

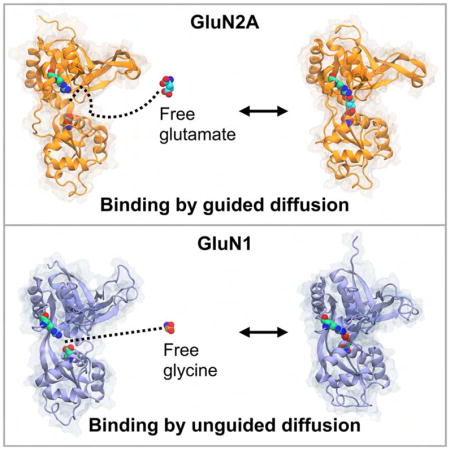
SUMMARY
At central nervous system synapses, agonist binding to post-synaptic ionotropic glutamate receptors (iGluRs) results in signaling between neurons. NMDA receptors are a unique family of iGluRs that activate in response to the concurrent binding of glutamate and glycine. Here, we investigate the process of agonist binding to the GluN2A (glutamate binding) and GluN1 (glycine binding) NMDA receptor subtypes using long-timescale unbiased molecular dynamics simulations. We find that positively charged residues on the surface of the GluN2A ligand-binding domain (LBD) assist glutamate binding via a “guided-diffusion” mechanism, similar in fashion to glutamate binding to the GluA2 LBD of AMPA receptors. Glutamate can also bind in an inverted orientation. Glycine, on the other hand, binds to the GluN1 LBD via an “unguided-diffusion” mechanism, whereby glycine finds its binding site primarily by random thermal fluctuations. Free energy calculations quantify the glutamate- and glycine-binding processes.
eTOC Blurb
In the NMDA receptor family of ionotropic glutamate receptors, the agonist glutamate and its co-agonist glycine bind to their respective subunits by different dynamic mechanisms. Glutamate binding is assisted by structural features on the receptor surface. Glycine binding does not receive such assistance.
INTRODUCTION
NMDA receptors are ligand-gated ion channels important for fast excitatory synaptic transmission, distributed throughout the central nervous system (Monyer et al., 1992; Moriyoshi et al., 1991; Paoletti et al., 2013). They are necessary for learning and memory and are drug targets for treating Alzheimer’s disease (McKeage, 2009), depression (Moskal et al., 2014), epilepsy (Hu et al., 2016), and schizophrenia (Balu, 2016). These receptors are obligate heterotetramers, typically comprising two glycine-binding GluN1 subunits and two glutamate-binding GluN2 subunits (Mayer et al., 1984). Each subunit contains an extracellular amino-terminal domain (ATD) and ligand-binding domain (LBD) in addition to a transmembrane domain (TMD) and an intracellular C-terminal domain (CTD) (Mayer, 2017). The LBDs possess an overall clamshell-like conformation in which a hinge separates two lobes of the LBD (Lobes 1 and 2). The TMDs of the four subunits surround a central pore to form the core of the ion channel, and in each subunit, the TMD is connected to Lobe 2 of the LBD by three short linkers.
Binding of two distinct agonists, glycine (or D-serine) and glutamate, to the individual glycine-binding and glutamate-binding LBDs activates the channel. Agonist binding triggers conformational change in the LBD, causing the two lobes to close around the ligand and is thought to provide the useful work to open the channel pore, which, together with a voltage-dependent unblock of magnesium, allows the entry of calcium into the postsynaptic cell and depolarization of the membrane potential (MacDermott et al., 1986; Mayer et al., 1984; Nowak et al., 1984). Crystallographic and cryo-electron microscopic studies have captured NMDA receptor LBDs bound to glycine and glutamate, as well as various other agonists and antagonists, shedding light on the molecular interactions involved in stabilizing the ligands and conformational change accompanying ligand binding (Inanobe et al., 2005; Furukawa et al., 2005; Yao et al., 2013; Jespersen et al., 2014; Hackos et al., 2016; Volgraf et al., 2016; Zhu et al., 2016; Tajima et al., 2016; Yi et al., 2016; Villemure et al., 2017; Lü et al., 2017; Romero-Hernandez and Furukawa, 2017; Lind et al., 2017). The precise molecular details of the dynamical processes associated with ligand binding, however, are unknown.
AMPA receptors are related ionotropic glutamate receptors (iGluRs) that mediate fast excitatory neurotransmission, and are also typically heteromeric ion channels, composed of GluA2 subunits in conjugation with GluA1, GluA3, or GluA4 subunits (Herguedas et al., 2016; Isaac et al., 2007). Like NMDA receptors, they have functionally modular domains separated into the ATDs, LBDs, TMD, and CTDs, but differ in that activation of the channel requires binding of only a single agonist, glutamate, to the separate LBDs. Recent computational and electrophysiology studies have elucidated the binding mechanisms of glutamate to the AMPA receptor and found binding intermediates that assist the process of ligand binding (Yu and Lau, 2017; Yu et al., 2018).
Molecular simulation has proven useful in describing the dynamics and energetics of NMDA receptor LBD cleft closure and the potentiating role of glycans (Yao et al., 2013; Dai and Zhou, 2013, 2015, 2016; Dutta et al., 2015; Omotuyi and Ueda, 2015; Sinitskiy and Pande, 2017; Sinitskiy et al., 2017). Here, using an aggregate of ~75 μs of unbiased all-atom molecular dynamics (MD) simulations, we investigate the molecular mechanisms of binding for both glutamate and glycine to the LBDs of the NMDA receptor subunits, GluN2A and GluN1. We directly simulate glutamate binding to the GluN2A LBD as well as glycine binding to the GluN1 LBD and subsequent cleft closure. Analysis of the cross-lobe interactions formed during cleft closure suggests these interactions may contribute to previously observed dynamical motions (Yao et al., 2013). Free energy landscapes indicate that the dynamic mechanisms responsible for glutamate binding are distinct from that of glycine binding in NMDA receptors.
RESULTS
Glutamate binding to the GluN2A LBD
Previously, glutamate was found to bind to the GluA2 LBD via a series of metastable interactions in which positively charged sidechains help guide the ligand into its binding pocket (Yu and Lau, 2017; Yu et al., 2018). Glutamate binding to the GluN2A LBD proceeds in a similar fashion (Figures 1 and 2). We simulated five trajectories involving GluN2A and 40 mM glutamate (23.5 μs), five trajectories involving GluN2A and 4 mM glutamate (11.5 μs), and three trajectories involving a GluN2A/GluN1 heterodimer with the following ligand concentrations: 20 mM glutamate and 20 mM glycine (5.2 μs), 20 mM glutamate and 18 mM glycine (5.3 μs), and 2 mM glutamate and 2 mM glycine (4.2 μs). In the heterodimer simulations, glycine binding to the GluN1 LBD was not observed, but glutamate bound seven times to the GluN2A LBD. In trajectory T1 (Movie S1), the glutamate ligand first contacts the LBD on the ξ1 side of the binding cleft (Figure 2B). The α-carboxylate interacts with a helix E arginine, R692 (Figure 2C), then the γ-carboxylate forms interactions with R518 (Figure 2D). The GluN2A LBD helix E is homologous to helix F on both the GluN1 and GluA2 LBDs. Protein-ligand contacts at the α- and γ-carboxylates are broken, and the ligand flips twice, trading interaction partners between the α- and γ-carboxylates (Figure 2D–G, Figure 2G–I). Next, contacts between helix E and glutamate are broken, and glutamate moves into the binding pocket in the inverted orientation while contacting R518 (Figure 2J). Once inside the binding pocket, the glutamate ligand contacts residues on Lobe 1 prior to interacting with residues on Lobe 2. The establishment of interactions with Lobe 1 before Lobe 2 is consistent with time-resolved vibrational spectroscopy experiments with the GluA2 LBD (Cheng et al., 2005).
Figure 1.

The glutamate-binding GluN2A and glycine-binding GluN1 LBDs. Lobes 1 and 2 of each LBD are indicated. ξ1 and ξ2 each refer to distances between Lobes 1 and 2 at the marked clusters of atoms. Disulfide bonds connecting the two lobes are colored red.
Figure 2.

Glutamate binding to the GluN2A LBD. (A) Prior to binding, the LBD is open and glutamate is in bulk solvent. Helix E is analogous to helix F in GluA2 and GluN1. (B) Glutamate contacts R695 on the ξ1side of the LBD. Residues involved in binding are labeled. (C) The ligand interacts with R692. (D) Glutamate forms a metastable salt bridge between R692 and its α-carboxylate, and R518 and its γ-carboxylate. (E) Contact with R692 is broken, and the ligand’s α-carboxylate swings upwards towards R518. (F) Contact with R518 is broken, and the ligand’s γ-carboxylate swings downward to interact with R692. (G) The ligand’s α- and γ-carboxylates have traded interaction partners from (D). In crystal structures, R518 is the binding partner for glutamate in the closed conformation. (H, I) Contacts are broken as the α- and γ-carboxylates trade interaction partners again. (J) Glutamate moves into the binding pocket, in a non-crystallographic, inverted conformation. Snapshots of the binding process were taken from a 524 ns interval. Simulation time is given relative to panel (A). See also Movie S1.
Crystal structures of glutamate-bound GluN2A show interactions between the ligand’s α-carboxylate and R518, the ligand’s amine and S511, T513, and H485, and the ligand’s γ-carboxylate with the backbone amines of S689 and T690 (Furukawa et al., 2005; Jespersen et al., 2014). During our simulations, glutamate forms contacts with R518 alternately at both the α- and γ-carboxylates, flipping between the inverted orientation and the orientation seen in crystal structures. The ligand’s amine is also able to interact with S511, T513, and H485, but these interactions are weaker in the inverted conformation. Vibrational spectroscopic studies with the GluA2 LBD indicate that partial agonists have weaker interactions at the α-amine than full agonists (Cheng and Jayaraman, 2004; Mankiewicz et al., 2007), which suggests that the inverted ligand conformation in GluN2A may elicit receptor behavior consistent with a partial agonist. In our trajectories, Lobe 2 interactions with the ligand are only made between S689 and the α-carboxylate while the γ-carboxylate contacts R518, resulting in an inverted conformation for the ligand and incomplete cleft closure. It is possible, though, that additional sampling in this state could lead to glutamate rotating to the orientation seen in crystal structures, consequently permitting full cleft closure, as observed in glycine binding to the GluN1 LBD (see below). This inverted bound conformation has been seen in glutamate binding to the GluA2 LBD, although the potential physiological relevance of this binding mode is currently unknown (Yu et al., 2018). For several trajectories in which the α-carboxylate contacts R518 (T1–5), the γ-carboxylate does not form the requisite Lobe 2 contacts for stable binding, and the ligand subsequently dissociates.
Since H485 contacts the glutamate ligand in crystal structures (Furukawa et al., 2005; Jespersen et al., 2014), it is possible that different protonation states of this residue might affect the ligand binding process. We therefore performed ligand-binding simulations in which either the ε-nitrogen or δ-nitrogen were protonated. The resulting pathways turned out to not be significantly different. In GluN1, the analogous residue that coordinates the glycine ligand is F484.
Glycine binding to the GluN1 LBD
Our atomistic simulations of glycine binding to the GluN1 LBD consist of five trajectories that total 25.1 μs. In contrast to glutamate binding into the GluN2A or GluA2 LBDs, glycine binding into the GluN1 LBD is not characterized by interactions with residues outside of the binding pocket. The homologous helix F arginines, R694 and R695, do not interact significantly with the ligand prior to entry into the pocket. In trajectory T6 (Movie S2, Table 1), glycine initially enters the cleft via the ξ1 side of the LBD and quickly docks to binding pocket residues on Lobe 1 of the LBD (Figure 3A,B). Protein-ligand interactions form between the open LBD and the ligand, first between R523 and the ligand’s α-carboxylate (Figure 3C) and then between F484 and the glycine carbons (Figure 3D,E). Notably, F484 rotates ~108° around its χ1 dihedral angle prior to recruiting the ligand (Figure 3B,C). Next, the ligand rotates so that its amine contacts the P516 carbonyl oxygen and the hydroxyl group of the T518 sidechain (Figure 3E,F). Once glycine forms these contacts, the ligand is in the same conformation seen in crystal structures (Furukawa and Gouaux, 2003; Furukawa et al., 2005; Jespersen et al., 2014); although at this stage, if the LBD does not immediately close, either water molecules in bulk solvent or free glycine ligands can displace the bound glycine from Lobe 1. After the LBD closes, we do not observe dissociation of glycine from the ligand-bound, closed conformation.
Table 1.
Trajectories of the simulation systems.
| Trajectory | System | Simulation Time (μs) |
|---|---|---|
| T1 | GluN2A LBD, 10 glutamates (H485, prot-εN) | 5 |
| T2 | GluN2A LBD, 10 glutamates (H485, prot-εN) | 5 |
| T3 | GluN2A LBD, 10 glutamates (H485, prot-δN) | 6.6 |
| T4 | GluN2A LBD, 10 glutamates (H485, prot-δN) | 4 |
| T5 | GluN2A LBD, 10 glutamates (H485, prot-δN) | 2.859 |
| T6 | GluN1 LBD, 10 glycines | 2 |
| T7 | GluN1 LBD, 10 glycines | 10 |
| T8 | GluN1 LBD, 10 glycines | 2 |
| T9 | GluN1 LBD, 1 glycine | 5.538 |
| T10 | GluN1 LBD, 1 D-serine | 5.549 |
| T11 | GluN2A LBD, 1 glutamate | 1.492 |
| T12 | GluN2A LBD, 1 glutamate | 1.492 |
| T13 | GluN2A LBD, 1 glutamate | 4.205 |
| T14 | GluN2A LBD, 1 glutamate | 1.497 |
| T15 | GluN2A LBD, 1 glutamate | 2.826 |
| T16 | GluN1/GluN2A LBD dimer, 10 glycines, 10 glutamates | 5.166 |
| T17 | GluN1/GluN2A LBD dimer, 9 glycines, 10 glutamates | 5.344 |
| T18 | GluN1/GluN2A LBD dimer, 1 glycine, 1 glutamate | 4.171 |
| Total | 74.739 |
Figure 3.

Glycine binding to the GluN1 LBD. (A) Prior to ligand binding, glycine diffuses in bulk solvent, and the LBD is open. Helices F and H are labeled. (B) Glycine enters via the ξ1 side of the binding cleft. Binding pocket residues are labeled. (C) R523 contacts the glycine carboxylate. (D) Glycine moves into the binding pocket. (E) F484 interacts with the ligand’s amine. (F) The ligand rotates such that its amine contacts the T518 sidechain carboxylate and the P516 carbonyl oxygen. (G) The glycine amine rotates out of the binding pocket to interact with the S688 sidechain. (H) The amine contacts the sidechain carboxylate of D732, partially closing the LBD to (ξ1, ξ2) = (10.8 Å, 12.7 Å). (I) The LBD reopens, and glycine makes Lobe 1 contacts prior to cleft closure. (J) The LBD closes around the ligand to (ξ1, ξ2) = (9.2 Å, 10.3 Å). Glycine is unable to dissociate from this conformation. Snapshots of the binding process were taken from a 448 ns interval. Simulation time is given relative to panel (A). See also Movies S2 and S3.
The ligand’s amine is more mobile than the glycine carboxylate, once attached to Lobe 1 residues. In several instances, contacts between the P516 carbonyl with the ligand’s amine and T518 with the ligand’s amine are broken, and the amine flips towards Lobe 2 of the LBD to briefly interact with S688 and D732, causing the LBD to partially close to (ξ1, ξ2) = (10.8 Å, 12.7 Å) (Figure 3G,H). Large conformational change occurs once the ligand’s amine stably contacts the D732 sidechain carboxylate, and the LBD closes to (ξ1, ξ2) = (9.2 Å, 10.3 Å) (Figure 3I,J). Further interdomain interactions are formed across the two lobes of the binding cleft that lock the LBD closed and contribute to the stability of the closed cleft conformation (Movie S3). Helix F residues R694 and R695 on lobe 2 contact E488 and E522 on lobe 1 at the ξ1 side of the binding pocket. Cross-lobe interactions also form between K483 and E712, and N499/G485 and Q686 as seen in crystal structures of partial agonists bound to GluN1 (Inanobe et al., 2005), although these interactions are more transient, and break and reform throughout the simulation.
After the LBD closes around the ligand, conformational change relative to the two lobes of the LBD is still possible. Helix H shifts upward towards Lobe 1 when R722 contacts E406, and Helix F shifts either closer to or away from Lobe 1 on the basis of the strength of the following interactions: R694 with E488 and R695 with E522. The formation and breakage of these interdomain interactions is likely the molecular basis for dynamical motions detected in prior simulations (Yao et al., 2013), and suggest conformational changes can occur in iGluR LBDs beyond simple cleft closure. The cross-lobe interactions positioned at helix F, including R694-E488 and R695-E522, and at helix H, including R722-E406, are more stable during the simulations than K483-E712 and N499/G485-Q686, identified by Inanobe et al. (Inanobe et al., 2005).
Potential of mean force for agonist binding to GluN2A and GluN1
We calculated the three-dimensional free energy landscape, or potential of mean force (PMF), for glutamate binding to the GluN2A LBD and glycine binding to the GluN1 LBD. The PMF for GluN2A features several binding pathways and multiple metastable minima (Figure 4A–C). The global free energy minimum contains three separate states of the ligand, which largely reflect interactions between the glutamate’s α- or γ-carboxylate and R518 (Figure 4A). In state 1, the carboxylate that does not interact with R518 forms contacts within the binding pocket, whereas in states 2 and 3, this carboxylate is stabilized by contacts with arginines on helix E, R692 and R695. At intermediate energetic contours (1.70 kcal/mol), significant ligand density is positioned in the cavity between helices C and E, suggesting that ligand entry to the binding pocket occurs on the ξ1 side of the binding cleft (Figure 4B). The PMF contoured to 2.15 kcal/mol shows continuous density from the surface of the LBD into the binding pocket, indicative of at least three binding pathways, two of which may involve metastable interactions between the ligand’s carboxylates and helix E arginines, R692 and R695, and R518 (Figure 4C). Glutamate binding to the GluN2A LBD can therefore be described as an “guided-diffusion” process, where binding assistance is provided by surface-exposed arginines. Error analysis is presented in Figure 5.
Figure 4.

The three-dimensional PMFs for glutamate and glycine binding to the GluN2A and GluN1 LBDs, respectively. (A–C) The PMF for glutamate binding is shown contoured at different free energy values. At a contour level of 0.6 kcal/mol (A), the free energy minimum contains 3 separate sites. Site 1 reflects interactions between the ligand and R518, once the ligand is bound, whereas Site 2 and Site 3 reflect energetically favorable contacts between R518 and glutamate prior to binding. The PMF contoured at 1.7 kcal/mol (B) indicates that residue-ligand interactions on the ξ1 side of the cleft are energetically favorable. At a 2.15 kcal/mol contour (C), the PMF shows interconnected minima, which indicates the ligand is guided from the periphery of the LBD into the binding pocket. Approximate binding pathways observed in the trajectory data are indicated with red arrows. (D–F) The PMF for glycine binding is contoured at different free energy values. At a contour level of 0.2 kcal/mol (D), the free energy minimum contains density for the glycine ligand inside the binding pocket. At higher free energy contours, 1.8 kcal/mol (E) and 2.8 kcal/mol (F), the free energy minimum remains disconnected from densities on other parts of the protein.
Figure 5.

The PMF for glutamate binding to the GluN2A LBD and associated error analysis. (A–F) The free energy landscape is contoured from 0.6 kcal/mol to 3.6 kcal/mol in increments of 0.6 kcal/mol. (G–L) The standard deviation is shown contoured from 0.9 kcal/mol to 1.65 kcal/mol in increments of 0.15 kcal/mol. Ligand positions close to bulk solvent (G) have low statistical uncertainty, with a standard deviation of < 0.9 kcal/mol. Ligand positions inside the binding cleft (I–L) have higher statistical uncertainty, with standard deviations ranging from 1.2 kcal/mol to 1.65 kcal/mol. Statistical uncertainty was calculated using the approach of block averaging.
The PMF for glycine binding to the GluN1 LBD is shown in Figure 4D–F. The global free energy minimum is set to 0 kcal/mol and is located within the binding pocket, proximal to residues that interact with the ligand in crystal structures of the bound complex (Furukawa and Gouaux, 2003; Furukawa et al., 2005; Jespersen et al., 2014) (Figure 4D). Contouring the PMF at higher values does not reveal significant metastable interactions proximal to the binding site (Figure 4E), and ligand densities surrounding the LBD, which reflect glycine-LBD interactions, are separated by free energy barriers (Figure 4F) similar to that of positions in bulk solvent (i.e., 3.6 kcal/mol). Overall, the PMF does not form continuous ligand densities into the global free energy minimum within the binding pocket. This indicates a lack of binding pathways. Glycine binding to the GluN1 LBD can therefore be described as an “unguided-diffusion” process, in which the ligand docks to Lobe 1 by random diffusion followed by LBD closure. Error analysis is presented in Figure 6.
Figure 6.

The PMF for glycine binding to the GluN1 LBD and associated error analysis. (A–F) The free energy landscape is contoured from 0.6 kcal/mol to 3.6 kcal/mol in 0.6 kcal/mol increments. (G–L) The three-dimensional standard deviation of the PMF is shown contoured from 0.75 kcal/mol to 2.0 kcal/mol in increments of 0.25 kcal/mol. Ligand positions in bulk solvent (G) have low statistical uncertainty, with a standard deviation of < 0.75 kcal/mol. Ligand positions within the binding cleft (I–L) have higher statistical uncertainty, with a standard deviation ranging from 1.25 to 2.00 kcal/mol. Statistical uncertainty was determined using the block averaging approach.
DISCUSSION
In the present study, we simulated glutamate binding to the GluN2A LBD and glycine binding to the GluN1 LBD. In our simulations, glycine binds in the same orientation seen in crystal structures, whereas glutamate binds in an inverted pose. This observation does not contradict glutamate binding in a non-inverted pose; it simply suggests an inverted pose is possible. Pre-binding intermediates for glutamate were able to interconvert between a crystal structure orientation, in which the α-carboxylate contacts R518 (in Lobe 1 of GluN2A), and an inverted orientation, in which the γ-carboxylate contacts R518. It is unknown whether, or to what extent, NMDA receptors can be activated by glutamate bound in the inverted conformation. In addition to the amount of cleft closure, the orientation of the agonist within the binding pocket may also be a factor in activation. This study complements prior studies that examined the free energy of conformational change between ligand-bound and apo GluN1 and GluN2A LBDs (Yao et al., 2013; Dai and Zhou, 2015).
The binding mechanisms in NMDA receptors for glutamate and its co-agonist, glycine, are distinct. Glutamate-binding PMFs for GluN2A and GluA2 feature continuous densities linking the periphery of the LBD to the binding pocket. The glycine-binding PMF for GluN1, on the other hand, features disconnected density between the periphery and the pocket. These distinct binding mechanisms serve as examples from two opposing paradigms for protein-ligand binding, one in which peripheral residue-ligand interactions may contribute substantially to the binding process, and the other in which protein-ligand interactions at the binding site are the only important factor in ligand binding. Glutamate binding to GluN2A is representative of the former; binding occurs along several preferred pathways, whereby positively charged residues mediate protein-ligand interactions at the periphery of the LBD and assist binding via a guided-diffusion mechanism. These binding intermediates are similar to those found in AMPA receptors, and electrophysiological recordings of AMPA receptors mutants lacking these metastable interactions exhibit slowed activation and deactivation kinetics (Yu et al., 2018). In contrast, glycine binding to GluN1 proceeds via an unguided-diffusion mechanism, where binding is largely a random diffusive process, and the most important protein-ligand interactions are those made between the Lobe 1 binding site residues and the ligand. It is possible that GluN2A LBDs behave similarly to AMPA receptors when metastable interactions between the protein and ligand are abolished, whereas GluN1 LBDs are more resistant to such alterations.
Conformational changes are observed in the LBDs after ligand binding, including partial cleft closure for GluN2A and complete cleft closure for GluN1. For GluN2A, additional conformational rearrangements in the protein and ligand may be required before the ligand can be accommodated within Lobe 2. For example, it is likely that the glutamate ligand must rotate such that its α-carboxylate interacts with Lobe 1 in order to allow full cleft closure (Figure 7). The predicted glutamate binding modes can be experimentally tested using NMR. The glutamate carboxylates sample vastly different electronic environments in the two binding modes, and signatures for these modes can be probed using various NMR methods (Palmer, 2014). For GluN1, our simulations reveal that the cross-lobe interactions that stabilize the closed state are actually quite dynamic. The formation and breakage of these interactions can give rise to a range of conformational dynamics in the LBD (Yao et al., 2013).
Figure 7.

Conformational states of the GluN2A LBD bound to a glutamate ligand. (1) The LBD is open with the ligand’s α-carboxylate tethered to Lobe 1 (at R518). (2) The LBD is partially closed with the ligand’s α-carboxylate tethered to Lobe 1. (3) The LBD is fully closed with the ligand bound in the crystallographically observed configuration. (4) the LBD is open with the ligand’s γ-carboxylate tethered to Lobe 1 (at R518). (5) The LBD is partially closed with the ligand’s γ-carboxylate tethered to Lobe 1; the LBD cannot fully close with the ligand in this configuration. Full cleft closure would require the system to transition to State 2 and then to State 3. States corresponding to pre-bound configurations, i.e., those involving metastable interactions outside the binding pocket, are not shown.
Why do glutamate and glycine bind to the receptor in such different ways? Given the overall structural similarity between the GluN2A and GluN1 LBDs, one might conclude that the LBDs also bind ligands via similar processes. NMDA receptors with engineered disulfide linkages that lock the GluN1 lobes shut conduct current with kinetic profiles similar to that of wild-type NMDA receptors, suggesting that glutamate is the primary neurotransmitter, whereas D-serine, the endogenous agonist for GluN1 at synapses, or glycine, play more modulatory roles (Kussius and Popescu, 2010; Mothet et al., 2000). We speculate that the distinct binding mechanisms of glutamate and glycine may reflect the differing physiological roles the ligands play during NMDA receptor activation. Differences in these binding mechanisms may inform strategies for the design of therapeutic agents.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Albert Lau (alau@jhmi.edu).
METHOD DETAILS
Simulation system preparation
Initial atomic models were prepared from crystal structures of the GluN1 LBD (PDB ID: 4KCC), GluN2A LBD (PDB ID: 2A5S), and GluN1/GluN2A LBD dimer (PDB ID: 2A5T). Missing amino acids were constructed using the Modloop server (Fiser and Sali, 2003), and missing sidechains were added using SCWRL4 (Krivov et al., 2009). Crystallographic waters in the binding cleft were included in the models. The solvated GluN1, GluN2A, and GluN1/GluN2A LBD systems contained a total of 46,869, 46,012, and 93,704 atoms, respectively, and were neutralized by adding Na+ and Cl− ions into bulk solution until the salt concentration was ~150 mM. Periodic boundary conditions were imposed on an orthorhombic cell with approximate dimensions of 88 Å × 78 Å × 72 Å for GluN1, 97 Å × 66 Å × 70 Å for GluN2A, and 105 Å × 99 Å × 88 Å for GluN1/GluN2A. The systems were energetically minimized and equilibrated for 4 ns under constant pressure and temperature (NPT) conditions of 1 atm and 310 K with a timestep of 2 fs. For all simulations, the all-atom potential energy function PARAM36 for proteins and ions (Best et al., 2012; MacKerell et al., 1998; Mackerell et al., 2004) and the TIP3P potential energy function for water (Jorgensen et al., 1983) were used.
For the GluN1 and GluN2A LBD monomers, targeted molecular dynamics simulations were performed to generate LBD conformations close to the energetic minimum of the apo LBD monomers by introducing harmonic biasing potentials with a force constant of 2 kcal/mol/Å2 along the ξ1 and ξ2 order parameters, as reported previously (Yao et al., 2013). 10 glycine, 10 glutamate, and (10 glycine and 10 glutamate) molecules were added to the GluN1, GluN2A, and GluN1/GluN2A systems, respectively, and initially placed at arbitrary positions and orientations in bulk solvent at least 8 Å from any non-water molecules. A salt concentration of ~150 mM NaCl was used, and the number of Na+ and Cl− ions was adjusted to provide neutrality. Energy minimization and system equilibration was performed using CHARMM (Brooks et al., 2009). Pre-production simulations were performed using NAMD 2.9 (Phillips et al., 2005). For energy minimization, system equilibration, and pre-production simulations, electrostatic interactions were computed using the particle-mesh Ewald (PME) algorithm, and short-range, non-bonded interactions were truncated at 12 Å. The initial protein configuration of each system was relaxed with Langevin dynamics in the presence of harmonic restraints under constant volume and temperature (NVT) conditions for 30 ps before switching to NPT conditions of 1 atm and 310 K for 4 ns of pre-production simulation.
MD simulations
All production runs used the NPT ensemble at 1 atm and 310 K. Bond lengths for hydrogen were constrained using the M-SHAKE algorithm (Kräutler et al., 2001). An r-RESPA integrator (Tuckerman et al., 1992) was used with a timestep of 2 fs, and long-range electrostatics were computed every 6 fs using the k-space Gaussian split Ewald method (Shan et al., 2005). Short-range interactions including van der Waals and short-range electrostatics were truncated at 9 Å. To remove overall rotational and translational motion of the protein, solvent and ligand molecules were wrapped into a simulation box with the origin at the center of mass of the protein using PBCTools in VMD (Humphrey et al., 1996), and all frames were RMSD-aligned with respect to the protein backbone. All production simulations were carried out on the special purpose Anton machines at the Pittsburgh Supercomputing Center, Anton1 (Shaw et al., 2009) and Anton2 (Shaw et al., 2014). A total of 18 trajectories were generated for an aggregate simulation time of 74.7 μs (Table 1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Ligand-binding PMFs
The three-dimensional free energy landscapes for ligand binding were computed using the Cartesian coordinates for the non-hydrogen atoms for glutamate or glycine as order parameters. Coordinates were recorded in 0.12 ns intervals. Trajectories containing the monomeric GluN1 LBD and monomeric GluN2A LBD systems with 10 ligands were used for increased sampling. Data from the dimer systems (GluN1/GluN2A LBDs) were not used to calculate the ligand binding PMF to avoid inter-subunit conformational change affecting the density calculation. Coordinates were RMSD-aligned with respect to the LBD protein backbone. The density of atomic positions, ρ(r⃗), was calculated using a hard sphere van der Waals approximation on a discretized grid with unit cell spacings of 0.5 Å × 0.5 Å × 0.5 Å. Standard Boltzmann weighting was used to produce the free energy maps, i.e., W(r⃗) = −kBT ln[ρ(r⃗)], where kB is Boltzmann’s constant and T is temperature. Statistical uncertainties were determined using the approach of block averaging (Zhu and Hummer, 2012) (Figures 5 and 6). Data were divided into 10 blocks, and a PMF was calculated for each block. Standard deviation in the 10 PMFs was calculated. Varying the number of blocks used from 5 to 15 all gave qualitatively similar results.
Supplementary Material
This movie corresponds to trajectory T1, a system consisting of the LBD monomer with 10 glutamate ligands. For clarity, the ligands that do not bind are not shown.
This movie corresponds to trajectory T6, a system consisting of the LBD monomer with 10 glycine ligands. For clarity, the ligands that do not bind are not shown.
This movie corresponds to trajectory T6 and is an extension of Movie S2. In the binding pocket, R523 (the anchor for the glycine ligand) is not shown so as to not obscure some cross-lobe interactions.
Highlights.
Glutamate binds to NMDA receptors via a guided-diffusion mechanism.
Glycine binds to NMDA receptors via an unguided-diffusion mechanism.
All-atom simulations locate metastable sites that assist glutamate binding.
Binding of glutamate can occur in two orientations.
Acknowledgments
We thank Dominique Frueh for helpful discussion. Anton computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health. The Anton2 and Anton machine at PSC were generously made available by D.E. Shaw Research. This study also used resourced provided by the Maryland Advanced Research Computing Center (MARCC) at Johns Hopkins University. This work was supported by the National Institutes of Health Grant R01GM094495 (to A.Y.L.).
Footnotes
AUTHOR CONTRIBUTIONS
A.Y. and A.Y.L. designed the project, analyzed the data, and wrote the manuscript. A.Y. performed the research.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balu DT. Advances in Pharmacology. Elsevier; 2016. The NMDA Receptor and Schizophrenia; pp. 351–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Jayaraman V. Chemistry and conformation of the ligand-binding domain of GluR2 subtype of glutamate receptors. J Biol Chem. 2004;279:26346–26350. doi: 10.1074/jbc.M403111200. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Du M, Ramanoudjame G, Jayaraman V. Evolution of glutamate interactions during binding to a glutamate receptor. Nat Chem Biol. 2005;1:329–332. doi: 10.1038/nchembio738. [DOI] [PubMed] [Google Scholar]
- Dai J, Zhou HX. An NMDA Receptor Gating Mechanism Developed from MD Simulations Reveals Molecular Details Underlying Subunit-Specific Contributions. Biophys J. 2013;104:2170–2181. doi: 10.1016/j.bpj.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Zhou HX. Reduced curvature of ligand-binding domain free-energy surface underlies partial agonism at NMDA receptors. Structure. 2015;23:228–236. doi: 10.1016/j.str.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Zhou HX. Semiclosed Conformations of the Ligand-Binding Domains of NMDA Receptors during Stationary Gating. Biophys J. 2016;111:1418–1428. doi: 10.1016/j.bpj.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Krieger J, Lee JY, Garcia-Nafria J, Greger IH, Bahar I. Cooperative Dynamics of Intact AMPA and NMDA Glutamate Receptors: Similarities and Subfamily-Specific Differences. Structure. 2015;23:1692–1704. doi: 10.1016/j.str.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, Gustafson A, Wallweber HJA, Volgraf M, Sellers BD, et al. Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron. 2016;89:983–999. doi: 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- Herguedas B, García-Nafría J, Cais O, Fernández-Leiro R, Krieger J, Ho H, Greger IH. Structure and organization of heteromeric AMPA-type glutamate receptors. Science. 2016:aad3873. doi: 10.1126/science.aad3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF. Human GRIN2B variants in neurodevelopmental disorders. J Pharmacol Sci. 2016;132:115–121. doi: 10.1016/j.jphs.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Furukawa H, Gouaux E. Mechanism of Partial Agonist Action at the NR1 Subunit of NMDA Receptors. Neuron. 2005;47:71–84. doi: 10.1016/j.neuron.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Isaac JTR, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Jespersen A, Tajima N, Fernandez-Cuervo G, Garnier-Amblard EC, Furukawa H. Structural insights into competitive antagonism in NMDA receptors. Neuron. 2014a;81:366–378. doi: 10.1016/j.neuron.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen A, Tajima N, Fernandez-Cuervo G, Garnier-Amblard EC, Furukawa H. Structural insights into competitive antagonism in NMDA receptors. Neuron. 2014b;81:366–378. doi: 10.1016/j.neuron.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kräutler V, van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22:501–508. [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius CL, Popescu GK. NMDA receptors with locked glutamate-binding clefts open with high efficacy. J Neurosci. 2010;30:12474–12479. doi: 10.1523/JNEUROSCI.3337-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind GE, Mou TC, Tamborini L, Pomper MG, Micheli CD, Conti P, Pinto A, Hansen KB. Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits. Proc Natl Acad Sci. 2017;114:E6942–E6951. doi: 10.1073/pnas.1707752114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü W, Du J, Goehring A, Gouaux E. Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science. 2017:eaal3729. doi: 10.1126/science.aal3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Mankiewicz KA, Rambhadran A, Du M, Ramanoudjame G, Jayaraman V. Role of the chemical interactions of the agonist in controlling alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor activation. Biochemistry. 2007;46:1343–1349. doi: 10.1021/bi062270l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. The Challenge of Interpreting Glutamate-Receptor Ion-Channel Structures. Biophys J. 2017;113:2143–2151. doi: 10.1016/j.bpj.2017.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- McKeage K. Memantine: a review of its use in moderate to severe Alzheimer’s disease. CNS Drugs. 2009;23:881–897. doi: 10.2165/11201020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Moskal JR, Burch R, Burgdorf JS, Kroes RA, Stanton PK, Disterhoft JF, Leander JD. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opin Investig Drugs. 2014;23:243–254. doi: 10.1517/13543784.2014.852536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc Natl Acad Sci. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Omotuyi OI, Ueda H. Molecular dynamics study-based mechanism of nefiracetam-induced NMDA receptor potentiation. Comput Biol Chem. 2015;55:14–22. doi: 10.1016/j.compbiolchem.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Palmer AG. Chemical exchange in biomacromolecules: past, present, and future. J Magn Reson. 2014;241:3–17. doi: 10.1016/j.jmr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Hernandez A, Furukawa H. Novel Mode of Antagonist Binding in NMDA Receptors Revealed by the Crystal Structure of the GluN1-GluN2A Ligand-Binding Domain Complexed to NVP-AAM077. Mol Pharmacol. 2017;92:22–29. doi: 10.1124/mol.116.107912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122:54101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. IEEE; 2014. pp. 41–53. [Google Scholar]
- Shaw David E, Dror Ron O, Salmon John K, Grossman JP, Grossman JP, Bank Joseph A, Young Cliff, Deneroff Martin M, Batson B, Bowers Kevin J, et al. Millisecond-Scale Molecular Dynamics Simulations on Anton. Proceedings of the ACM/IEEE Conference on Supercomputing (SC09); Portland, Oregon. 2009. [Google Scholar]
- Sinitskiy AV, Pande VS. Simulated Dynamics of Glycans on Ligand-Binding Domain of NMDA Receptors Reveals Strong Dynamic Coupling between Glycans and Protein Core. J Chem Theory Comput. 2017;13:5496–5505. doi: 10.1021/acs.jctc.7b00817. [DOI] [PubMed] [Google Scholar]
- Sinitskiy AV, Stanley NH, Hackos DH, Hanson JE, Sellers BD, Pande VS. Computationally Discovered Potentiating Role of Glycans on NMDA Receptors. Sci Rep. 2017;7:44578. doi: 10.1038/srep44578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz-Avalos R, Grigorieff N, Furukawa H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- Villemure E, Volgraf M, Jiang Y, Wu G, Ly CQ, Yuen P, Lu A, Luo X, Liu M, Zhang S, et al. GluN2A-Selective Pyridopyrimidinone Series of NMDAR Positive Allosteric Modulators with an Improved in Vivo Profile. ACS Med Chem Lett. 2017;8:84–89. doi: 10.1021/acsmedchemlett.6b00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgraf M, Sellers BD, Jiang Y, Wu G, Ly CQ, Villemure E, Pastor RM, Yuen P, Lu A, Luo X, et al. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J Med Chem. 2016;59:2760–2779. doi: 10.1021/acs.jmedchem.5b02010. [DOI] [PubMed] [Google Scholar]
- Yao Y, Belcher J, Berger AJ, Mayer ML, Lau AY. Conformational analysis of NMDA receptor GluN1, GluN2, and GluN3 ligand-binding domains reveals subtype-specific characteristics. Structure. 2013b;21:1788–1799. doi: 10.1016/j.str.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Mou TC, Dorsett KN, Volkmann RA, Menniti FS, Sprang SR, Hansen KB. Structural Basis for Negative Allosteric Modulation of GluN2A-Containing NMDA Receptors. Neuron. 2016;91:1316–1329. doi: 10.1016/j.neuron.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Lau AY. Energetics of Glutamate Binding to an Ionotropic Glutamate Receptor. J Phys Chem B. 2017;121:10436–10442. doi: 10.1021/acs.jpcb.7b06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Salazar H, Plested AJR, Lau AY. Neurotransmitter Funneling Optimizes Glutamate Receptor Kinetics. Neuron. 2018;97:139–149. e4. doi: 10.1016/j.neuron.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Hummer G. Convergence and error estimation in free energy calculations using the weighted histogram analysis method. J Comput Chem. 2012;33:453–465. doi: 10.1002/jcc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, Mchaourab HS, Gouaux E. Mechanism of NMDA Receptor Inhibition and Activation. Cell. 2016;165:704–714. doi: 10.1016/j.cell.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This movie corresponds to trajectory T1, a system consisting of the LBD monomer with 10 glutamate ligands. For clarity, the ligands that do not bind are not shown.
This movie corresponds to trajectory T6, a system consisting of the LBD monomer with 10 glycine ligands. For clarity, the ligands that do not bind are not shown.
This movie corresponds to trajectory T6 and is an extension of Movie S2. In the binding pocket, R523 (the anchor for the glycine ligand) is not shown so as to not obscure some cross-lobe interactions.