 µm. (E) Actin dissociates from β-catenin at the lateral junction (z=2 µm) in myosin-1c-knockdown cells. Immunofluorescence of actin (phalloidin) and β-catenin at the lateral junction (z=2 µm) of myosin-1c-knockdown cells. Red arrowheads mark the junctional region with actin dissociated from β-catenin. Line graphs show intensities of actin (phalloidin) and β-catenin across the lateral junctions. The bar graphs shows the mean±s.e.m. of the half-maximal width of actin (phalloidin) across 12 junctions in wild-type (WT) and myosin-1c-knockdown cells. P<0.001 for phalloidin between WT and myosin KD junctions. Data are representative of 10 sets of images from one experiment out of three independent experiments. Scale bar: 2 µm.
In polarized MDCK cells, knockdown of myosin-1c resulted in a gross disruption of the apical actin meshwork (Fig. S4A). Quantification of actin intensities revealed that the distribution of actin on the apical plane was dramatically altered by myosin-1c depletion (Fig. S4B). Instead of a single peak in the actin intensity histogram, myosin-1c-knockdown cells show two peaks, corresponding to a lower intensity and a higher intensity population, indicating that some areas are depleted of actin but other areas have increased actin accumulation. By organizing the apical actin meshwork, myosin-1c supports force transmission on the apical plane and contributes to tension generation at the apical junction.
Myosin-1c knockdown decreases E-cadherin tension at the cell junction
To further investigate whether myosin-1c plays a role in tension regulation at cell–cell contacts, we generated a new E-cadherin tension sensor (E-cadherin_sstFRET) using a spectrin-repeat as the force sensor and a cerulean fluorescent protein (CeFP)–Venus fluorescent protein (VFP) pair as the Förster resonance energy transfer (FRET) reporter (Fig. 4A). The spectrin-repeat FRET module was originally designed as an α-actinin force sensor that reports on mechanical stress at focal adhesions (Meng and Sachs, 2011). Previous calibration of the spectrin-repeat FRET module shows a 50% decrease in FRET in response to ∼5–7 pN force (Meng and Sachs, 2011). We inserted the tension sensor module between the transmembrane and the extracellular domains of E-cadherin (Fig. 4A). When the E-cadherin tension sensor was expressed in MDCK cells it shows strong localization at the cell–cell interface (Fig. 4B). By using a dual-view beam splitter to simultaneously obtain donor (CeFP) emission and acceptor (VFP) emission, we found that the FRET index decreases from ∼0.8 in a young monolayer (1-day confluent) to ∼0.45 in a mature monolayer at ∼7 days (Fig. 4C), indicating an increase in tension at E-cadherin during junction maturation to slightly above ∼5–7 pN. In older monolayers at >4 weeks in culture, the FRET index reached a steady-state minimum of ∼0.3 (Fig. 4C), indicating that E-cadherin experiences ≫7 pN of force in these very old monolayers. In a mature monolayer at ∼7 days in culture, endogenous whole-cell contraction resulted in a slight decrease in the FRET index (Fig. 4D; Movie 4). These observations indicate that the extracellular domain of E-cadherin experiences tension constantly and additional force can be exerted on the extracellular domain during endogenous contractions (Fig. 4D,E).
Fig. 4.

Myosin-1c knockdown decreases E-cadherin tension at the cell junction. (A) Construction of a new E-cadherin tension sensor by inserting a tension sensor module [spectrin repeat flanked by CeFP and VFP between the extracellular (EC1–5) and the transmembrane (TM) domains of E-cadherin]. Full-length E-cadherin tension sensor (E-cadherin_sstFRET) contains a cytoplasmic juxtamembrane domain (JXM) and the β-catenin-binding domain (CBD). A decrease in FRET would reveal the generation of tension at E-cadherin extracellular domain. (B) Dual-view live-cell image of CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) of E-cadherin_sstFRET in young (1-day confluent) and mature (7-day confluent) MDCK monolayers showing its localization on the lateral cell–cell interface and the decrease in FRET in mature junctions. Image is representative of eight image sets from one experiment out of four independent experiments. Scale bar: 10 µm. (C) Quantification of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) in young (1-day confluent), mature (7-day confluent) and old (28-day confluent) cell monolayers expressing the E-cadherin force sensor. The FRET index is expressed as FRET/(CeFP+FRET). Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between young, mature and old junctions. Data are representative of eight image sets from one experiment out of four independent experiments. (D) Measurement of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) of E-cadherin_sstFRET showing a decrease in FRET index during whole-cell contraction (Movie 4). Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between pre-contraction and contracting junction. Data are representative of three image sets from one experiment out of three independent experiments. (E) A diagram showing how whole-cell contraction that pulls on the lateral membrane can exert force on cell–cell adhesion extracellular domains to decrease FRET on E-cadherin. (F) E-cadherin tension sensor missing the β-catenin-binding domain (CBD) could experience pulling force on the extracellular domain if the lateral membrane was coupled to the actin cytoskeleton by myosin-1c. (G) Quantification of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) in cells expressing the full-length E-cadherin force sensor (FL E-Cad_sstFRET) or the E-cadherin tension sensor missing the β-catenin-binding domain (deleted CBD), and in cells expressing FL E-Cad_sstFRET that had been pre-treated with 10 µM ROCK inhibitor Y-27632 for 2 h. The FRET index is expressed as FRET/(CeFP+FRET). Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between FL E-cadherin_sstFRET and deleted CBD or Y-27632. Data are representative of eight image sets from one experiment out of three independent experiments. (H) Dual-view live-cell image of CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) of E-Cad_sstFRET in the mature myosin-1c-knockdown cell monolayer showing localization on the lateral cell–cell interface and high FRET index. Image set is representative of three image sets from one experiment out of three independent experiments. Scale bar: 10 µm. (I) Measurement of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) showing an increase in E-cadherin FRET index at the junctions of myosin-1c-knockdown (KD) cells. Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between vector control and myosin-1c knockdown. Data are representative of three image sets from one experiment out of three independent experiments. (J) Measurement of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) of E-cadherin_sstFRET in clonal cell lines expressing a control vector (clonal lines #4 and #9) or shRNA for myosin-1c (clonal lines #22 and #23). Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between control and myosin-1c KD clonal lines. Data are representative of three image sets from one experiment out of three independent experiments.
Our current results corroborate those of previous studies using a spider silk-based elastic tension sensor, which showed that the E-cadherin intracellular domain is under constitutive tension (Borghi et al., 2012). In that study, deletion of the cytoplasmic domain of E-cadherin increased the FRET index from ∼0.29 to ∼0.4 but did not abolish tension, indicating that additional unknown factors are responsible for creating tension on E-cadherin in the absence of direct actin binding. We deleted the β-catenin-binding domain in our E-cadherin tension sensor (Fig. 4F) and found that the tension on the extracellular domain of E-cadherin decreased only slightly from a FRET index of ∼0.45 to ∼0.52 in mature junctions (7-day confluent) (Fig. 4G). In addition, inhibition of Rho-dependent contractility with the ROCK inhibitor Y-27632 (10 µM for 2 h) only resulted in an increase of FRET index from ∼0.45 to ∼0.53 (Fig. 4G). These results are in agreement with the previous E-cadherin tension sensor study showing that the FRET index only slightly changed (<5%) upon depletion of α-catenin with shRNA, actin disruption with cytochalasin D, or myosin II inhibition with ML-7 (Borghi et al., 2012). Our current study and the previous findings predict the presence of other factor(s) that can regulate E-cadherin tension.
To determine whether myosin-1c contributes to E-cadherin tension, we expressed the E-cadherin_sstFRET tension sensor in myosin-1c-knockdown cells. We found that knockdown of myosin-1c dramatically increased the FRET index from ∼0.46 to 0.83 in mature cell monolayers (7-day confluent) (Fig. 4H). In clonal cell lines, myosin-1c-knockdown cells exhibited a FRET index of ∼0.79–0.84 (clonal lines #22 and #23) whereas vector control cells exhibited a FRET index ∼0.45–0.48 (clonal lines #4 and #9) (Fig. 4J). These findings indicate that, by coupling the lateral membrane to actin, myosin-1c contributes to the generation of tension on the extracellular domain of E-cadherin.
A myosin-1c force sensor reveals that there is an increase in myosin-1c tension during junction maturation and cell contraction
To further investigate the role of myosin-1c in force transmission, we performed live-cell imaging of EFGP–myosin-1c in MDCK cells (Fig. 5A,B; Movie 5). We found that myosin-1c is tethered on the lateral plasma membrane during endogenous cellular contractions (Fig. 5B,C; Movie 5). Measurements of cell width on the z-axis indicate that cellular contraction is associated with an inward movement of myosin-1c along the entire lateral cell–cell interface (Fig. 5C). These observations indicate that myosin-1c is stably tethered on the lateral membrane and can potentially play a role in force transmission between the actomyosin II cytoskeleton and the lateral cell–cell interface. To test this hypothesis, we generated a myosin-1c tension sensor (myo1c_sstFRET) using the same spectrin-repeat FRET-based module as described above. We inserted the tension sensor module between the lipid-binding domain and the actin-binding region of myosin-1c (Fig. 5D). When the myo1c_sstFRET was expressed in MDCK cells it showed a strong localization to the lateral membrane (Fig. 5E). We found that the FRET index decreases from ∼0.85 in young monolayer (1 day confluent) to ∼0.6 in the mature monolayer at ∼7 days (Fig. 5F), indicating an increase in tension on myosin-1c during junction maturation. These results indicate that myosin-1c experiences <5 pN of force on the lateral membrane. During endogenous contractions, there is a slight decrease in the FRET index of the myosin-1c tension sensor to ∼0.5 (Fig. 5G), indicating that additional force can be exerted on myosin-1c during endogenous contractions. These results are in agreement with a previous single-molecule study showing that myosin-1c can hold onto actin filament under stress of up to ∼4–5 pN of force (Greenberg et al., 2012).
Fig. 5.

The myosin-1c force sensor reveals that there is an increase in myosin-1c tension during junction maturation and cell contraction. (A) A diagram showing the regions of EGFP–myosin-1c (EGFP_myo1c), which contains an actin-binding motor, an IQ domain, and a phosphatidylinositol-binding (PIPs-binding) domain attached to EGFP at the C-terminus. (B) Time-lapse frames showing EGFP–myosin-1c along the entire lateral cell–cell interface (from apical z=6 µm to basal z=0 µm) during whole-cell contractions (pink and blue asterisks). Data are representative of eight sets of live-cell time-lapse movies. Scale bar: 5 µm. (C) Measurements of cell width on the apical (z=6 µm) and lateral (z=4 µm) junction (marked by pink and blue lines in B) showing inwards movement of the lateral membrane during whole-cell contraction. Data are representative of eight sets of live-cell time-lapse movies. (D) Construction of a new myosin-1c tension sensor (myo1c_sstFRET) by inserting a tension sensor module (spectrin repeat flanked by CeFP and VFP) between the IQ-containing and the phosphatidylinositol-binding domains of myosin-1c. (E) Dual-view live-cell image of CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) of myo1c_sstFRET showing localization on the lateral cell-cell interface. Image is representative of eight sets of images from one experiment out of four independent experiments. Scale bar: 10 µm. (F) Quantification of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) in young (1-day confluent) and mature (7-day confluent) cell monolayers expressing myo1c_sstFRET. The FRET index is expressed as FRET/(CeFP+FRET). Horizontal lines represent mean fluorescence intensities. P<0.001 for FRET index between young and mature junctions. Data are representative of eight image sets from one experiment out of four independent experiments. (G) Measurement of junctional CeFP (excitation CeFP, emission CeFP) and FRET (excitation CeFP, emission VFP) showing the decrease in FRET index during whole-cell contraction. Data are representative of three image sets from one experiment out of three independent experiments.
Myosin-1c knockdown compromises the coupling between actomyosin II contractility and the lateral membrane
To examine whether myosin-1c is required for the coupling between actomyosin II contractility and the lateral cell–cell interface, we depleted myosin-1c in cells expressing Venus–α-actinin. Depletion of myosin-1c compromises Venus–α-actinin localization at the cell junction (Fig. 6A,B) and alters the behavior of cellular contractions (Movies 6,7). Instead of pulling the lateral membrane inwards, the myosin-1c-knockdown cells expanded during endogenous contraction, resulting in an increase in the cell area (Fig. 6A; Movies 6,7). Moreover, myosin-1c-knockdown cells constantly undergo blebbing on the lateral membrane (Movie 8) and are unable to generate a proper cell height, resulting in shorter cells that are more spread (Fig. 6B). These observations indicate that myosin-1c is necessary for the coupling between the lateral membrane and the actin cytoskeleton to maintain membrane integrity and to facilitate tension-dependent processes on the cell–cell adhesion interface.
Fig. 6.

Myosin-1c knockdown compromises the coupling between actomyosin contractility and the lateral plasma membrane. (A) Live-cell images of Venus–α-actinin in the myosin-1c-knockdown (Myo1c KD) cell monolayer from Movie 7 showing pre-contraction (purple outline), contraction (orange outline) and post-contraction (blue outline) cell boundaries. The graph plots the area of the cell. Purple, orange and blue asterisks correspond to the cells outlined in purple, orange and blue, respectively. Data are representative of six sets of live-cell time-lapse movies. (B) Live-cell images of Venus–α-actinin in mature wild-type (WT MDCK) and myosin-1c-knockdown cell monolayers. x-z, y-z and x-y images were used to calculate the cell height, spread area and cell volume. Image set is representative of 16 sets of images from one experiment out of four independent experiments. Scale bars: 10 µm. Bar graphs show measurements of cell height, spread area and cell volume of wild-type and myosin-1c knockdown cells. Results are mean±s.e.m. of 16 measurements. P<0.001 for height and area between WT and myosin-1c KD. Data are representative of one experiment out of four independent experiments. (C) Wide-field immunofluorescence of α-actinin-4 in wild-type, myosin-1c knockdown and synaptopodin (Synpo) knockdown (KD) cell monolayers. Image set is representative of 16 sets of images from one experiment out of three independent experiments. Scale bar: 10 µm. (D) Cell spread area, cell density and cell height of wild-type, myosin-1c knockdown and synaptopodin knockdown monolayers. Bar graphs show the mean±s.e.m. of 16 measurements. P<0.001 for height, area and density between WT and myosin-1c KD. Data are representative of one experiment out of four independent experiments. (E) Immunoprecipitation of active Rho using the Rhotekin Rho-binding domain (RBD) followed by western blotting for Rho showing same level of Rho activities in MDCK wild-type and myosin-1c knockdown cells. Data are representative of one experiment out of three independent experiments. (F) Western blot for myosin light chain Thr18/Ser19 phosphorylation showing the same level in MDCK wild-type and myosin-1c knockdown cells. Data are representative of one experiment out of three independent experiments.
The disruption of α-actinin localization upon myosin-1c depletion phenocopies the lack of junctional α-actinin-4 in synaptopodin-knockdown cells (Kannan and Tang, 2015). To assess whether the depletion of synaptopodin leads to a decrease in cell height, we compared synaptopodin- and myosin-1c-knockdown cell monolayers (Fig. 6C,D). We found that synaptopodin and myosin-1c depletion both abolished the accumulation of junctional α-actinin-4 but depletion of synaptopodin did not result in a defect in cell height generation. These findings indicate that the inability to generate a proper lateral membrane in myosin-1c-knockdown cells is not due to the lack of junctional α-actinin-4.
The disruption of tension-dependent α-actinin recruitment to the cell junction could be a result of a decrease in contractility in myosin-1c-knockdown cells. To determine whether myosin-1c knockdown compromises actomyosin II activities, we assessed two indicators of cellular contractility, active RhoA and myosin light chain (MLC2, also known as MYL2) phosphorylation (Fig. 6E,F). By using a Rhotekin pull-down assay, we found that the levels of active RhoA in wild-type and myosin-1c-knockdown cells are the same (Fig. 6E). In addition, western blot analysis using phospho-specific antibodies shows that the levels of myosin light chain phosphorylation in wild-type and myosin-1c-knockdown cells are the same (Fig. 6F). These results indicate that the lack of junctional α-actinin in myosin-1c-knockdown cells is not caused by a decrease in cellular contractility but rather a disruption in force transmission and tension generation at the cell junction.
Myosin-1c knockdown abolishes junction recruitment of α-actinin-4 and weakens cell–cell cohesion
To investigate the relationship between myosin-1c and junctional proteins, we compared their localization in wild-type and myosin-1c-knockdown cells. As shown in Fig. 6B,C, knockdown of myosin-1c results in shorter cells, which makes comparison between wild-type and myosin-1c-knockdown cells problematic due to the difference in the z-height of their lateral junctions. To make a fair assessment, we standardized a protocol that compels the myosin-1c-knockdown cells to exhibit a lateral membrane similar to that of the wild-type cells. When myosin-1c-knockdown cells are plated onto a Transwell insert cup at 100% confluent density (that of wild-type cells), they are forced to squeeze among themselves and create a lateral membrane. By using this protocol, we found that none of the canonical adhesion proteins (E-cadherin, α-catenin, β-catenin and p120-catenin) is affected by myosin-1c knockdown (Fig. 7A–D). However, myosin-1c has an indirect role in the stabilization and the accumulation of junctional proteins over time. We found that, upon confluency, the cellular levels of junctional proteins gradually increase to a steady-state mature level over several days [Fig. 7D, young, Y (1 day) and mature, M (7 days)]: E-cadherin from 0.5 to 1 (normalized), α-catenin from 0.6 to 1 (normalized), β-catenin from 0.6 to 1 (normalized), p120-catenin from 0.56 to 1 (normalized), synaptopodin from 0.45 to 1 (normalized) and myosin IIB from 0.35 to 1 (normalized). Knockdown of myosin-1c retards the accumulation of E-cadherin (from 1 to 0.75) and synaptopodin (1 to 0.7) over this maturation period (Fig. 7D). Interestingly, myosin-1c knockdown did not affect the accumulation of α-catenin and β-catenin during junction maturation, indicating that mechanism(s) other than binding to E-cadherin, such as binding to other cadherin(s) may be responsible. These findings support an indirect role for myosin-1c in the accumulation of adhesion proteins during epithelial maturation.
Fig. 7.

Myosin-1c knockdown abolishes junction recruitment of α-actinin-4 and weakens cell-cell cohesion. (A) OS-SIM images of myosin-1c (Myo1c), α-catenin, p120-catenin and E-cadherin at the apical plane of heterogeneous myosin-1c knockdown (KD) cell monolayers. Data are representative of 12 sets of images from one experiment out of three independent experiments. (B) OS-SIM images of myosin-1c, β-catenin, synaptopodin (synpo) and α-actinin-4 at the apical plane of heterogeneous myosin-1c knockdown cell monolayers. Data are representative of 12 sets of images from one experiment out of three different independent experiments. (C) Quantification of junctional myosin-1c, α-catenin, p120-catenin, E-cadherin, β-catenin, synaptopodin and α-actinin-4 immunofluorescence in wild-type (WT) and myosin-1c knockdown (Myo1cKD) cells. Bar graphs show the normalized mean±s.e.m. intensity of 24 junctions. P<0.001 for myosin-1c and α-actinin-4 between WT and myosin-1c KD. Data are representative of one experiment out of three independent experiments. (D) Western blots for myosin-1c, synaptopodin, E-cadherin (E-cad), α-catenin, β-catenin, p120-catenin, myosin IIB (myoIIB), myosin IIA (myoIIA), α-actinin-4 and vinculin in total cell lysates from mature (M, 7-day confluent) and young (Y, 1-day confluent) monolayers of wild-type (W) and myosin-1c knockdown (K) cells. Data are representative of one set of experiments out of four independent experiments. Red lines indicate the position of molecular mass markers at 135, 100 and 75 kDa; green lines indicate the position of molecular mass markers at 180, 135 and 100 kDa. (E) Maximum intensity projections from a 2.5 µm z-stack of 0.2 µm OS-SIM z-images showing immunofluorescence of myosin-1c, synaptopodin and α-actinin-4 in the mature cell monolayer with heterogeneous knockdown of myosin-1c (Myo1c KD). DNA (Hoechst 33528) is shown in merge image with α-actinin-4. Image set is representative of 12 image sets from one experiment out of six independent experiments. Scale bar: 10 µm. (F) Constrained iterative deconvolved microscopy showing immunofluorescence of myosin-1c, α-actinin-4 and β-catenin in a young monolayer with heterogeneous myosin-1c knockdown (Myo1c KD) that has been untreated (control) or subjected to cyclic tension [30 cycles per min (cpm)]. Orange arrows show unperturbed junctions that have myosin-1c at the junction; white arrows show broken junctions in myosin-1c-depleted cells. Image set is representative of eight image sets from one experiment out of four independent experiments. Scale bars: 10 µm.
In a mature cell monolayer that has heterogeneous knockdown of myosin-1c, cell–cell interactions between myosin-1c-knockdown cells often show spontaneous breakage (Fig. 7E). To directly test whether mysoin-1c knockdown affects cohesion between cells, we applied stress to cell–cell adhesions (Kannan and Tang, 2015). We found that mechanical force that usually does not disrupt wild-type cell junctions (∼20 nN per cell at 30 cycles per min for 10 min) causes a separation of the cell–cell interface at myosin-1c and α-actinin-4-negative junctions (Fig. 7F, white arrows). These observations support our hypothesis that myosin-1c is required for junction maturation and adhesion strengthening in epithelial cell monolayers.
Exogenous force rescues junction recruitment of α-actinin-4 in myosin-1c-knockdown cells
We have provided evidence that myosin-1c plays an essential role in tension generation at E-cadherin (Fig. 5) and supports tension-dependent recruitment of α-actinin-4 to the cell junction (Fig. 6). We have shown that myosin-1c is stably tethered on the lateral membrane during cellular contraction and experiences force at the cell–cell interface (Fig. 4). These findings are consistent with our hypothesis that myosin-1c provides a direct linkage between actin and the lateral membrane that is necessary for force transmission to the cell junction. If the function of myosin-1c is to promote tension generation at cell–cell adhesions, we should be able to rescue α-actinin-4 junction recruitment by applying force to the cell junction in myosin-1c-knockdown cells (Fig. 8A). However, myosin-1c-knockdown cells are prone to damage and this rescue experiment proved to be quite challenging. After empirically testing a series of force magnitudes and durations, we were able to find conditions that rescued the localization defect of α-actinin-4 in myosin-1c-knockdown cells. We found that a low level of pulsatile force (∼7 nN per cell at eight cycles per min for 90 min) was able to trigger a full recovery of α-actinin-4 at cell junctions that had almost no myosin-1c (Fig. 8B,C). These observations indicate that the molecular mechanism that support tension-dependent junction maturation is completely intact in myosin-1c-knockdown cells.
Fig. 8.
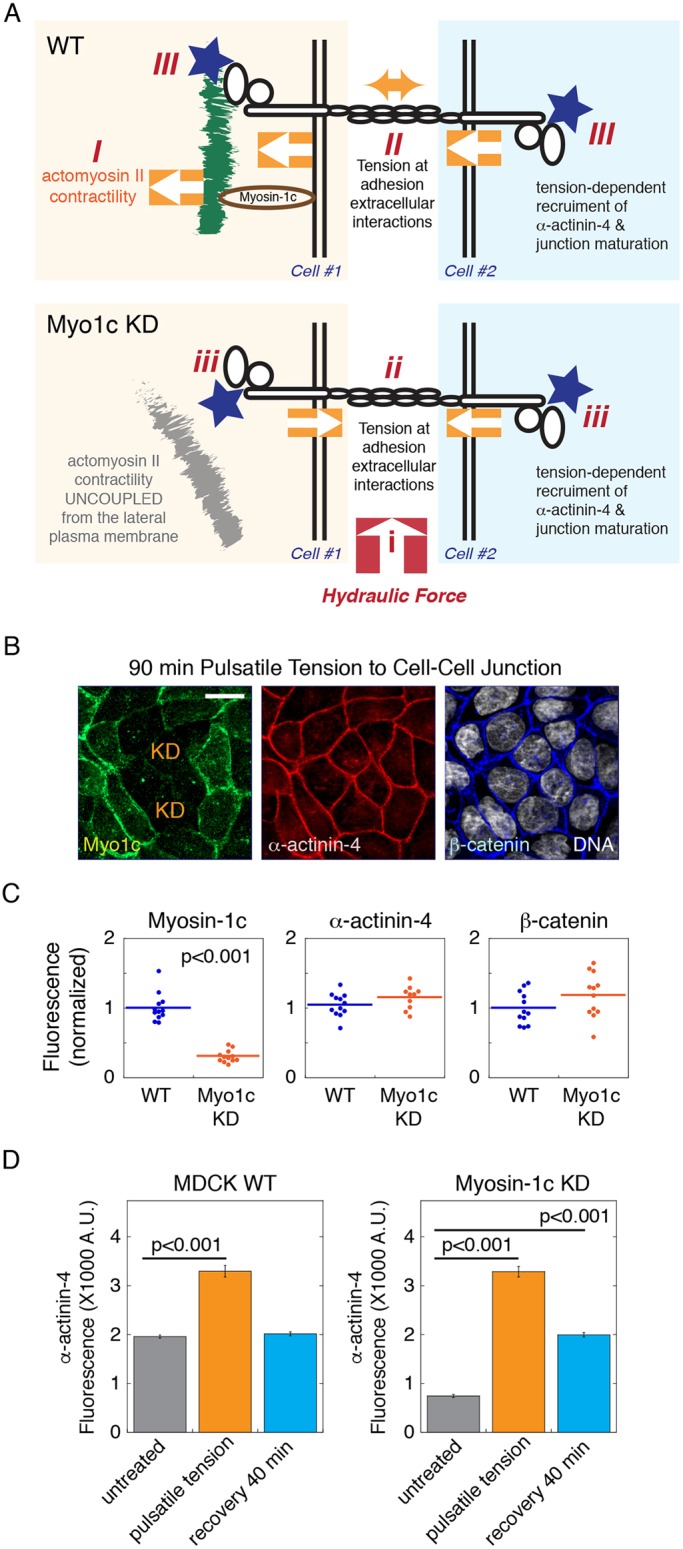
Exogenous force rescues junction recruitment of α-actinin-4 in myosin-1c-knockdown cells. (A) A diagram showing how myosin-1c facilitates force transmission from actomyosin contractility to the lateral membrane and promotes actin compaction onto cell–cell adhesion. (I) Whole-cell contraction from actomyosin II contractility pulls on cortical actin. (II) Force is transmitted from the lateral membrane to create tension at the extracellular domains of adhesion molecules. (III) Intercellular tension stimulates tension-dependent recruitment of α-actinin-4 to the cell junction. In the absence of myosin-1c (Myo1c KD), actomyosin II contractility is uncoupled from the lateral membrane. However, (i) application of hydraulic force can (ii) create tension at the extracellular domains of cell–cell adhesions to (iii) induce tension-dependent recruitment of α-actinin-4. (B) Maximum intensity projections from a 2.5 µm z-stack of 0.2 µm OS-SIM z-images showing myosin-1c, α-actinin-4 and β-catenin at the apical junction of monolayers with heterogeneous knockdown of myosin-1c (KD) subjected to 90 min of pulsatile tension. Data are representative of one experiment out of six independent experiments. Scale bar: 10 µm. (C) Quantification of junctional myosin-1c, α-actinin-4 and β-catenin immunofluorescence in wild-type (WT) and myosin-1c knockdown monolayers subjected to pulsatile tension. Horizontal lines represent mean fluorescence intensities. P<0.001 for myosin-1c between WT and myosin-1c KD. Data are representative measurements from one experiment out of six independent experiments. (D) Quantification (A.U., arbitrary units) of junctional α-actinin-4 immunofluorescence in wild-type and myosin-1c knockdown monolayers before (untreated) and immediately after pulsatile tension, and 40 min after recovery (recovery 40 min) from the 90 min pulsatile tension. Bar graphs show the mean±s.e.m. of 36 junctions. P<0.001 for α-actinin-4 between untreated cells and cells subjected to pulsatile tension (for both wild-type and myosin-1c-knockdown cells), and between untreated cells and cells after recovery for the myosin-1c-knockdown cells. Data are representative of one experiment out of three independent experiments.
To further characterize the α-actinin-4 tension response in myosin-1c-knockdown cells, we used mature myosin-1c-knockdown monolayers (7 days) that had gradually accumulated α-actinin-4 at the junction although at a much lower level (∼30%) than in the wild-type cells (Fig. 8D). Under these conditions, myosin-1c-knockdown cell monolayers can withstand slightly higher mechanical stress than cells with almost no myosin-1c (Fig. 8B). We found that application of pulsatile force (∼10 nN per cell at 20 cycles per min for 60 min) to wild-type and myosin-1c-knockdown monolayers elicited a similar α-actinin-4 tension response (Fig. 8D). Interestingly, upon tension release, the level of α-actinin-4 returns to a pre-tension level in wild-type junctions whereas the level of α-actinin-4 returns to a higher level in myosin-1c knockdown junctions, similar to the level at WT cell junctions (Fig. 8D). These results demonstrate that the recruitment and retention of junctional α-actinin-4 does not require the presence of myosin-1c on the lateral membrane. Thus, the dependency of α-actinin-4 on myosin-1c is purely biophysical in nature, which is supported by the force transmission function of myosin-1c.
Myosin-1c is recruited to the lateral membrane in a tension-sensitive manner
We have noticed in one of the above experiments that myosin-1c responded to mechanical force to accumulate at the cell junction (Fig. 7F). To further investigate the tension response of myosin-1c, we applied a low level of cyclic force (∼7 nN per cell at 20 cycles per min for 60 min) to young monolayers (Fig. S5A). Quantification of z-images shows that myosin-1c and α-actinin-4 were strongly induced at the apical and lateral junctions (Fig. S5B,C) whereas β-catenin was only weakly affected (Fig. S5D). Importantly, myosin-1c was upregulated to a greater extent at the lateral junction than at the apical junction (Fig. S5B, 2–6 µm z-focal plane). By contrast, α-actinin-4 was upregulated to a greater extent at the apical junction than at the lateral junctions (Fig. S5C, 1 µm z-focal plane).
To determine whether constant tension is required to maintain the accumulation of myosin-1c and α-actinin-4 at the junction, we applied cyclic force (∼7 nN per cell at 20 cycles per min for 60 min) to cell monolayers and measured their levels over a recovery period of 40 min (Fig. S5E–G). We found that both myosin-1c and α-actinin-4 were strongly induced at the apical junction (z=1 µm) immediately after force application but their levels were gradually reset to new steady-state levels (Fig. S5E,F). Whereas the lag time for myosin-1c to reach the new steady-state level is ∼15 min, the lag time for α-actinin-4 to reach the new steady-state level is ∼30 min. For a control, we measured the level of β-catenin at the apical junction (z=1 µm) and found that it was unchanged throughout (Fig. S5G). These observations indicate that myosin-1c and α-actinin-4 can respond to the same level of force but do not behave as a complex on the lateral membrane.
Tension-induced junctional myosin-1c is independent of synaptopodin and α-actinin-4
To further investigate the tension responses of myosin-1c, synaptopodin and α-actinin-4, we compared their junctional levels after application of cyclic force (∼10 nN per cell at 20 cycles per min for 60 min) (Fig. S6A–D). We found that myosin-1c strongly correlated with synaptopodin and α-actinin-4 at the apical junction after force application (Fig. S6A, white arrows). By contrast, myosin-1c showed no correlation with β-catenin levels (Fig. S6B–D). These results indicate that myosin-1c, synaptopodin and α-actinin-4 can respond to the same level of force.
To determine whether the tension response of myosin-1c is dependent on synaptopodin and α-actinin-4, we compared junctional myosin-1c before and after application of cyclic force (∼10 nN per cell at 20 cycles per min for 60 min) in synaptopodin- and α-actinin-4-knockdown cells. We found that junctional myosin-1c and its tension response are completely unaffected by synaptopodin and α-actinin-4 knockdown (Fig. S6E–H). Moreover, in mature junctions (7 days) with a steady-state level of synaptopodin, a high level of cyclic force (∼50 nN per cell at 30 cycles per min for 60 min) can induce additional myosin-1c recruitment to the junction without eliciting a synaptopodin tension response (Fig. S6I,J). Therefore, the mechanotransduction mechanism that regulates myosin-1c continues to exist after junction maturation.
Actin-binding function of myosin-1c is required for membrane integrity, epithelial cohesion and α-actinin-4 recruitment
Myosin-1c mutants that are ATPase-defective (Tokuo and Coluccio, 2013) and a motorless truncation of myosin-1c (Maravillas-Montero et al., 2011) have previously been used to study its actin-binding and motility function. Here, we generated cell lines expressing these myosin-1c mutants: EGFP–myosin-1c-R162A, which has defective actin binding (Shimada et al., 1997; Furch et al., 1999; Li et al., 1998), EGFP–myosin-1c-G389A, which has weak actin binding (Kambara et al., 1999; Sasaki et al., 1998), and EGFP–myosin-1c motorless, which has the motor domain deleted. We found that expression of EGFP–myosin-1c-R162A and the motorless EGFP–myosin-1c mutant dramatically disrupted the cell–cell interface, causing formation of large blebs on the lateral membrane (Fig. S7A, orange circles). These membrane blebs are reminiscent of the membrane blebs seen in myosin-1c-knockdown cells (Movies 6–8). Live-cell imaging of cells expressing wild-type EGFP–myosin-1c and EGFP–myosin-1c-G389A using wide-field microscopy (Movies 9,10) and OS-SIM (Movies 11,12) revealed dynamic and sporadic blebbing at the cell boundary of EGFP–myosin-1c-G389A-expressing cells but not the wild-type EGFP–myosin-1c-expressing cells (Fig. S7B, orange circles). These findings indicate that the actin-binding function of myosin-1c plays an essential role in the overall integrity and stability of the lateral cell-cell adhesion interface in epithelial monolayers.
To assess whether cell–cell adhesion is affected by myosin-1c mutants, we applied mechanical force to cell monolayers expressing wild-type myosin-1c (WT Myo1c) or myosin-1c mutants (R162A, G389A, Motorless tail) (Fig. S8A). We found that a moderate level of cyclic force (∼30 nN per cell at 20 cycles per min for 5 min) that usually does not affect wild-type junctions can induce large holes in mutant-expressing monolayers (Fig. S8A, yellow asterisks). The motorless myosin-1c mutant consistently showed the most striking disruption of membrane integrity and monolayer cohesion (Figs S7A, S8A). Expression of the motorless EGFP–myosin-1c mutant reduced the accumulation of α-actinin-4 and synaptopodin at the cell junction (Fig. S8B,C). These results are consistent with the phenotypes seen in myosin-1c-knockdown cells (Figs 6,7), indicating that the motorless tail mutant can act as a dominant negative to endogenous myosin-1c.
In summary, we have identified myosin-1c as a membrane-associated protein that localizes in close proximity to the cell–cell adhesion complex to support the generation of E-cadherin tension and tension-sensitive recruitment of α-actinin-4 to the epithelial cell junction.
DISCUSSION
Actomyosin II contractility plays an essential role in the development of cell–cell contacts (Bajpai et al., 2008; Kannan and Tang, 2015; Le Duc et al., 2010; Barry et al., 2014; Thomas et al., 2013; Liu et al., 2010; Rozbicki et al., 2015; Delanoe-Ayari et al., 2004). A repertoire of molecular mechanisms involved in this process has been characterized, including force-induced actin loading onto α-catenin (Buckley et al., 2014), force-induced conformational change in α-catenin (Yonemura et al., 2010), force-induced binding of vinculin to α-catenin (Le Duc et al., 2010), and force-induced accumulation of α-actinin-4 and synaptopodin at the cell junction (Kannan and Tang, 2015). Here, we have discovered that the membrane-associated actin-binding motor myosin-1c is an upstream regulator of α-actinin-4.
We propose that global contraction of the actomyosin II cytoskeleton results in an inward movement of the lateral membrane that is coupled to actin by myosin-1c. The inward movement of the lateral membrane causes cell–cell adhesions to pull onto each other and mechanically stimulates tension-dependent changes at the cell junction. We showed that myosin-1c is tethered on the lateral cell membrane during whole-cell contraction, experiences tension on the lateral cell membrane and is required for pulling the lateral cell membrane inwards during whole-cell contraction. We showed that myosin-1c promotes the association of actin with the E-cadherin complex, facilitates tension generation at E-cadherin and is required for the recruitment of tension-dependent α-actinin-4 to the cell junction. Our current study supports a novel mechanism of tension regulation at cell–cell adhesion and implicates a non-junctional component in the development of the epithelial junction.
In our previous study, we found that synaptopodin knockdown compromised junction accumulation of α-actinin-4 (Kannan and Tang, 2015). Here, we show that myosin-1c knockdown also compromised the accumulation α-actinin-4 at the cell junction. Because the recruitment of α-actinin-4 is a tension-dependent process, there are at least two possible mechanisms that can prevent its localization to the cell junction: (1) disruption of α-actinin-4 binding to its junctional receptor, or (2) disruption of the force transmission pathway at cell–cell adhesion. To distinguish between these two possibilities, we applied exogenous force to cell–cell adhesions in synaptopodin-knockdown cell monolayers (Kannan and Tang, 2015). We found that, in the absence of synaptopodin, application of force was unable to rescue α-actinin-4 recruitment, suggesting that synaptopodin is necessary for targeting α-actinin-4 to the junction (Kannan and Tang, 2015). Here, we show that application of force to cell–cell adhesion in myosin-1c-depleted cells fully rescued α-actinin-4 localization, indicating that the mechanism for α-actinin-4 recruitment is intact in myosin-1c-knockdown cells. Once α-actinin-4 is targeted to the junction, its accumulation is maintained independently of myosin-1c. Thus, the dependency of α-actinin-4 on myosin-1c is indirect and strictly due to the force transmission function of myosin-1c.
In this study, we have discovered that myosin-1c can respond to exogenously applied force and accumulates at the cell–cell adhesion interface. We found that tension-induced myosin-1c correlates with the accumulation of junctional synaptopodin and α-actinin-4. However, knockdown of synaptopodin or α-actinin-4 does not affect myosin-1c localization nor its tension response. In addition, tension-induced myosin-1c accumulation is most prominent on the lateral membrane whereas tension-induced α-actinin-4 accumulation is most prominent at the apical junction. These findings indicate that myosin-1c is regulated differently from synaptopodin and α-actinin-4. Since myosin-1c localization on the lateral membrane requires its phosphoinositide-binding site (Tokuo and Coluccio, 2013), a force-sensitive mechanism may exist to facilitate myosin-1c targeting via the regulation of phosphoinositide levels. We have previously shown that mechanical force applied to cell–cell adhesion can induce Akt phosphorylation (Kannan and Tang, 2015), which is a downstream effector of phosphoinositide 3-kinase (PI3K) signaling. Therefore, a mechanotransduction mechanism that stimulates the production of phosphoinositides on the lateral membrane would be able to promote myosin-1c accumulation. Future experiments to study tension regulation of lipid pathways will provide a better mechanistic understanding of myosin-1c tension response on the lateral membrane.
After junction maturation, myosin-1c remains sensitive to mechanical force and continues to show strong localization to the lateral cell membrane. Therefore, myosin-1c is likely to have important functions in force-dependent processes in mature epithelial cell sheets. By linking the force-generating actomyosin II cytoskeleton to the plasma membrane, myosin-1c positions itself directly in the force transmission pathway. In addition, myosin-1c has been shown to participate in force production (Pyrpassopoulos et al., 2012, 2016). Thus, myosin-1c is an ideal candidate for force modulation. Myosin-1c interacts with actin in a force-sensitive manner and would decrease its detachment rate under load (Greenberg et al., 2012). Consequently, myosin-1c can hold onto an actin filament longer if the actin filament is pulled. Myosin-1 has been shown to promote the generation of resting membrane tension (Dai et al., 1999; Nambiar et al., 2009; Venit et al., 2016) and facilitate actin compression on the plasma membrane (Sokac et al., 2006; Kittelberger et al., 2016). Moreover, the interaction of myosin-1c with actin can be fine-tuned by intracellular Ca2+ (Lieto-Trivedi and Coluccio, 2008; Lu et al., 2015; Manceva et al., 2007; Phillips et al., 2006; Lewis et al., 2012). By regulating myosin-1c through calcium, the biophysical characteristics of force, such as the magnitude, duration, or wave-form, may be adjusted. In conclusion, myosin-1c is likely to serve multiple functions by acting as a linker molecule that possesses the ability to transmit and modulate force on the lateral cell-cell adhesion interface.
MATERIALS AND METHODS
Antibodies and reagents
Rabbit polyclonal antibodies against synaptopodin were raised against a synthetic peptide corresponding to amino acids (aa) 899–918 of human synaptopodin (SPRAKQAPRPSFSTRNAGIE) that had been coupled to KLH (Pacific Immunology). Rabbit polyclonal antibodies against α-actinin-4 were raised in house against a synthetic peptide corresponding to aa 8–24 of human α-actinin-4 (NQSYQYGPSSAGNGAGC) that had been coupled to KLH. Antibodies against myosin-1c (sc-136544, mouse), α-actinin-4 (sc-49333, goat), β-catenin (sc-7963, mouse and sc-7199, rabbit), synaptopodin (sc-50459, rabbit, sc-21536 and sc-21537, goat), α-catenin (sc-1495, goat and sc-9988, mouse), p120 (sc-13957, rabbit), vinculin (sc-5573, rabbit), Myh9 (sc-98978, rabbit) and Myh10 (sc-47204, goat) were purchased from Santa Cruz Biotechnology. Antibodies against myosin-1c (ab94539, rabbit) was purchased from Abcam. Antibodies against E-cadherin (24E1, rabbit) were purchased from Cell Signaling Technology, Inc. Antibodies against phospho-myosin light chain 2 (Thr18/Ser19) were purchased from Cell Signaling Technology (#3674, rabbit). Secondary antibodies were purchased from Bio-Rad Laboratories [horseradish peroxidase (HRP)-conjugated goat anti-rabbit-IgG], Santa Cruz Biotechnology (HRP-conjugated rabbit anti-goat-IgG and goat anti-mouse-IgG), Life Technologies/Invitrogen (FITC and Cy3 goat anti-mouse-IgG, FITC and Cy3 goat anti-rabbit-IgG, Alexa Fluor 488-conjugated donkey anti-mouse, Alexa Fluor 568-conjugated donkey anti-rabbit-IgG, Alexa Fluor 647 donkey anti-goat-IgG). Alexa Fluor 647–phalloidin, was purchased from Life Technologies/Invitrogen. Leupeptin, Pefabloc, E-64, antipain, aprotinin, bestatin, and calpain inhibitors I and II were purchased from A.G. Scientific, Inc. Phosphatase inhibitor cocktail (Simple Stop 1 #GB-450) was purchased from Gold Biotechnology. ROCK inhibitor Y-27632 (#10005583) was purchased form Cayman Chemical, Inc.
DNA constructs
shRNAs for myosin-1c (5′-AACTTCACCAGTGAGGCAGCCTTCATTGA-3′ and 5′-AATGACAAGAGTGACTGGAAGGTCGTCAG-3′) were synthesized and subcloned into blasticidin-selectable pGFP-B-RS vector (Origene). Knockdowns of α-actinin-4 and synaptopodin were performed as published (Kannan and Tang, 2015). Briefly, shRNAs for α-actinin-4 (5′-AGGTCCTGTTCCTCTGACTCGGTATCTAT-3′ and 5′-ACACAGATAGAGAACATCGACGAGGACTT-3′) were synthesized and subcloned into blasticidin-selectable pRS vector and shRNA for canine synaptopodin (5′-GAGGTGAGATCCAGCACACTTCTGATTGA-3′) was synthesized and subcloned into puromycin-selectable pRS vectors (Origene). Lyn-D3cpV cDNA was kindly provided by Amy Palmer and Roger Tsien (Departments of Chemistry and Biochemistry, HHMI and the University of California, San Diego, La Jolla, CA 92093, USA) (Palmer et al., 2006). Venus–α-actinin cDNA (this construct uses the α-actinin-1 isoform) was kindly provided by Fanjie Meng and Frederick Sachs (Department of Physiology and Biophysics, State University of New York, Buffalo, NY 14214, USA) (Meng and Sachs, 2011). pcDNA3 EGFP–myosin-1c constructs (EGFP–myosin-1c wild-type full length, EGFP–myosin-1c R162A, EFGP-myosin-1c G389A, and EFGP–myosin-1c motorless tail were kindly provided by Hiroshi Tokuo and Lynne Coluccio (Department of Physiology and Biophysics, Boston University of Medicine, Boston, MA 02118, USA) (Tokuo and Coluccio, 2013).
The E-cadherin tension sensor was constructed by inserting the spectrin-repeat FRET module consists of a spectrin-repeat flanked by CeFP and VFP (sequence available from corresponding author upon request) between aa 699 and 700 of human E-cadherin (P12830.3). The sequence was constructed and cloned into pcDNA3.1(+)_myc_HisA by Genscript. The E-cadherin tension sensor without the catenin-binding site (deleted C-terminal 90 aa) was constructed and cloned into pcDNA3.1(+)_myc_HisA by Genscript.
The myosin-1c tension sensor was constructed by inserting a force sensor FRET module [a spectrin repeat flanked by CeFP and VFP between aa 863 and 864 of human myosin-1c isoform a (NP_001074248)]. The sequence was synthesized and cloned into pcDNA3.1(+)_myc_HisA using XhoI/XbaI cloning sites by Genscript.
Cell culture and transfection
Madin–Darby canine kidney (MDCK) cells were originally from Kai Simons laboratory (EMBL, Germany) and a gift from Barry Gumbiner (University of Washington, Seattle, WA). The cells were authenticated by staining of E-cadherin and ZO-1 using antibodies that only recognize the canine proteins, RR1 for E-cadherin and R40.76 for ZO-1. The cells are free from mycoplasma contamination as determined by the original source. MDCK cells were maintained in MEM/Earle's balanced salt solution supplemented with 25 mM HEPES and 5% fetal bovine serum. For transfection, cells were incubated in Opti-MEM (Invitrogen) with a 1:1 mixture of DNA/polyethyleimine and selected for 10 days using G418, puromycin or blasticidin. Clonal cell lines of α-actinin-4 and synaptopodin knockdown were obtained as published previously (Kannan and Tang, 2015). Briefly, antibiotic resistant clonal cell lines were expanded and assessed for knockdown efficiency by western blotting and immunofluorescence. Clonal cell lines with homogeneous knockdown phenotype were used for a second round of transfection with shRNA. Secondary clonal cell lines were expanded and assessed for knockdown efficiency by western blotting and immunofluorescence. Clonal cell lines with a knockdown efficiency of >70% were used for western blotting and live-cell imaging studies.
For analysis of a mixed population of myosin-1c knockdown, antibiotic-resistant cells were pooled and used for experiments at 14–21 days post-transfection. For analysis of a homogenous population of myosin-1c knockdown cells, three clonal cell lines were expanded and assessed for knockdown efficiency by western blotting and immunofluorescence.
For live-cell imaging of Venus–α-actinin (G418 selection), clonal cell lines were expanded and assessed as described previously (Kannan and Tang, 2015). For myosin-1c knockdown in Venus–α-actinin-expressing cells, Venus–α-actinin cells were transfected with shRNA for myosin-1c and selected using blasticidin. Three clonal myosin-1c-knockdown cell lines were expanded and assessed for knockdown efficiency by western blotting and immunofluorescence. For live-cell imaging of EGFP–myosin-1c, two clonal cell lines expressing EGFP–myosin-1c constructs (EGFP–myosin-1c wild-type, EGFP–myosin-1c R162A, EFGP–myosin-1c G389A and EFGP–myosin-1c motorless tail) were expanded and assessed for expression efficiency by live-cell microscopy. For live-cell imaging of Lyn-D3cpV, clonal cell lines were expanded and assessed for expression efficiency by live-cell microscopy. For E-cadherin tension sensor and myosin-1c tension sensor expressions, transfected cells were treated with G418, and mixed cultures and two clonal cell lines were expanded and used for experiments.
Western blotting
For comparison of young and mature monolayers, confluent monolayers of wild-type and myosin-1c MDCK cells were trypsinized and re-plated at high confluent density (107 cells per 10 cm). Cells were allowed to form cell–cell interactions for 2 days (young) or 7 days (mature). Total cell lysates were obtained by solubilizing the cells in SDS-PAGE sample buffer containing 25 mM dithiothreitol, 2% SDS, 50 mM Tris-HCl, 5% glycerol, pH 8.8 and protease inhibitors (10 µg/ml Leupeptin, 1 mg/ml Pefabloc, 10 µg/ml E-64, 2 µg/ml antipain, 2 µg/ml aprotinin, 50 µg/ml bestatin, 20 µg/ml calpain inhibitors I and 10 µg/ml calpain inhibitor II). The Bio-Rad DC detergent-compatible protein assay was used to determine the total protein concentration in cell lysates. Equal protein amounts of cell lysates were used for comparison of junctional protein by western blotting using 0.1 µg/ml primary antibodies.
For western blotting of phosphorylated myosin light chain, cell extraction was carried out as above with the addition of phosphatase inhibitor cocktails (Simple Stop 1).
Active Rho detection
Rho activity was assayed using the Active Rho Detection Kit (Cell Signaling Technology #8820). Briefly, confluent monolayers of MDCK and myosin-1c-knockdown cells were lysed and the supernatants were used for immunoprecipitation using the GST-tagged Rhotekin Rho-binding domain. The immunoprecipitated protein complexes were eluted in SDS sample buffer and analyzed on a SDS-PAGE gel. Rho levels in the immunoprecipitates were detected by performing a western blot using anti-Rho antibodies.
Force application in cell monolayer
Cells grown on Transwell-Clear (Corning) were used in tension experiments as described previously (Kannan and Tang, 2015). Briefly, Transwell filter cups were mounted onto a custom-made pressure chamber instrument. Hydraulic pressure was applied to the basal compartment by pumping medium into the basal chamber. Pressure was monitored through an outlet from the basal pressure chamber to a pressure gauge. An apical adaptor is mounted onto the apical chamber of the Transwell cup that is filled with cell culture medium and sealed with an O-ring. An outlet from the apical adaptor is either exposed to ambient pressure or connected via a bifurcation to a pressure gauge and a syringe hooked up to a syringe pump. Cyclic or pulsatile pressures were applied to cell monolayers by means of a programmable infuse/withdrawal syringe pump (Lagato SPLG270). All experiments were performed in a 37°C room.
Immunofluorescence of cells
Cells grown on Transwell-Clear (Corning) were used in localization studies. For immunofluorescence, cells were rinsed twice in 20 mM HEPES pH 7.8, 150 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, and fixed in 1% formaldehyde in the same buffer at 4°C for 2 h. The reaction was quenched with Tris-HCl in quenching buffer (20 mM HEPES pH 8.0, 0.05% Triton X-100, 50 mM Tris-HCl, 100 mM NaCl) for 3 h. After rinsing in staining buffer (20 mM HEPES, pH 7.5, 0.01% Triton X-100, 100 mM NaCl), the cells were incubated with primary antibodies (1 µg/ml) in staining buffer overnight. After rinsing in staining buffer three times, the cells were incubated in secondary antibodies in staining buffer for 90 min. Then, the cells were rinsed three times in staining buffer and incubated with fluorescently labeled phalloidin or Hoechst 33528 for 60 min. Finally, the cells were rinsed three times in staining buffer and post-stain fixed with 1% formaldehyde in staining buffer for 60 min. Transwell filters were excised using a razor blade and mounted on glass slides with ProLong Diamond antifade (Invitrogen).
Image acquisition of fixed cells
For Figs 2, 3, 6, 7, 8, and Figs S2, S3, S4, S8, images were collected in 200-nm steps using an Axio Imager.Z2m microscope equipped with Apotome.2 (Zeiss) and X-cite 120 LED (Lumen Dynamics). For optical sectioning structured illumination microscopy (OS-SIM), seven phases/images were collected for each constructed image using an alpha Plan-Apochromat 100×/1.46 oil DIC M27 objective (Zeiss), Apotome.2 and a 4K ORCA-Flash4.0 V2 digital CMOS camera (ORCA-ER; Hamamatsu Photonics). Wide-field optical z-images were deconvolved using the Zen2 pro deconvolution module (nearest neighbor, fast iterative, or constrained iterative algorithms as stated in figure legends). For Fig. S8A, low-magnification wide-field images were collected using a Plan Apochromat 20×/0.8 objective (Zeiss) and a 2K Optimos digital cmos camera (Qimaging).
For Figs S5 and S6, images were collected in 200-nm steps using an inverted microscope (IX-71; Olympus), a 1K charge-coupled device camera (Cool SNAp HQ, Applied Precision), a 60×/1.42 NA oil objective with a 1.6× auxiliary magnification and SoftWorx DMS software (Applied Precision). Wide-field optical z-images were deconvolved using the enhanced ratio constrained iterative deconvolution algorithm with ten iteration cycles (Applied Precision).
Live-cell imaging setup
For live-cell imaging, glass coverslips were soaked in 100% ethanol and sterilized under UV for 60 min. Sterilized coverslips were coated with 20 μg/ml collagen IV in phosphate-buffered saline for 60 min and used immediately for plating of cells. Cells grown on collagen-coated glass coverslips were mounted upside down onto an in-house fabricated glass slide chamber. The chamber was assembled by attaching a 1.5×1.5 cm medical-grade silicone gel (PediFix) onto a sterilized glass slide. The center of the silicone gel was excised and used as a sink for media during imaging. Live-cell imaging was performed in FluoroBrite/DMEM (Gibco) containing 1% fetal bovine serum and 10 mM HEPES, pH 7.5. Sample temperature was maintained at 35°C on a heated stage and with an objective heater (PeCon) mounted onto the Axio Imager.Z2m microscope.
Live-cell imaging of Venus–α-actinin, EGFP–myosin-1c and Lyn-D3cpV
For live-cell wide-field imaging of Venus–α-actinin and EGFP–myosin-1c, images were collected using a FLUAR 40×/1.3 NA oil objective (Zeiss) and the ORCA-Flash4.0 V2 camera. For live-cell OS-SIM, five phases/images were collected for each constructed image using an alpha Plan-Apochromat 100×/1.46 NA oil DIC M27 objective (Zeiss), Apotome.2 (Zeiss), and a 2K Optimos camera (Qimaging) mounted onto an Axio Imager.Z2m microscope (Zeiss) equipped with X-cite 120 LED light source (Lumen Dynamics).
Intracellular Ca2+ was monitored using a lipid-modifiable Ca2+ sensor protein, Lyn-D3cpV (CFP–cpVenus FRET pair, cpVenus is cyclic-permutated Venus). Changes in Ca2+ concentration were monitored by excited emission of Lyn-D3cpV (excitation of CFP and emission of cpVenus) and cpVenus (excitation of cpVenus and emission of cpVenus with YFP filter). For Fig. 1G and Movie 3, images were collected with a FLUAR 40×/1.3 NA oil objective (Zeiss) using a Axio Imager.Z2m microscope (Zeiss) equipped with X-cite 120 LED light source (Lumen Dynamics).
Live-cell imaging of FRET-based tension sensors
E-cadherin and myosin-1c tension sensors were imaged using the Gemini dual-view system (Hamamatsu Photonics) equipped with an excitation filter for Cerulean fluorescent protein (CeFP) and emission filters for Cerulean fluorescent protein (CeFP) and Venus fluorescent protein (VFP). Simultaneous acquisition of images for CeFP and VFP emission were obtained using a Plan-Apochromat 40×/1.3 NA oil DIC M27 objective (Zeiss) mounted onto an Axio Imager.Z2m microscope (Zeiss) equipped with a large format c-MOS ORCA-Flash4.0 V2 camera (ORCA-ER; Hamamatsu Photonics). For treatment with ROCK inhibitor Y-27632 (Cayman Chemicals), cells were incubated in 10 µM of Y-27632 (diluted from 1000× stock in DMSO) for 2 h prior to image acquisition.
Junctional intensities of CeFP and VFP is used to calculate the FRET index (EmVFP)/(EmCeFP+EmVFP), which is shown as FRET/(CeFP+FRET). Briefly, the CeFP and VFP channels were overlaid on top of each other using a macro written within the Zen2 software (Zeiss). Each junctional region is outlined manually with a freehand drawing tool in the Zen2 imaging tool (Zeiss). The intensities of the junctional signal were measured and subtracted from the background signal (an area with no cells) before being used for the calculation of the FRET index.
Image analysis
All images were corrected for chromatic shift on the x, y and z-axes for each fluorescence channel before being used for analysis. Quantification of immunofluorescence intensity was performed in ImageJ. All measured intensities were subtracted from background signal (an area with no cells) before being used for statistical analyses and calculation of intensity ratios. Each junctional region is outlined manually with a freehand drawing tool. The mean pixel intensity of each defined junctional region is used for comparison of junction localization of individual junctional protein. For calculation of the Pearson's correlation coefficient R, intensities of individual pixel within the defined junctional region were used and each pixel corresponds to 45×45 nm of the imaged sample. Line intensity graphs were generated in Excel (Microsoft) using pixel intensities from original images.
For measurement of cell width, a line tool was use to draw lines from one side of the cell to the other. The position of the line was determined empirically by moving and rotating the line 360° so that the line is as perpendicular to the junctions as possible.
Image processing
For figure generation, images were cropped, contrasted and scaled using Photoshop software (Adobe) before being importing into Illustrator (Adobe). For movie generation, individual images of cropped cells were imported into QuickTime (Apple) to generate movie files. Composite images were generated using ImageJ (NIH) or Photoshop (Adobe).
Data analysis and statistics
All experiments were repeated at least three times. At least six data sets from each experiment were collected. All P-values were calculated using non-paired Student t-tests. Linear regression lines were fitted using original data points. All analyses were performed using KaleidaGraph software (Synergy).
Supplementary Material
Acknowledgements
We thank Lydia Lee for propagation of cell lines, Kevin Huang for western blotting and Nilmani Singh for DNA work.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: V.W.T.; Methodology: N.K., V.W.T.; Software: N.K.; Validation: N.K., V.W.T.; Formal analysis: V.W.T.; Investigation: N.K.; Resources: N.K.; Data curation: N.K.; Writing - original draft: V.W.T.; Visualization: V.W.T.; Supervision: V.W.T.; Funding acquisition: V.W.T.
Funding
Funding was provided by the National Institutes of Health (NIH) (R01 DK098398) to V.W.T. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.211334.supplemental
References
- Adams C. L., Nelson W. J. and Smith S. J. (1996). Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 135, 1899-1911. 10.1083/jcb.135.6.1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif E., Wagner M. C., Johnstone D. B., Wong H. N., George B., Pruthi P. A., Lazzara M. J. and Nihalani D. (2011). Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol. Cell. Biol. 31, 2134-2150. 10.1128/MCB.05051-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai S., Correia J., Feng Y., Figueiredo J., Sun S. X., Longmore G. D., Suriano G. and Wirtz D. (2008). α-Catenin mediates initial E-cadherin-dependent cell-cell recognition and subsequent bond strengthening. Proc. Natl. Acad. Sci. USA 105, 18331-18336. 10.1073/pnas.0806783105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry A. K., Tabdili H., Muhamed I., Wu J., Shashikanth N., Gomez G. A., Yap A. S., Gottardi C. J., De Rooij J., Wang N. et al. (2014). alpha-catenin cytomechanics–role in cadherin-dependent adhesion and mechanotransduction. J. Cell Sci. 127, 1779-1791. 10.1242/jcs.139014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begnaud S., Chen T., Delacour D., Mège R.-M. and Ladoux B. (2016). Mechanics of epithelial tissues during gap closure. Curr. Opin. Cell Biol. 42, 52-62. 10.1016/j.ceb.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas K. H., Hartman K. L., Yu C.-, Harrison O. J., Song H., Smith A. W., Huang W. Y. C., Lin W.-C., Guo Z., Padmanabhan A. et al. (2015). E-cadherin junction formation involves an active kinetic nucleation process. Proc. Natl. Acad. Sci. USA 112, 10932-10937. 10.1073/pnas.1513775112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois P. R. J., Borgon R. A., Vonrhein C. and Izard T. (2005). Structural dynamics of alpha-actinin-vinculin interactions. Mol. Cell. Biol. 25, 6112-6122. 10.1128/MCB.25.14.6112-6122.2005 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Bois P. R. J., O'Hara B. P., Nietlispach D., Kirkpatrick J. and Izard T. (2006). The vinculin binding sites of talin and alpha-actinin are sufficient to activate vinculin. J. Biol. Chem. 281, 7228-7236. 10.1074/jbc.M510397200 [DOI] [PubMed] [Google Scholar]
- Borghi N., Sorokina M., Shcherbakova O. G., Weis W. I., Pruitt B. L., Nelson W. J. and Dunn A. R. (2012). E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 109, 12568-12573. 10.1073/pnas.1204390109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley C. D., Tan J., Anderson K. L., Hanein D., Volkmann N., Weis W. I., Nelson W. J. and Dunn A. R. (2014). Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 346, 1254211 10.1126/science.1254211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby L. M. and Waters C. M. (2010). Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L715-L731. 10.1152/ajplung.00361.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J., Ping Ting-Beall H., Hochmuth R. M., Sheetz M. P. and Titus M. A. (1999). Myosin I contributes to the generation of resting cortical tension. Biophys. J. 77, 1168-1176. 10.1016/S0006-3495(99)76968-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delanoe-Ayari H., Al Kurdi R., Vallade M., Gulino-Debrac D. and Riveline D. (2004). Membrane and acto-myosin tension promote clustering of adhesion proteins. Proc. Natl. Acad. Sci. USA 101, 2229-2234. 10.1073/pnas.0304297101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn D. and Franz H. (1986). Identification of actin-, alpha-actinin-, and vinculin-containing plaques at the lateral membrane of epithelial cells. J. Cell Biol. 102, 1843-1852. 10.1083/jcb.102.5.1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furch M., Fujita-Becker S., Geeves M. A., Holmes K. C. and Manstein D. J. (1999). Role of the salt-bridge between switch-1 and switch-2 of Dictyostelium myosin. J. Mol. Biol. 290, 797-809. 10.1006/jmbi.1999.2921 [DOI] [PubMed] [Google Scholar]
- Gohy S. T., Hupin C., Pilette C. and Ladjemi M. Z. (2016). Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin. Exp. Allergy 46, 529-542. 10.1111/cea.12712 [DOI] [PubMed] [Google Scholar]
- Greenberg M. J., Lin T., Goldman Y. E., Shuman H. and Ostap E. M. (2012). Myosin IC generates power over a range of loads via a new tension-sensing mechanism. Proc. Natl. Acad. Sci. USA 109, E2433-E2440. 10.1073/pnas.1207811109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokanson D. E. and Ostap E. M. (2006). Myo1c binds tightly and specifically to phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate. Proc. Natl. Acad. Sci. USA 103, 3118-3123. 10.1073/pnas.0505685103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokanson D. E., Laakso J. M., Lin T., Sept D., Ostap E. M. and Parent C. (2006). Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol. Biol. Cell 17, 4856-4865. 10.1091/mbc.e06-05-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys B. D. (2014). Kidney injury, stem cells and regeneration. Curr. Opin Nephrol. Hypertens. 23, 25-31. 10.1097/01.mnh.0000437332.31418.e0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N., Tanaka N., Abe K., Yang Y. J., Abbas Y. M., Umitsu M., Nagar B., Bueler S. A., Rubinstein J. L., Takeichi M. et al. (2013). An autoinhibited structure of alpha-catenin and its implications for vinculin recruitment to adherens junctions. J. Biol. Chem. 288, 15913-15925. 10.1074/jbc.M113.453928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. P. and Craig S. W. (1995). F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature 373, 261-264. 10.1038/373261a0 [DOI] [PubMed] [Google Scholar]
- Kambara T., Rhodes T. E., Ikebe R., Yamada M., White H. D. and Ikebe M. (1999). Functional significance of the conserved residues in the flexible hinge region of the myosin motor domain. J. Biol. Chem. 274, 16400-16406. 10.1074/jbc.274.23.16400 [DOI] [PubMed] [Google Scholar]
- Kannan N. and Tang V. W. (2015). Synaptopodin couples epithelial contractility to alpha-actinin-4-dependent junction maturation. J. Cell Biol. 211, 407-434. 10.1083/jcb.201412003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D. F., Taylor D. W., Bakolitsa C., Bobkov A. A., Bankston L., Liddington R. C. and Taylor K. A. (2006). Structure of the alpha-actinin-vinculin head domain complex determined by cryo-electron microscopy. J. Mol. Biol. 357, 562-573. 10.1016/j.jmb.2005.12.076 [DOI] [PubMed] [Google Scholar]
- Kim T.-J., Zheng S., Sun J., Muhamed I., Wu J., Lei L., Kong X., Leckband D. E. and Wang Y. (2015). Dynamic visualization of alpha-catenin reveals rapid, reversible conformation switching between tension states. Curr. Biol. 25, 218-224. 10.1016/j.cub.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelberger N., Breunig M., Martin R., Knölker H.-J. and Miklavc P. (2016). The role of myosin 1c and myosin 1b in surfactant exocytosis. J. Cell Sci. 129, 1685-1696. 10.1242/jcs.181313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen K. A., Soler A. P., Johnson K. R. and Wheelock M. J. (1995). Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J. Cell Biol. 130, 67-77. 10.1083/jcb.130.1.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremerskothen J., Plaas C., Kindler S., Frotscher M. and Barnekow A. (2005). Synaptopodin, a molecule involved in the formation of the dendritic spine apparatus, is a dual actin/alpha-actinin binding protein. J. Neurochem. 92, 597-606. 10.1111/j.1471-4159.2004.02888.x [DOI] [PubMed] [Google Scholar]
- Le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D. and De Rooij J. (2010). Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189, 1107-1115. 10.1083/jcb.201001149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. H., Greenberg M. J., Laakso J. M., Shuman H. and Ostap E. M. (2012). Calcium regulation of myosin-I tension sensing. Biophys. J. 102, 2799-2807. 10.1016/j.bpj.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-D., Rhodes T. E., Ikebe R., Kambara T., White H. D. and Ikebe M. (1998). Effects of mutations in the gamma-phosphate binding site of myosin on its motor function. J. Biol. Chem. 273, 27404-27411. 10.1074/jbc.273.42.27404 [DOI] [PubMed] [Google Scholar]
- Lieto-Trivedi A. and Coluccio L. M. (2008). Calcium, nucleotide, and actin affect the interaction of mammalian Myo1c with its light chain calmodulin. Biochemistry 47, 10218-10226. 10.1021/bi8011059 [DOI] [PubMed] [Google Scholar]
- Liu Z., Tan J. L., Cohen D. M., Yang M. T., Sniadecki N. J., Ruiz S. A., Nelson C. M. and Chen C. S. (2010). Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 107, 9944-9949. 10.1073/pnas.0914547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q., Li J., Ye F. and Zhang M. (2015). Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat. Struct. Mol. Biol. 22, 81-88. 10.1038/nsmb.2923 [DOI] [PubMed] [Google Scholar]
- Manceva S., Lin T., Pham H., Lewis J. H., Goldman Y. E. and Ostap E. M. (2007). Calcium regulation of calmodulin binding to and dissociation from the myo1c regulatory domain. Biochemistry 46, 11718-11726. 10.1021/bi700894h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravillas-Montero J. L., Gillespie P. G., Patino-Lopez G., Shaw S. and Santos-Argumedo L. (2011). Myosin 1c participates in B cell cytoskeleton rearrangements, is recruited to the immunologic synapse, and contributes to antigen presentation. J. Immunol. 187, 3053-3063. 10.4049/jimmunol.1004018 [DOI] [PubMed] [Google Scholar]
- Mcgregor A., Blanchard A. D., Rowe A. J. and Critchley D. R. (1994). Identification of the vinculin-binding site in the cytoskeletal protein alpha-actinin. Biochem. J. 301, 225-233. 10.1042/bj3010225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcneill H. Ryan T. A., Smith S. J. and Nelson W. J. (1993). Spatial and temporal dissection of immediate and early events following cadherin-mediated epithelial cell adhesion. J. Cell Biol. 120, 1217-1226. 10.1083/jcb.120.5.1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F. and Sachs F. (2011). Visualizing dynamic cytoplasmic forces with a compliance-matched FRET sensor. J. Cell Sci. 124, 261-269. 10.1242/jcs.071928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar R., Mcconnell R. E. and Tyska M. J. (2009). Control of cell membrane tension by myosin-I. Proc. Natl. Acad. Sci. USA 106, 11972-11977. 10.1073/pnas.0901641106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieset J. E., Redfield A. R., Jin F., Knudsen K. A., Johnson K. R. and Wheelock M. J. (1997). Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin. J. Cell Sci. 110, 1013-1022. [DOI] [PubMed] [Google Scholar]
- Palmer A. E., Giacomello M., Kortemme T., Hires S. A., Lev-Ram V., Baker D. and Tsien R. Y. (2006). Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 13, 521-530. 10.1016/j.chembiol.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Park S., Gonzalez D. G., Guirao B., Boucher J. D., Cockburn K., Marsh E. D., Mesa K. R., Brown S., Rompolas P., Haberman A. M. et al. (2017). Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nat. Cell Biol. 19, 155-163. 10.1038/ncb3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips K. R., Tong S., Goodyear R., Richardson G. P. and Cyr J. L. (2006). Stereociliary myosin-1c receptors are sensitive to calcium chelation and absent from cadherin 23 mutant mice. J. Neurosci. 26, 10777-10788. 10.1523/JNEUROSCI.1847-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers J. G., Higham C., Broussard K. and Phillips T. J. (2016). Wound healing and treating wounds: chronic wound care and management. J. Am. Acad. Dermatol. 74, 607-625; quiz 625-6 10.1016/j.jaad.2015.08.070 [DOI] [PubMed] [Google Scholar]
- Pyrpassopoulos S., Feeser E. A., Mazerik J. N., Tyska M. J. and Ostap E. M. (2012). Membrane-bound myo1c powers asymmetric motility of actin filaments. Curr. Biol. 22, 1688-1692. 10.1016/j.cub.2012.06.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrpassopoulos S., Arpağ G., Feeser E. A., Shuman H., Tüzel E. and Ostap E. M. (2016). Force Generation by Membrane-Associated Myosin-I. Sci. Rep. 6, 25524 10.1038/srep25524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan E. S. and Izard T. (2012). The cytoskeletal protein alpha-catenin unfurls upon binding to vinculin. J. Biol. Chem. 287, 18492-18499. 10.1074/jbc.M112.351023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R., Metzger M., Knyphausen P., Ramezani T., Slanchev K., Kraus C., Schmelzer E. and Hammerschmidt M. (2016). Re-epithelialization of cutaneous wounds in adult zebrafish combines mechanisms of wound closure in embryonic and adult mammals. Development 143, 2077-2088. 10.1242/dev.130492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozbicki E., Chuai M., Karjalainen A. I., Song F., Sang H. M., Martin R., Knölker H.-J., MacDonald M. P. and Weijer C. J. (2015). Myosin-II-mediated cell shape changes and cell intercalation contribute to primitive streak formation. Nat. Cell Biol. 17, 397-408. 10.1038/ncb3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon J., Gaston C., Magescas J., Duvauchelle B., Canioni D., Sengmanivong L., Mayeux A., Michaux G., Campeotto F., Lemale J. et al. (2017). Contractile forces at tricellular contacts modulate epithelial organization and monolayer integrity. Nat. Commun. 8, 13998 10.1038/ncomms13998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N., Shimada T. and Sutoh K. (1998). Mutational analysis of the switch II loop of Dictyostelium myosin II. J. Biol. Chem. 273, 20334-20340. 10.1074/jbc.273.32.20334 [DOI] [PubMed] [Google Scholar]
- Schilders K. A. A., Eenjes E., van Riet S., Poot A. A., Stamatialis D., Truckenmüller R., Hiemstra P. S. and Rottier R. J. (2016). Regeneration of the lung: lung stem cells and the development of lung mimicking devices. Respir. Res. 17, 44 10.1186/s12931-016-0358-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples E. J. (2007). Acute kidney injury: stimulation of repair. Curr. Opin Crit. Care 13, 652-655. 10.1097/MCC.0b013e3282f1be4b [DOI] [PubMed] [Google Scholar]
- Shaw T. J. and Martin P. (2016). Wound repair: a showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 42, 29-37. 10.1016/j.ceb.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Shimada T., Sasaki N., Ohkura R. and Sutoh K. (1997). Alanine scanning mutagenesis of the switch I region in the ATPase site of Dictyostelium discoideum myosin II. Biochemistry 36, 14037-14043. 10.1021/bi971837i [DOI] [PubMed] [Google Scholar]
- Sokac A. M., Schietroma C., Gundersen C. B. and Bement W. M. (2006). Myosin-1c couples assembling actin to membranes to drive compensatory endocytosis. Dev. Cell 11, 629-640. 10.1016/j.devcel.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang V. W. and Brieher W. M. (2012). alpha-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J. Cell Biol. 196, 115-130. 10.1083/jcb.201103116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas W. A., Boscher C., Chu Y.-S., Cuvelier D., Martinez-Rico C., Seddiki R., Heysch J., Ladoux B., Thiery J. P., Mege R.-M. et al. (2013). alpha-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 288, 4957-4969. 10.1074/jbc.M112.403774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuo H. and Coluccio L. M. (2013). Myosin-1c regulates the dynamic stability of E-cadherin-based cell-cell contacts in polarized Madin-Darby canine kidney cells. Mol. Biol. Cell 24, 2820-2833. 10.1091/mbc.e12-12-0884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschumperlin D. J., Oswari J. and Margulies A. S. (2000). Deformation-induced injury of alveolar epithelial cells. Effect of frequency, duration, and amplitude. Am. J. Respir. Crit. Care. Med. 162, 357-362. 10.1164/ajrccm.162.2.9807003 [DOI] [PubMed] [Google Scholar]
- Van Der Gracht E., Zahner S. and Kronenberg M. (2016). When insult is added to injury: cross talk between ILCs and intestinal epithelium in IBD. Mediators Inflamm. 2016, 9765238 10.1155/2016/9765238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V., Bauer C., Yin M. and Fuchs E. (2000). Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100, 209-219. 10.1016/S0092-8674(00)81559-7 [DOI] [PubMed] [Google Scholar]
- Venit T., Kalendová A., Petr M., Dzijak R., Pastorek L., Rohožková J., Malohlava J. and Hozák P. (2016). Nuclear myosin I regulates cell membrane tension. Sci. Rep. 6, 30864 10.1038/srep30864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsstock D. H., Wilkins J. A. and Lin S. (1987). Specific interaction of vinculin with alpha-actinin. Biochem. Biophys. Res. Commun. 146, 554-560. 10.1016/0006-291X(87)90564-X [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M., Uchida N., Imamura Y., Nagafuchi A., Fujimoto K., Uemura T., Vermeulen S., Van Roy F., Adamson E. D. and Takeichi M. (1998). alpha-Catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J. Cell Biol. 142, 847-857. 10.1083/jcb.142.3.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss E. E., Kroemker M., Rüdiger A.-H., Jockusch B. M. and Rüdiger M. (1998). Vinculin is part of the cadherin-catenin junctional complex: complex formation between alpha-catenin and vinculin. J. Cell Biol. 141, 755-764. 10.1083/jcb.141.3.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M. and Wieschaus E. (2016). Myosin-dependent remodeling of adherens junctions protects junctions from Snail-dependent disassembly. J. Cell Biol. 212, 219-229. 10.1083/jcb.201508056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. K., Budnar S., Yap A. S. and Gomez G. A. (2014). Pulsatile contractility of actomyosin networks organizes the cellular cortex at lateral cadherin junctions. Eur. J. Cell Biol. 93, 396-404. 10.1016/j.ejcb.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Yamada A., Irie K., Fukuhara A., Ooshio T. and Takai Y. (2004). Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in MDCK cells. Genes Cells 9, 843-855. 10.1111/j.1365-2443.2004.00768.x [DOI] [PubMed] [Google Scholar]
- Yao M., Qiu W., Liu R., Efremov A. K., Cong P., Seddiki R., Payre M., Lim C. T., Ladoux B., Mège R.-M. et al. (2014). Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat. Commun. 5, 4525 10.1038/ncomms5525 [DOI] [PubMed] [Google Scholar]
- Yonemura S. (2011). Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 23, 515-522. 10.1016/j.ceb.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Yonemura S., Itoh M., Nagafuchi A. and Tsukita S. (1995). Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J. Cell Sci. 108, 127-142. [DOI] [PubMed] [Google Scholar]
- Yonemura S., Wada Y., Watanabe T., Nagafuchi A. and Shibata M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12, 533-542. 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
- Zhang J., Betson M., Erasmus J., Zeikos K., Bailly M., Cramer L. P. and Braga V. M. (2005). Actin at cell-cell junctions is composed of two dynamic and functional populations. J. Cell Sci. 118, 5549-5562. 10.1242/jcs.02639 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.