

Abstract
Co-translational N-terminal protein acetylation regulates many protein functions including degradation, folding, interprotein interactions, and targeting. Human NatA (hNatA), one of six conserved metazoan N-terminal acetyltransferases, contains Naa10 catalytic and Naa15 auxiliary subunits, and associates with the intrinsically disordered Huntingtin Yeast two-hybrid Protein K (HYPK). We report on the crystal structures of hNatA and hNatA/HYPK, and associated biochemical and enzymatic analyses. We demonstrate that hNatA contains unique features: a stabilizing inositol hexaphosphate (IP6) molecule and a metazoan-specific Naa15 domain that mediates high-affinity HYPK binding. We find that HYPK harbors intrinsic hNatA-specific inhibitory activity through a bipartite structure: a Ubiquitin Associated domain that binds a hNaa15 metazoan-specific region and an N-terminal loop-helix region that distorts the hNaa10 active site. We show that HYPK binding blocks hNaa50 targeting to hNatA, likely limiting Naa50 ribosome localization ribosome in vivo. These studies provide a model for metazoan NAT activity and HYPK regulation of N-terminal acetylation.
Keywords: NatA, HYPK, N-terminal acetylation, Huntington interacting protein, protein complex, X-ray crystallography
eTOC Blurb
Co-translational N-terminal acetylation is catalyzed by N-terminal acetyltransferases (NATs). Gottlieb et al. describe the structure of the human NatA, its activity and uncover the basis for regulation by its binding partner, HYPK.
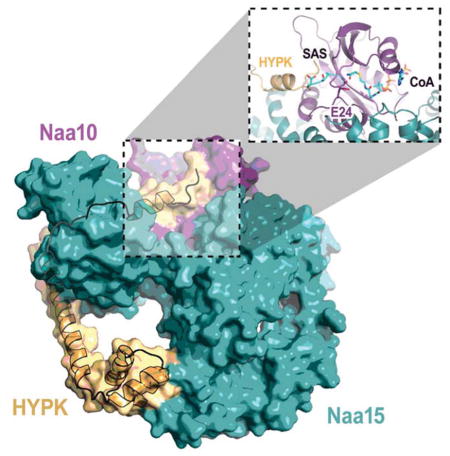
Introduction
The irreversible N-terminal (Nt) acetyl mark occurs on ~60% and ~85% of the yeast and human proteomes, respectively (Arnesen et al., 2009; Van Damme et al., 2011b). This modification effects a range of cellular phenotypes and protein activities, including cellular morphology and phosphorylation, sister chromatid cohesion, protein degradation, and protein-protein interactions (Helbig et al., 2010; Kim et al., 2014; Rong et al., 2016; Scott et al., 2011; Van Damme et al., 2012). Not surprisingly, studies have emerged suggesting that the aberrant activity of N-terminal acetyltransferases (NATs) and NAT substrates are correlated with human diseases such as cancer, developmental and neurological disorders, demonstrating the importance of Nt-acetylation (Fluge et al., 2002; Kalvik and Arnesen, 2013; Neri et al., 2017; Van Damme et al., 2014; Yu et al., 2009).
The five conserved eukaryotic enzymes (NatA-E) are each minimally composed of a catalytic subunit, and up to two auxiliary subunits (NatA-C) (Aksnes et al., 2016). The auxiliary subunits of NatA-C have been shown to contribute to enzymatic activity and selectivity of the corresponding catalytic subunit, as well as to mediate ribosomal targeting to facilitate co-translational Nt-acetylation. NatD binds directly to the ribosome and Naa50 is brought to the ribosome through direct interactions with the NatA complex via the formation of the ternary NatE complex (Gautschi et al., 2003; Hole et al., 2011; Magin et al., 2017; Starheim et al., 2008; Starheim et al., 2009). By contrast, the metazoan NatF localizes to the golgi apparatus for post-translational activity (Aksnes et al., 2015). The recently identified plant NatG localizes in chloroplasts, while NatH only functions post-translationally (Dinh et al., 2015; Drazic et al., 2018). The NAT enzymes are differentiated from one another by their substrate specificities. The NatA complex, with the broadest range of substrates, canonically targets nascent peptides containing an N-terminal Ala-, Cys-, Gly-, Ser-, Thr-, and Val-residue (Aksnes et al., 2016). However, as a monomer, the Naa10 catalytic subunit of the NatA only acetylates Glu- and Asp-N-termini; although the physiological relevance of this activity, which acts off the ribosome is still under investigation (Van Damme et al., 2011a). NatE, NatF, NatB and NatC complexes Nt-acetylate N-termini containing a Met-followed by a second residue that determines specificity. NatD has been reported to exclusively modify H2A and H4 (Ser-) substrates (Van Damme et al., 2011a).
While NatA contains an evolutionarily conserved eukaryotic auxiliary subunit, Naa15, higher eukaryotes and some fungi contain a third associated protein called Huntingtin Yeast two-hybrid Protein K (HYPK) (Arnesen et al., 2010). HYPK was first identified as a protein that interacts with the Huntingtin protein, which is associated with the autosomal neurodegenerative Huntington’s disease (Faber et al., 1998). More recently, HYPK was shown to interact with numerous proteins, including NatA, leading to the stabilization of HYPK and optimal NatA enzymatic activity towards some substrates (Arnesen et al., 2010). HYPK is an intrinsically disordered protein that contains homology to the Nascent Polypeptide-Associated Complex alpha (NACα) within its C-terminal region, which contains a Ubiquitin Associated (UBA) domain (Raychaudhuri et al., 2008a). Consistent with this homology, HYPK has also been shown to interact with nascent proteins (Raychaudhuri et al., 2014).
Recent structural and enzymatic studies have shed light on the molecular basis for NatA substrate specificity and its association with HYPK (Liszczak et al., 2013; Weyer et al., 2017). A structure of the S. pombe Naa10/Naa15 complex reveals that binding of the Naa15 auxiliary subunit induces a conformational change in the Naa10 catalytic subunit, allowing for enhanced canonical NatA complex substrate binding selectivity (Liszczak et al., 2013). A more recent structure of Naa10/Naa15/HYPK complex from the thermophilic fungus Chaetomium thermophilum, reveals that HYPK associates with both the Naa10 and Naa15 subunits to negatively regulate NatA activity (Weyer et al., 2017).
In this study, we set out to determine the molecular mechanism of metazoan NatA and its regulation by HYPK. To do so, we determined the crystal structure of the human NatA complex with and without HYPK bound, characterized the biophysical and enzymatic properties of the NatA and NatA/HYPK complexes, and assessed the NatA-HYPK interaction by structure-guided mutational analysis studies. These studies reveal unique features of metazoan NatA and HYPK-mediated regulation of NatA including a structural inositol hexaphosphate (IP6) molecule that appears to stabilize the Naa10-Naa15 interaction and a metazoan-specific Naa15 domain that participates in high-affinity HYPK binding. We also show that HYPK inhibits the acetyltransferase activity of human NatA, but not the activity of S. pombe NatA, other NATs or histone acetyltransferases (HATs); and does so through a bipartite structure that includes the tight binding of its UBA domain to the metazoan-specific Naa15 region and an N-terminal loop-helix region that interacts with Naa10 to distort the active site. We also provide biochemical evidence that HYPK binding blocks the targeting of Naa50 to NatA, likely limiting the localization of Naa50 to the ribosome for co-translational Nt-acetylation in vivo. Together, these studies provide a model for the mechanism of metazoan NatA activity and its regulation by HYPK for protein Nt-acetylation.
Results
Overall structure of human NatA reveals metazoan-specific features
In order to biochemically and structurally characterize the human NatA complex, we overexpressed the recombinant heterodimeric complex using baculovirus-infected Sf9 cells since previous attempts to co-express the human NatA complex in bacteria cells were unsuccessful. We found that co-expression of the C-terminally truncated Naa10 subunit (1–160 out of 235 residues) and full-length N-terminally 6xHis-tagged Naa15 (residues 1–866) allowed for the production of soluble, active protein that could be purified to homogeneity and crystallized. We were able to obtain crystals that formed in the P212121 space group, which contained two heterodimers in the asymmetric unit. Although, we used sedimentation equilibrium studies to demonstrate that one heterodimer exists in solution (Fig S1). The 2.80 Å resolution structure was determined using molecular replacement with the S. pombe (Sp) NatA complex (PDB: 4KVO (Liszczak et al., 2013)) as the initial search model, and refined to final Rwork and Rfree values of 18.8% and 24.2%, respectively, with good geometry (Fig 1A and 1B, Table 1).
Figure 1. Overall Structure of Human NatA.

(A) hNaa10 (light blue) and hNaa15 (dark blue) shown in cartoon overlayed with S. pombe (Sp) NatA (dark grey) (pdb: 4KVO). Electron density for α33 and α34 helices, annotated with an asterisk (*), was not resolved in the human structure.
(B) A 90° view rotation of A. with bound inositol hexaphosphate (IP6) in stick format and annotated by a dashed box.
(C) Zoom view depicting key residues involved in interactions with IP6.
(D) Zoom view of the hydrophobic pocket formed at the interface of the hNaa15 α22–α23 helices and C-terminal α44–45 helices.
(E) Sedimentation velocity of hNatA mutants of Y834 (yellow in D). WT, wild-type.
(F) Differential scanning calorimetric analysis of hNatA mutants.
(G) Bar graph showing the initial velocities of mutants (Py834F=0.0001 and PY834A<0.0001 by Student’s unpaired, two-tailed t-test). Assays performed in triplicate; error bars S.E.M.
Table 1.
hNatA and hNatA/HYPK Complex Data Collection and Refinement Statisticsa
| Crystal | hNatA | hNatA/HYPK |
|---|---|---|
| PDB | 6C9M | 6C95 |
| Resolution range (Å)b | 48.43 - 2.8 (2.9–2.8) | 49.04 – 3.152 (3.265–3.152) |
| Space groupb | P21 21 21 | P63 |
| Unit cell dimensions | ||
| a, b, c (Å)b | 95.11, 171.83, 178.69 | 182.32, 182.32, 86.07 |
| α, β, γ (°)b | 90, 90, 90 | 90, 90, 120 |
| Total reflectionsc | 807,478 | 119,010 |
| Unique reflectionsb | 72666 | 24588 |
| Multiplicityc | 11.1 (11.3) | 4.84 (4.91) |
| Completeness (%)b | 99.94 (99.96) | 95.23 (95.94) |
| Mean I/σc (I) | 20.8 (1.60) | 11.43 (1.2) |
| Wilson B-factorb | 82.42 | 87.76 |
| R-mergeb | 0.009 (0.229) | 0.013 (0.148) |
| R-workb | 0.1880 (0.2938) | 0.2207 (0.3762) |
| R-freeb | 0.2424 (0.3605) | 0.2552 (0.4348) |
| RMSb (Å) | 0.009 | 0.005 |
| RMSb (°) | 1.05 | 0.71 |
| Ramachandran favoredb (%) | 96.92 | 96.96 |
| Ramachandran allowedb (%) | 2.92 | 2.75 |
| Ramachandran outliersb (%) | 0.16 | 0.29 |
| Rotamer outliersb (%) | 0.06 | 0 |
| Clashscoreb | 11.08 | 9.41 |
| Average B-factorb | 91.41 | 95.16 |
numbers provided in parenthesis correspond to the highest resolution shell.
numbers generated in Phenix.
numbers retrieved from XDS log file.
While the human structure displays a high degree of structural conservation to the SpNatA structure (1.433 Å R.M.S. deviation over 608 common Cα atoms), there are several noteworthy features. The hNaa15 auxiliary subunit is composed of 45 α-helices forming a total of 13 tetracopeptide repeat (TPR) motifs and a C-terminal Sel1-like repeat (SLR) (Karpenahalli et al., 2007; Zimmermann et al., 2017). TPR and SLR segments often promote protein-protein interactions (Blatch and Lassle, 1999; Mittl and Schneider-Brachert, 2007), which is consistent with previous studies that have demonstrated that the Naa15 TPR motifs mediate interactions with Naa50 and HYPK (Neubauer, 2012; Weyer et al., 2017). Using the helical numbering of the S. pombe structure, we did not observe electron density corresponding to the hNatA α33 and α34 helices, which are presumably disordered, and the loop-α40-loop segment of the hNaa15 C-terminus is only resolved in one of the protomer subunits due to stabilization by crystal packing. We also found that the hNaa10 catalytic subunit was N-terminally acetylated and features an additional C-terminal α-helix (α5) relative to SpNatA (Fig 1A and B).
Unexpectedly, we observed electron density for inositol hexaphosphate (IP6) between hNaa10 and hNaa15 bound by a series of electropositive and hydrophilic interactions within the hNaa10 β2-loop-β3 and β4-loop-α3 segments as well as the hNaa15 α19, α24, and α25 helices (Fig 1C, Fig S2). The hNatA complex primarily forms hydrogen bonds via positively charged residues to make direct (Naa15: K416, K419, H420, K447, K450; and Naa10: K78) and water-mediated hydrogen bonds (Naa15: R330), as well as electrostatic interactions (Naa15: K454 and Naa10: K78) to engage with IP6. Nearly all of the hNatA residues that make IP6 interactions are well conserved in metazoan Naa15 and Naa10 (Fig S2–4), supporting its biological relevance.
hNaa15 also contains an extended C-terminus (α39-α45) relative to SpNaa15 (Fig S3). This divergent region is comprised of the Sel1-like repeats (residues 798–833), which extends to wrap back and pack up against the hNaa15 core bottom, forming an additional ring scaffold. This extended region of hNaa15 appears to play a key role in complex formation and stability. This is consistent with our observation that deletion of the C-terminus results in the destabilization of the hNatA complex (Fig S5). We also note that Y834 within a distal loop of hNaa15 interacts with a hydrophobic pocket formed by P390, S391, I417, Y418 and A421 between the α22-α23 helices and P833, A835, L836 surrounding Y834 within the loop-α45 helix segment (Fig 1D). Notably, these residues are highly conserved among metazoan Naa15 proteins (Fig S3), but are charged residues in the α22 of the SpNaa15 auxiliary subunit. To investigate the role of Y834 in the structural stability and enzymatic activity of hNatA, we mutated Y843 to F and A and showed that although the Y843F and Y843A mutants did not significantly change the shape of the hNatA complex, as measured by sedimentation velocity experiments (Fig 1E), they did diminish complex stability, as measured by differential scanning calorimetry, as well as its enzymatic activity (Fig 1F–G). We observed that wild-type hNatA melted at 46.47 ± 0.011°C, while Y8 34F and Y834A melted at 45.41 ± 0.0084°C and 41.98 ± 0.012°C, respectively. We also observed a reduction in enzymatic velocity of the Y834F and Y834A mutants relative to wild-type by 38.6% and 72.7%, respectively. Together, these studies demonstrate that the metazoan-specific extended C-terminal segment of hNaa15 makes important contributions to hNatA activity.
HYPK specifically inhibits human NatA
In order to determine the effects of human HYPK on NatA enzymatic activity, we incubated hNatA with bacterially-produced recombinant human HYPK and measured hNatA activity against H4 peptide in an in vitro radioactive acetyltransferase assay. Surprisingly, incubation of human HYPK with the hNatA complex resulted in a significant reduction in hNatA activity (Fig 2A). To assess the specificity of HYPK to inhibit other acetyltransferases, we extended our in vitro enzymatic assays to include SpNatA complex as well as other human NATs (NB, D, E and F) and histone acetyltransferases (hMOF and hPCAF). We found that HYPK did not inhibit the other acetyltransferases, although hNatF and hPCAF showed a marginal but significant level of activation (Fig 2A), likely attributed to the known protein chaperone-like activity of HYPK (Raychaudhuri et al., 2008b; Szolajska and Chroboczek, 2011). Previous studies also showed that HYPK does not interact with NatB or NatC when subjected to immunoprecipitation (Arnesen et al., 2010), consistent with the enzymatic studies. Also consistent with the enzymatic results, we used MBP pull-down studies employing MBP-HYPK to demonstrate that HYPK directly interacts with hNatA but not SpNatA (Fig 2B), consistent with the hypothesis that HYPK inhibits hNatA through residues that are divergent with the S. pombe enzyme at the C-terminus of hNaa15 (Fig S3).
Figure 2. Specific Binding and Inhibition by HYPK to Human NatA.

(A) Bar graph showing the relative effect of HYPK presence on Nt- (NAT) and Histone acetyltransferase (HAT) activity (PhNatA=0.0003, PhPCAF=0.0148, and PhNatF=0.0253, by Student’s unpaired, two-tailed t-test). Reactions were performed in triplicate; error bars S.E.M.
(B) MBP pull-down assay comparing the ability of HYPK to bind to the human and SpNatA complexes.
Overall structure of the human NatA/HYPK complex reveals a bipartite mode of HYPK inhibition of NatA
To elucidate the molecular basis for HYPK-mediated hNatA complex inhibition, we overexpressed the recombinant ternary human NatA/HYPK complex using baculovirus-co-infected Sf9 cells. We were initially able to obtain crystals of the full-length HYPK bound to the hNatA complex, but these crystals diffracted poorly. Using a combination of sequence conservation (Fig 3A), limited proteolysis and LC-MS we identified an N-terminally truncated HYPK construct (Δ1–33), which yielded hNatA/HYPK crystals that diffracted to 3.15 Å resolution (Fig 3B–D, and Table 1). The crystals formed in the P63 space group and contained one hNatA/HYPK complex in the asymmetric unit. We determined the structure of the hNatA/HYPK complex using hNatA as a molecular replacement model and refined the model to Rwork and Rfree values of 22.1% and 25.5%, respectively, with good geometry (Table 1).
Figure 3. Structure of HYPK-Bound Human NatA.

(A) Sequence alignment of HYPK with Nascent polypeptide-Associated Complex (NAC) proteins from M. thermautotrophicus (Mt); S. cerevisiae (Sc); H. sapiens (Hs); and S. pombe (Sp). Secondary structure determined from hNatA/HYPK structure shown with hNaa10 (black square) and hNaa15 contacts (red circle) in the middle. Dashed line indicates construct used for crystallization. Ubiquitin-Associated domain (UBA) indicated by black bar.
(B) HYPK-bound hNaa10 (purple) and hNaa15 (dark teal) shown bound to HYPK (light orange) in cartoon with bound IP6 (stick format).
(C) HYPK-bound hNatA complex shown in cartoon superimposed with hNaa10 (light cyan) and hNaa15 (dark blue). hNaa10 α4 is annotated in yellow.
(D) A 90° view rotation of C. (E) Zoom view of the interface between the HYPK UBA domain (90–129) and the hNaa15 α38 and α40 helices.
(F) Zoom view of HYPK-bound hNaa15 in E. in tube and stick format depicting residues involved in HYPK-hNaa15 interaction.
(G) A 180° view rotation of F.
(H) Zoom view of the hNaa10 active site demonstrating the obstruction and rearrangement of key active site residues by HYPK.
(I) Zoom view of Y49.
(J) Zoom view of E74.
The structure of hNatA/HYPK overlays well with the hNatA structure with an R.M.S. deviation of 0.617 Å over the common Cα atoms of 776 residues and, like hNatA, also contains electron density corresponding to IP6. HYPK is composed of five α-helices that engage with hNatA by forming a clamp around the hNaa15 auxiliary subunit (Fig 3B–D). This clamping causes significant conformational changes to accommodate HYPK binding (Fig 3C–E), specifically in helices α38/α40 and α1–3 of hNaa15.
The C-terminal α3–5 helices of HYPK adopts a classical UBA (ubiquitin-associated) fold (Hofmann and Bucher, 1996; Mueller and Feigon, 2002) that facilitates binding to the hNaa15 C-terminus via the α38/α40 helices and burying a total surface area of 643.18 Å2 (Fig 3D and E), shifting and stabilizing the hNaa15 α40 helix by ~6 Å. The contacts made between the HYPK UBA domain and the hNaa15 C-terminus primarily consist of a network of van der Waals interactions between HYPK (L97, E101, M102, E103, V121, L124, I125, and T128) and hNaa15 (L689, L690, Q693, T729, T732, V733, Q736, and E737) as well as a set of hydrogen bonds between the backbone of T100 of HYPK and K687 of hNaa15, and E101 of HYPK and K685 and a water-mediated hydrogen-bond with E655 of hNaa15. A network of hydrogen bonds between N129 and T128 of HYPK with R697 and K696/E737 of hNaa15 caps the hNaa15-UBA domain interaction (Fig 3A, F, and G). The largely hydrophobic protein interaction properties of the HYPK C-terminus are typical of proteins featuring a UBA fold, which has been shown to facilitate the protein-protein interactions mitigated by other UBA-fold containing proteins, such as the UV excision protein Rad23A with HIV-1 Vpr (Mueller and Feigon, 2002).
The hNaa15 N-terminal α1–3 helices move by ~2–3 Å to accommodate the apex of the N-terminal HYPK α1 helix (Fig 3A, C, D, and I). There are only a few contacts with HYPK that facilitates the interaction between both N-termini. hNaa15-H23 shifts by ~3 Å in order to form a Π-Π interaction with HYPK-Y49 (Fig 3I). hNaa15-R531, which is already in position for engagement with HYPK in the hNatA structure without HYPK, makes hydrogen-bond interactions with the backbone carbonyl oxygen atom of HYPK V46. Additional residues, such as hNaa15 Y528, F525, and D524 shift slightly to accommodate the HYPK N-terminal helix. In addition, the HYPK N-terminal helix appears to lay on top of the hNaa10 α2 helix within a hydrophobic pocket, where several hNaa10 residues including Y30, K29, and Y33 make van der Waals contacts with HYPK V46 and L43, and hNaa10-Y26 forms a hydrogen bond with HYPK-H35. The N-terminal loop preceding the HYPK α1 helix, and HYPK-H35 in particular, appears to reorient the positions of hNaa10 E24, Y26, H110, R112, Y137, and Y138 (Fig 3H) The reorientation of residues Y137 and E24, the corresponding residues of which were previously reported to be involved in SpNatA-mediated catalysis (Liszczak et al., 2013), likely plays a particularly important role in HYPK-mediated inhibition of hNatA activity.
A 27-residue-long HYPK α2 helix serves to bridge the bipartite interactions between the α3–5 helices with hNaa15 and the α1 helix with hNaa15 and hNaa10. The HYPK α2 helix is largely free of hNatA contact except for the N-terminal end, which packs against the hNaa15 α8–9 helices (Fig 3B and D). HYPK-R71 engages with hNaa15 E135 and Y138 via the formation of a hydrogen bond and a cation-Π interaction, respectively. HYPK-E74 forms a hydrogen bond with hNaa15-Y158 and an electrostatic interaction with hNaa15-R134, while the flanking HYPK-R70 forms a salt bridge with hNaa15-D163 in the α9-loop-α10 segment. The distal HYPK-R81 forms a charged-charged interaction with hNaa15-E131. A number of van der Waals contacts close the gap between the HYPK α2 helix (L60, M64, I67, and R70) with hNaa15 (Y138, L141, W151, Y154, and M166) (Fig 3J). Together, the HYPK α2 helix, as a feature that distinguishes HYPK from NAC proteins, serves the purpose of directing the HYPK N-terminus into the hNaa10 active site. The α1-loop-α2 segment threads between the hNaa15 helices, which hold the loop in place by forming a combination of hydrophobic interactions (HYPK-I55 with hNaa15 F174 and T177), hydrogen bonds and charged-charged interactions (HYPK K53, E54, and Q56 with hNaa15 R144, R148, and T180).
HYPK increases NatA thermal stability and deletion and mutational analysis is consistent with a bipartite mode of NatA inhibition
To quantify the elaborate contacts within hNatA and the impact of HYPK binding to hNatA, we compared the thermostability of hNatA and hNatA/HYPK using differential scanning calorimetry. We found that while the hNatA complex melted at 46.47 ± 0.011°C, the binding of HYPK caused a marked increa se of ~6.1°C with a melting temperature of 52.57 ± 0.049°C (Fig 4A).
Figure 4. Molecular and Thermodynamic Details of HYPK-hNatA Interaction.

(A) Differential scanning calorimetric analysis comparing hNatA and hNatA/HYPK complexes.
(B) MBP pull-down assay evaluating the effects of mutations on critical HYPK residues or HYPK truncation on the hNatA binding.
(C) Bar graph evaluating the effect of mutant and truncation HYPK constructs on hNatA inhibition potency. Assay performed in triplicate; error bars S.E.M. WT, wild type.
To probe the functional importance of the crystallographically observed hNatA–HYPK interactions, we prepared a series of HYPK truncation constructs (Δ1–59, Δ60–129, Δ1–33, Δ1–39, Δ1–45, and Δ1–89) and site-directed mutations in the α1 helix (H36A and L43A/V46A), α2 helix (E74A), and UBA domain (E101A/E103A and V121A/I125A) for hNatA pull-down and enzymatic studies. The MBP-HYPK pull-down studies revealed that all the HYPK constructs with the exception of HYPKΔ60–129 deletion construct, which lacks the α2 helix and the UBA domain, were able to pull-down the hNatA complex (Fig 4B). This suggests that most of the binding energy between HYPK and hNatA is mediated by the α3–α5 UBA domain of HYPK. The observation that none of the double point mutants in the UBA domain had a discernible effect on HYPK-hNatA complex pull-down, highlights the strength of this interaction.
Evaluation of the ability of the HYPK constructs described above to inhibit hNatA activity, were consistent with the observations made in the crystal structure of the hNatA/HYPK complex. We observed that substantial relief of inhibition arose from HYPK mutants H36A, the double L43A/V46A point mutant, the deletion of the loop region before the N-terminal helix (Δ1–39), and the deletion of either the first or second half of HYPK (Δ1–59 or Δ60–129) (Fig 4C). Based on this data, it appears that the primary mode of inhibition arises from interactions within the hNaa10 active site with the HYPK loop-α1 region (H36 and L43/V46). This is underscored by the release of inhibition upon deletion of the N-terminal loop or the first half of HYPK, resulting in an increase in hNatA activity of either ~44% or ~65%, respectively. The observation that HYPKΔ1–59 shows drastically reduced inhibition activity reveals that the HYPK α2 helix and the UBA domain, both contained within this construct, do not contribute directly to inhibition. However, given that the UBA domain (residues 90–129) provides most of the binding energy of HYPK to hNatA, the UBA domain and α2 helix play key roles in HYPK inhibition of hNatA by bringing HYPK to hNatA and appropriately orienting the loop-α1 region of HYPK into the hNaa10 active site, respectively.
HYPK is a tight binding (Morrison-type) hNatA inhibitor with properties of partial uncompetitive and noncompetitive inhibition with respect to acetyl-CoA and peptide, respectively
NATs proceed using a bi-bi ordered mechanism where acetyl-CoA binds the NAT followed by the peptide substrate to form a ternary complex prior to catalysis (Evjenth et al., 2009; Liszczak et al., 2013). In order to determine the mechanism of HYPK-mediated inhibition, we characterized the kinetic parameters of the hNatA and hNatA/HYPK complexes with respect to H4 peptide and acetyl-CoA using an in vitro acetyltransferase assay (Fig 5A and B, and Table 2). We found that HYPK-binding decreased H4 peptide kcat ~12-fold while KM remained the same. By contrast, HYPK binding resulted in a reduction in kcat by ~15-fold and KM ~14-fold with respect to acetyl-CoA. It is interesting to note that HYPK improves the ability of acetyl-CoA to bind hNatA (lowers KM) such that the concentration is within a physiological range (~2–13 μM) (Lee et al., 2014), suggesting that HYPK acts to potentiate hNatA for acetyl-CoA binding and potentially sensitize hNatA towards acetyl-CoA flux similar to other acetyltransferases (Pietrocola et al., 2015). Based on this mechanistic data and the incomplete inhibition pattern, we propose that binding of HYPK to hNatA results in partial uncompetitive inhibition with respect to acetyl-CoA and partial noncompetitive inhibition with respect to peptide.
Figure 5. Mechanism of NatA Inhibition.

Michaelis-Menten kinetics of hNatA and hNatA/HYPK complexes (10 nM) with respect to
(A) H4
(B) Acetyl-CoA
(C) Dose-response curve corresponding to titration of full-length MBP-HYPK recombinant protein, HYPK peptide (containing the residues 35–48), and the non-specific gag peptide.
(D) Overlay of the SASE-CoA bisubstrate inhibitor (pdb: 4KVM, CMK stick format) bound in the hNatA-HYPK active site.
(E) Morrison inhibition plot of the dose-response curve corresponding solely to the titration of full-length MBP-HYPK recombinant protein.
Assays performed in triplicate; error bars correspond to the S.E.M. for each point.
Table 2.
Catalytic Parameters for wild-type hNatA and hNatA/HYPK complexes
| Substrate | Complex | kcat (min−1) | KM (μM) | kcat/KM (μM min−1) |
|---|---|---|---|---|
| Acetyl-CoA | hNatA | 150 ± 8.9 | 27 ± 3.9 | 5.5 ± 0.86 |
| hNatA/HYPK | 10 ± 0.39 | 1.7 ± 0.31 | 6.1 ± 1.1 | |
| H4 peptide | hNatA | 200 ± 8.1 | 30 ± 3.6 | 6.8 ± 0.90 |
| hNatA/HYPK | 16 ± 1.6 | 31 ± 9.5 | 0.52 ± 0.10 |
Errors represent S.E.M. (n=3)
The changes in kinetic parameters observed upon binding of HYPK to hNatA are consistent with the details observed in the structure. The HYPK N-terminal loop displaces the characteristic NAT β-hairpin (β6-loop-β7) that participates in substrate protein binding (Liszczak et al., 2011) by ~3–4 Å, likely causing the altered shape of the hNaa10 α4 helix and, consequently, the acetyl-CoA binding pocket (Fig 3C, 3H, and 5C). Furthermore, we found that hNaa10 R82 and R83, the corresponding residues of which participate in acetyl-CoA binding in the SpNatA structure, are well-ordered in the hNatA structure but are disordered and, in turn, potentially more accommodating for acetyl-CoA binding in the hNatA/HYPK structure. The repositioning of the NAT β-hairpin likely also contributes to the inability of the active site to promote catalysis. The restructuring of the peptide binding pocket upon binding of HYPK to hNatA by the repositioning of the key residues that contribute to catalysis, H110, R112, and Y138 (as demonstrated by studies using SpNatA (Liszczak et al., 2013)), likely contributes to a decrease in the kcat. Additionally, HYPK-K34 makes backbone hydrogen bonds with hNaa10-Y137 and HYPK-H35 forms a hydrogen bond with hNaa10-Y26, fastening the HYPK N-terminus in place. Consequently, HYPK H35 sterically clashes with the hNatA general base residue, E24, causing the reorientation of the carboxylic acid moiety. Superposition of the human hNatA/HYPK structure with SpNatA bound to a bisubstrate inhibitor reveals that the N-terminal loop of HYPK sterically clashes with only residue four of the peptide moiety (Fig 5C), does not engage with any of the peptide binding residues, and all of the peptide binding residues maintain their same position (Fig 3H). This observation demonstrates that HYPK binding to hNatA does not perturb peptide binding and is consistent with the noncompetitive nature of HYPK and peptide binding to hNatA.
To quantify and parse out the details HYPK-mediated hNatA inhibition, we conducted inhibition studies using either full-length recombinant MBP-tagged HYPK, a synthetic HYPK peptide corresponding to the N-terminal loop-α1 helix (residues 35–48), or a non-specific gag peptide corresponds to the first seven residues of the sequence of the S. cerevisiae NatC substrate (Fig 5D). We found that we were unable to fit a half-maximum concentration curve (IC50) for the recombinant HYPK due to the tight-binding properties of the interaction between HYPK and the hNatA complex. However, we were able to determine that the HYPK35–48 peptide has a modest IC50 of 420 ± 24 μM, which, taken together with the pull-down of HYPK truncations and mutants, is consistent with the mechanism of binding primarily arising from the UBA domain.
Due to the tight-binding affinity between HYPK and hNatA, we performed enzymatic assays under stoichiometric binding conditions. Using a tight-binding equation modified to incorporate a partial noncompetitive mode of inhibition (see Methods), we calculated a Ki of 0.7 ± 0.3 nM (Fig 5D). Consistent with the observed tight-binding capacity of HYPK with hNatA, other studies have found that the hydrophobic patch of the UBA domain is responsible for as low as low-nanomolar (VanderLinden et al., 2017; Wilkinson et al., 2001) to micromolar dissociation constants (Hurley et al., 2006).
HYPK likely indirectly inhibits NatE activity in vivo
In order to determine whether hNatA could form a complex concurrently with HYPK and hNaa50, we superimposed the hNatA and hNatA/HYPK structures with the S. cerevisiae (Sc) NatA/Naa50 structure (PDB: 4XPD (Neubauer, 2012)). Interestingly, we observed that the hNaa15 helical bundle that contacts HYPK shifts towards HYPK (α38–α40), while it shifts away from HYPK (in the opposite direction) in ScNaa15 relative to the corresponding helical bundle in hNatA alone (Fig 6A). This difference in helical position appears to result in different hNaa15 surfaces for hNaa50 binding, which is dependent on HYPK binding to hNatA (Fig 6B), suggesting that HYPK and hNaa50 binding to hNatA may be mutually exclusive. To test this hypothesis, we performed competition pull-down studies by incubating either MBP-HYPKΔ1–33 crystallization construct or GST-hNaa50 with hNatA and competing with either untagged hNaa50 or untagged HYPKΔ1–33, respectively. We found that hNaa50 did not pull-down with MBP-HYPKΔ1–33 preincubated with hNatA. In contrast, we observed an appreciable decrease in hNatA pull-down by GST-tagged hNaa50 in the presence of HYPKΔ1–33 (Fig 6C), which suggests that the order of binding influences the composition of hNatA-mediated complex formation. Taken together, this observation indicates a preferential order of binding of HYPK to hNatA. This is likely a result of HYPK tight-binding in conjunction with the incompatibility of the HYPK-induced conformational change with the changes necessary for simultaneous HYPK/Naa50 binding and Naa50 recruitment to the ribosome for NatE-mediated co-translational catalysis. This is consistent with previous studies indicating that the majority of endogenous hNaa50 does not associate with hNatA (Hou et al., 2007).
Figure 6. HYPK Binding Reduces Human NatA Capacity to Bind Naa50.

(A) Overlay of S. cerevisiae (Sc) Naa50-bound NatA (pdb: 4XPD; ScNatA: green; ScNaa50: magenta) with hNatA (dark blue) and HYPK (light orange) bound to hNatA (dark teal). Helices are illustrated in cylindrical cartoons and enumeration of human NatA helices are indicated. Arrows indicate the direction of Naa15 conformational change induced upon HYPK (dark teal) or Naa50 (green) binding.
(B) A 90° view rotation of A.
(C) Competition pull-downs of HYPK and hNaa50 binding to hNatA with MBP-HYPKΔ1–33 as bait in the presence of amylose resin (left) and GST-hNaa50 as bait in the presence of glutathione resin (right).
Discussion
While the molecular basis for the function of SpNatA has previously been described (Liszczak et al., 2013), we set out to understand the molecular mechanism of regulation of its more elaborate metazoan counterpart, which associates with the intrinsically disordered protein, HYPK. To do this, we determined the X-ray crystal structure of hNatA in the absence and presence of HYPK and carried out associated biochemical and enzymatic studies to make structure-function correlations. We found two unique features of hNatA relative to SpNatA: an additional ~190 residue C-terminal segment in the hNaa15 regulatory subunit that contributes to hNatA stability and acetyltransferase function, and a bound IP6 molecule at the hNaa10-hNaa15 interface that also appears to contribute to hNatA stability. While inositol phosphates such as IP6, more typically play roles in protein signaling cascades and regulating other cellular processes (Shamsuddin and Bose, 2012), there are a handful of other examples of their roles in mediating protein stability and enzymatic function. This includes the presence of IP6 in the mammalian adenosine deaminase ADAR1 (Macbeth et al., 2005) and the HopZ1a acetyltransferase from P. syringae, as well as the presence of IP4 in the histone deacetylase HDAC3-SMART/DAD complex (Watson et al., 2012).
This study also describes the molecular basis for interaction between hNatA complex with its binding partner, HYPK. Our findings show that HYPK specifically inhibits hNatA over SpNatA, as well as other NATs and HATs. To our knowledge, this is the first protein with intrinsic acetyltransferase inhibitory activity. Due to the tight and stabilizing nature of the hNatA-HYPK interaction with a calculated Ki of 0.7 nM, our findings suggest that the metazoan NatA and HYPK form an obligate ternary complex in vivo. This is consistent with earlier studies showing that siRNA-mediated hNatA or HYPK knockdown in HeLa cells leads to similar phenotypes of cell death and cellular accumulation in the G1/G0 phase (Arnesen et al., 2010). We find that HYPK uses a bipartite mode of hNatA inhibition in which a C-terminal UBA three-helix bundle (α3–α5) mediates high affinity binding to the hNaa15 auxiliary subunit while an N-terminal loop-helix segment (loop-α1) interacts with hNaa15 and the hNaa10 active site to interfere with catalysis. The partial uncompetitive and noncompetitive modes of HYPK inhibition with respect to acetyl-CoA cofactor and N-terminal peptide sequence, respectively, assures that HYPK promotes acetyl-CoA binding so that hNatA interacts with acetyl-CoA in the physiological range of ~ 1 μM, and is also able to be displaced by peptide substrate that emerges from the ribosome exit tunnel. While HYPK inhibits the intrinsic activity of NatA in vitro, it likely plays a more nuanced regulatory role in vivo. Taken together, we hypothesize that HYPK may serve as a hNatA substrate proofreading mechanism for hNatA activity.
Unexpectedly, a superposition of the human hNatA/HYPK structure reported here with a ScNatA/Naa50 structure, reveals that although HYPK and Naa50 bind to different NatA surfaces, they mediate NatA conformational changes that are incompatible with simultaneous binding of HYPK and Naa50 to NatA, which we confirm using competition pull-down studies. These findings suggest that HYPK may indirectly inhibit co-translational NatE (NatA/Naa50) activity in cells since Naa50 is targeted for co-translational Nt-acetylation through its interaction with NatA at the ribosome. In contrast to these findings, a previous study in D. melanogaster reported that Naa50 co-immunoprecipitates with HYPK and the NatA complex (Rathore et al., 2016). In light of this finding, it is possible that an additional cellular protein could facilitate the simultaneous binding of HYPK and Naa50 to NatA. An additional superposition of the hNatA/HYPK complex with NatA/HYPK structure from the thermophilic fungus Chaetomium thermophilum, reveals a similar mode of bipartite HYPK binding to NatA, further reinforcing a degree of evolutionary conservation in the mode of intrinsic HYPK inhibition of NatA (Weyer et al., 2017) and potentially also NatE.
Although we demonstrate that HYPK uses a bipartite mechanism for robust hNatA inhibition (Ki of 0.7 nM), we are able to show that a peptide harboring the N-terminal loop-helix (residues 35–48) segment of HYPK is able to inhibit hNatA with a modest but significant IC50 of ~400 μM. This result clearly highlights the importance of the UBA domain in HYPK inhibition of NatA, but also points to the possibility of elaborating on the N-terminal loop-helix HYPK segment for the development of small molecule hNatA inhibitors.
HYPK contains a Nascent polypeptide-Associated Complex alpha (NACα) domain (residues 90–129, α3–α5 (Raychaudhuri et al., 2014)). The NAC complex interacts with the ribosome, potentially interacts with the chaperone Hsp70, and contacts nascent peptides (Andersen et al., 2007; Koplin et al., 2010). Unlike the NAC proteins, however, HYPK contains additional α-helices (α1–α2) while NAC proteins contains N-terminal β-sheets, which are responsible for NAC complex dimerization (Liu et al., 2010). Since our study suggests that HYPK is likely constitutively associate with NatA, which is known to associate with the ribosome and nascent polypeptides, we propose that HYPK also interacts with these molecules as well, possibly to facilitate Nt-acetylation fidelity. HYPK has also been reported to interact with Huntingtin protein and to reduce Huntingtin aggregation (Arnesen et al., 2010; Raychaudhuri et al., 2008b). Interestingly, the N-terminal sequence of ATLEK-found in Huntingtin protein, would be predicted to be a substrate for NatA and, by consequence, a possible target for the N-end pathway (Varshavsky, 2011). Whether these observations are just coincidence or functionally connected will require further investigation. Taken together, these studies redefine the composition of the hNatA complex in metazoan species and describe the unique intrinsic NAT inhibitory properties of HYPK, providing a model for HYPK regulation of NatA-mediated protein N-terminal acetylation.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronen Marmorstein (marmor@upenn.edu).
Experimental Model
We used E.coli Rosetta (DE3)pLysS cells for recombinant expression of MBP-tagged HYPK for biochemical and biophysical experiments. The cells were cultured using standard practices in LB media.
We used Spodoptera frugiperda (Sf9) cells cultured in SFM II medium for the recombinant expression of hNatA and co-expression with HYPK for biochemical, biophysical, and X-ray crystallography experiments.
Method Details
Construction of E. coli Expression Vectors
MBP-HYPK was N-terminally truncated (Δ1–59, Δ60–129, Δ1–33, Δ1–39, Δ1–45, and Δ1–89) and mutated (H36A, E74A, L43A/L46A, E101A/E103A, and V121A/I125A) using ‘round the horn’ site directed mutagenesis with the pET-M41 HYPK vector as a template (IDT). MBP-HYPKΔ60–129 was constructed through the introduction of a TGA mutation into HYPK by the Stratagene QuikChange protocol.
Construction of Baculoviruses
A C-terminal truncation construct coding for the human Naa101–160 and full-length Naa15 (866 residues) was engineered using a pFastBac dual vector. A full-length tobacco etch virus (TEV)-cleavable 6xHis-tagged human NatB construct, consisting of Naa20 (178 residues) and Naa25 (972 residues), was also engineered using a pFastBac dual vector. Studies were performed with a hNaa15 subunit that contained either an uncleavable N-terminal 6xHis-tag or a TEV-cleavable 6xHis-tag, as indicated below. Truncation of hNaa15 (hNaa15Δ672–866) was generated by introduction of a TGA mutation and the mutant, hNaa15Y834F, was generated by introduction of a TTT mutation, all using Stratagene QuikChange protocol with the uncleavable 6xHis-tag vector. MBP-tagged full-length human HYPK was subcloned from the pET-m41 expression vector into a pFastBac 1 vector. MBP-HYPKΔ1–33 was generated using ‘round the horn’ site directed mutagenesis with the pFastBac 1 MBP-HYPK vector as a template. For each of these constructs, a bacmid was generated by transposition into DH10 bac competent E. coli cells using the bac-to-bac system (Invitrogen). Spodoptera frugiperda (Sf9) cells cultured in SFM II medium were transfected with the bacmid using cellfectin reagent (Invitrogen). The resulting baculovirus was amplified until reaching a high titer.
Expression and Purification of MBP-tagged HYPK Constructs
All MBP-tagged HYPK constructs were expressed in Rosetta (DE3)pLysS competent E. coli cells. Cells were grown in LB-media at 37°C to OD 600 0.6–0.7 prior to inducing protein expression with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18°C for ~16 hrs. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation and lysed in lysis buffer containing 25 mM Tris, pH 8.0, 150 mM NaCl, 10 mM β-mercaptoethanol (β-ME), 10 μg/ml phenylmethanesulfonylfluoride (PMSF), and DNase (Invitrogen). The lysate was clarified by centrifugation and incubated with amylose agarose resin (New England Biolabs) for 1 hr. before washing the resin with ≥80 column volumes (CV) of lysis buffer and then eluted with 10 CV of lysis buffer supplemented with 20 mM maltose by batch elution. Eluted full-length and truncated MBP-HYPK constructs were loaded onto a 5 mL HiTrap Q ion-exchange column (GE Healthcare). The protein was eluted in the same buffer with a salt gradient (150 mM – 1 M NaCl) over the course of 20 CV. Peak fractions were pooled and concentrated to a volume of 500 μl (50 kDa concentrator; Amicon Ultra, Millipore), and loaded onto and run on a Superdex 200 Increase 10/300 GL gel filtration column (GE Healthcare) in gel filtration buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, and 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP). Peak fractions were pooled and concentrated to ~20–40 mg/mL by UV280 (Nanodrop 2000; Thermo Fisher Scientific), and flash-frozen for storage at −80°C until use. Later studies were performed using samples prepared from peak fractions that had been pooled after ion exchange, dialyzed into gel filtration buffer and flash-frozen. Initially, pull-down and inhibition studies were performed using samples purified by gel filtration chromatography, however, results between gel filtration-purified and ion exchange-purified samples were identical. HYPK mutants and truncation constructs were purified following the same protocols with the following changes. Subsequent to elution from the amylose affinity column, the eluent was immediately purified by gel filtration chromatography to confirm proper folding.
Expression and Purification of 6xHis-tagged hNatA constructs
Sf9 cells were grown to a density of 1×106 cells/ml and infected using the amplified Naa101–160/Naa15 baculovirus to an MOI (multiplicity of infection) of ~1–2. The cells were grown at 27°C and harvested 48 hours post-infection. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation and lysed in lysis buffer containing 25 mM Tris, pH 8.0, 500 mM NaCl, 10 mM Imidazole, 10 mM β-ME, 10 μg/ml PMSF, DNase, and complete, EDTA-free protease inhibitor tablet (Roche). The lysate was clarified by centrifugation and incubated with nickel resin (Thermo Scientific) for 1 hr. before washing the resin with ~125 CV of lysis buffer and then eluted with 10 CV of elution buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 200 mM Imidazole, 10 mM β-ME) by batch elution. Eluted protein was diluted to a final salt concentration of 200 mM NaCl and loaded onto a 5 mL HiTrap SP ion-exchange column (GE Healthcare). The protein was eluted in the same buffer with a salt gradient (200mM-1 M NaCl) over the course of 20 CV. Peak fractions were pooled, concentrated to a volume of 500 μl (100 kDa concentrator), and loaded onto and run on a Superdex 200 Increase 10/300 GL gel filtration column in sizing buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, and 1 mM TCEP. Peak fractions were pooled, concentrated to 4.8 mg/mL, as measured by UV280, and flash-frozen for storage at −80°C until use. Mutants and hNatA truncations were prepared as described for wild-type hNatA.
Expression and Purification of hNatA/HYPK
Sf9 cells were grown to a density of 1×106 cells/ml and co-transfected with an equal volume of the MBP-HYPK and TEV-cleavable 6xHis-tagged hNatA baculoviruses to an MOI of ~1–2. The cells were grown at 27°C and harvested 48 hours post-infection. All purification steps were carried out at 4°C and are identical to the protocol implemented for the binary hNatA complex, with the following changes for the production of ternary complex for X-ray diffraction studies. After elution from the nickel column, the eluted protein was diluted to a final NaCl concentration of ~267 mM and incubated overnight with TEV protease to cleave the MBP- and 6xHis-tags. The MBP-cut protein was then diluted to the final NaCl concentration of 200 mM and loaded onto the SP ion-exchange column.
Expression and Purification of hNatB
hNatB was produced from Sf9 cells, as described above for hNatA. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation and lysed in lysis buffer containing 25 mM Tris, pH 8.0, 300 mM NaCl, 10 mM Imidazole, 10 mM β-ME, 10 μg/ml PMSF, DNase, and complete, EDTA-free protease inhibitor tablet. The lysate was clarified by centrifugation and incubated with nickel resin for 1 hr. before washing the resin with ~125 CV of lysis buffer supplemented with a final concentration of 25 mM Imidazole and then eluted with 10 CV of elution buffer (25 mM Tris, pH 8.0, 300 mM NaCl, 200 mM Imidazole, 10 mM β-ME) by batch elution. The resulting eluent was pooled, cut overnight with TEV protease to remove the 6xHis-tag, and dialyzed into 25 mM Tris pH8 300 mM NaCl 5 mM Imidazole 10 mM β-ME. The protein was then incubated on nickel column for an hour, after which the flow-through was collected and dialyzed overnight into 25 mM HEPES pH 7.5 50 mM NaCl 10 mM β-ME. The protein was loaded onto an SP ion exchange column and eluted in the same buffer with a salt gradient (50–750 mM NaCl) over the course of 20 CV. Peak fractions were pooled, concentrated to a volume of 500 μl (100 kDa concentrator), and loaded onto and run on a Superdex 200 Increase 10/300 GL gel filtration column in the same sizing buffer as hNatA. Peak fractions were pooled, concentrated to ~6 mg/mL, as measured by UV280, and flash-frozen for storage at −80°C until use.
Expression and Purification of hNatD
Full-length hNatD (237 residues) was engineered into a pCDF vector containg an N-terminal TEV-cleavable 6xHis-tag. hNatD was expressed using Rosetta (DE3)pLysS competent E. coli cells, which were grown at 37°C to an OD 600 of ~0.7–0.9 and induced by addition of 0.5 mM IPTG at 16°C for 16 hr. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation, lysed by soication in lysis buffer containing 25 mM Tris, pH 8.0, 1 M NaCl, 10 mM β-ME and 10 mg/mL PMSF. The lysate was clarified by centrifugation and passed over nickel resin, which was subsequently washed with >20 CV of lysis buffer supplemented with 25 mM imidazole. The protein was eluted in lysis buffer supplemented with 300 mM imidazole by batch elution. The 6xHis-tag was cleaved overnight by addition of 6His-tagged TEV-protease during dialysis into dialysis buffer containing 25 mM Tris, pH 8.5, 200 mM NaCl, 10 mM β-ME This solution was passed through an additional nickel column to remove TEV protease as well as any uncut hNatD. The resin was then washed with approximately 7 CV of dialysis buffer supplemented with 25 mM imidazole, which was pooled with the initial flow through. This solution was dialyzed into ion exchange buffer containing 25 mM Tris, pH 8.5, 50 mM NaCl and 1 mM dithiothreitol (DTT) and loaded onto a 5 ml HiTrap Q ion exchange column. The protein was eluted in the same buffer with a salt gradient (50–750 mM NaCl) over the course of 20 CV. Peak fractions were pooled and dialyzed into sizing buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, and 1 mM DTT for ~16 hours. This was concentrated to a volume of 500 ml (10 kDa concentrator), and loaded onto and run on an Superdex 75prep gel filtration column (GE Healthcare) in sizing buffer. Peak fractions were concentrated to 10 mg/ml as measured by UV280 and flash-frozen for storage at −80°C until use (Magin et al., 2015).
Expression and Purification of hNaa50
The full-length human Naa50 pETM-GST vector was engineered into a pETM-GST vector with an N-terminal TEV-cleavable GST tag. hNaa50 was expressed using BL21(DE3) cells, which were grown at 37°C to an OD600 of ~0.7 and induced by addition of 0.5 mM IPTG at 18°C for 16 hr. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation and lysed by sonication in lysis buffer containing 25mM HEPES pH 7.5, 100 mM NaCl, and 10 mM β-ME. The supernatant was isolated and passed over GST-binding resin (Gold Biotechnology). Unbound proteins were washed off the resin with lysis buffer, and TEV was added directly to the resin and allowed to incubate overnight at room temperature. Untagged hNaa50 was washed off the resin with lysis buffer, collected, dialyzed into low-salt buffer (25mM HEPES pH 7.5, 50 mM NaCl, and 10 mM β-ME) and bound to a 5-ml HiTrap SP ion exchange column. The protein was eluted in the same buffer using a salt gradient of 50–750 mM NaCl and further purified to homogeneity using a S75 gel filtration column in storage buffer (25 mM HEPES pH 7.5, 100 mM NaCl, and 10 mM DTT). Peak fractions were concentrated to ~12 mg/ml using a 10-kDa protein concentrator and stored at −80°C until use (Liszczak et al., 2011). GST-tagged protein was produced following the same protocol with the following exceptions. Glutathione-resin bound hNaa50 was eluted from the resin using lysis buffer supplemented with 20 mM glutathione. The eluent was then dialyzed for ion exchange overnight.
Expression and Purification of hNatF
A C-terminal truncation construct coding for 6xHis-SUMO-hNaa60 (5–184) was expressed in Rosetta (DE3) pLys competent cells overnight at 17°C using 0.5 mM IPTG. Cells were lys ed by sonication in lysis buffer (25 mM Tris-HCl pH 8.0, 1 M NaCl, 10 mM β-ME, and 10 mg/ml PMSF), the cell lysate was clarified by centrifugation, and overexpressed His-tagged protein was isolated by passing the supernatant over nickel resin. Subsequent purification steps were performed at 4°C. The resin was washed with wash buffer (25 mM Tris-HCl pH 8.0, 250 mM NaCl, 20 mM imidazole, 10 mM β-ME) and bound proteins eluted by slowly increasing the imidazole concentration to 300 mM. The 6xHis-SUMO tag was cleaved by adding SUMO protease (Ulp-1) to the eluted protein while the protein was dialyzed into a buffer without imidazole (25 mM Tris-HCl pH 8.0, 250 mM NaCl, and 10 mM β-ME), and the tag was removed from the sample by reverse nickel purification. The flowthrough was collected and dialyzed into size-exclusion buffer (25 mM HEPES pH 7.5, 300 mM NaCl, and 1 mM DTT). The dialyzed sample was concentrated to a final volume of 1 ml and loaded on a HiLoad Superdex 200 size exclusion chromatography column. Fractions containing hNaa605–184 were collected and concentrated to ~20 mg/mL and flash-frozen for storage at −80°C until use (Støve et al., 2016).
Expression and Purification of spNatA
The full-length S. pombe Naa15 (729 residues) was eingeered into MCSI of a pETDUET vector containing a TEV protease-cleavable 6XHis-tag with MCSII containing Naa10 (1–156). The construct was transformed into Rosetta (DE3)pLysS competent E. coli cells, grown to an OD600=0.7 and induced with 0.5 mM IPTG at 16°C for ~16 hours. Cells were isolated by centrifugation and lysed by sonication in a buffer containing 25 mM Tris pH 8.0, 500 mM NaCl, 10 mM β-ME and PMSF. Subsequent purification steps were performed at 4°C. The solution was isolated and passed over nickel resin, which was subsequently washed with >20 CV of lysis buffer supplemented with 25 mM imidazole. The protein was eluted using batch elution with buffer containing 200 mM imidazole and TEV protease was added to fractions containing the target protein for the duration of a 14-hour dialysis into lysis buffer. The solution was passed through an additional nickel column to remove TEV protease and 6xHis-tagged spNatA. The resin was then washed with ~ 7 CV of lysis buffer supplemented with 25 mM imidazole, which was pooled with the initial flow through. This solution was dialyzed into a buffer containing 25mM sodium citrate monobasic pH 5.5, 100 mM NaCl and 2 mM DTT and loaded onto a 5 mL HiTrap SP ion exchange column. The first of two peaks was concentrated to 1mL, and loaded onto an s200prep gel filtration column (GE Healthcare) in a buffer containing 25 mM HEPES pH 7.0, 200 mM NaCl and 1 mM TCEP. This protein was concentrated to 15 mg/mL as measured by UV280 until use (Liszczak et al., 2013).
Expression and Purification of hMOF
The HAT domain of hMOF (residues 174–449) was engineered into a pRSF vector with an N-terminal 6xHis-tag separated by a TEV protease recognition. The hMOF plasmid was transformed into E, coli BL21(DE3) codon plus RIL (Stratagene) cells and the protein was overexpressed in TB media, induced with 1mM IPTG at OD600 ~0.8–1.5, and grown at 15°C overnight. The cells were harvested and lysed by sonication in lysis buffer (50 mM HEPES pH 7.5, 0.5 M NaCl, 2 mM β-ME, 5 mM imidazole, 5% glycerol, 0.1% CHAPS, and PMSF), and the lysate was clarified by centrifugation. Subsequent purification steps were performed at 4°C. The lysate supernatant was loaded onto nickel resin equilibrated with lysis buffer. The resin was washed with 10 CV of lysis buffer containing 25 mM imidazole. The protein was eluted with lysis buffer containing 250 mM imidazole buffer in 10-ml fractions. The eluent was concentrated and injected onto an FPLC using a HiLoad Superdex 75 16/60 gel filtration column using sizing buffer (20mM HEPES pH 7.5, 0.5 M NaCl). Peak fractions were concentrated to ~5–15 mg/mL (10-kDa protein concentrator) as measured by UV280 and flash-frozen for storage at −80°C until use (McCullough et al., 2016).
Expression and Purification of hPCAF
The HAT-domain of hPCAF (residues 443–657) was engineered into a pET-28 and expressed in Rosetta (DE3) pLys competent cells overnight at 18°C using 0.5 mM IPTG. Cells were lys ed by sonication in lysis buffer containing phosphate buffer pH 74 (Thermo Fisher) containing 10 mM β-ME, and 10 mg/ml PMSF, the cell lysate was clarified by centrifugation, and overexpressed His-tagged protein was isolated by passing the supernatant over nickel resin. The resin was washed with lysis buffer supplemented with 25 mM imidazole and bound proteins eluted by batch elution using lysis buffer supplemented with 200 mM imidazole. The eluent was dialyzed into low-salt buffer (20 mM Sodium Citrate pH 6.0, 20 mM NaCl, and 10 mM β-ME) and bound to a 5-ml HiTrap SP ion exchange column. The protein was eluted in the same buffer using a salt gradient of 20–1 M NaCl and further purified to homogeneity using a S75 gel filtration column in storage buffer (20 mM Sodium Citrate pH 6.0, 150 mM NaCl, and 1 mM TCEP). Peak fractions were concentrated to ~2 mg/ml using a 10-kDa protein concentrator and stored at −80°C until use.
Binary hNatA Crystallization and Structure Determination
6xHis-tagged binary hNatA (4.5 mg/ml) was incubated with acetyl-CoA (Sigma-Aldrich) at a 1:3 molar ratio (although acetyl-CoA was not resolved in the final crystal structure). Crystals were obtained after 6–7 days with hanging-drop vapor diffusion in a drop containing a 1:1 mixture of protein to a 500 μL well solution containing 16% PEG 3350, 10% tascimate, pH 7.5. An additive screen (Hampton Research) revealed that diffraction could be improved when 2% acetone was present as an additive in the drop at a ratio of 2:1.6:0.4 of protein:well solution:additive. All crystals were cryoprotected by transferring them to their respective well solutions supplemented with 20% glycerol (EMD Millipore) before being flash-frozen in liquid nitrogen. Data for the binary hNatA complex were collected at beamline 24ID-C at the Advanced Photon Source (Argonne National Laboratory). All data was processed using XDS (Kabsch, 2010).
For phasing, a total of 720 degrees (0.5° per frame) were collected on a single crystal to reach desirable completeness and redundancy in the P212121 space group, and the data set was processed to 2.80 Å resolution using 600 frames. SpNatA (4KVO), where the N-terminus (5–90) and the C-terminus (560–731) in Naa15p were deleted, was used as a search model in order to generate a solution for the hNatA molecular data set. Refinement of the binary complex structure was carried out in Phenix using Phenix.refine and, until final rounds of refinement, two-fold NCS restraints. Manual refinement and additional model building were performed with Coot. Initially, the N-terminus and C-terminus of Naa15p were built as polyalanine chains. The final models were checked for errors with a simulated annealing composite omit map generated by AutoBuild in the Phenix suite.
Table 1 statistics for all data sets was generated using Phenix, with the exception of Rmerge, redundancy, and mean I/α, which were retrieved from the XDS log file (Adams et al., 2010; Kabsch, 2010).
All distance calculations, as well as three-dimensional alignment R.M.S. deviations and graphics were generated for publishing in PyMOL (http://www.pymol.org/) (Schrodinger, 2015). All surface area calculations were performed using PDBePISA (Proteins, Interfaces, Structures and Assemblies) (http://www.ebi.ac.uk/pdbe/pisa/) (Krissinel and Henrick, 2007).
hNaa15 TPR predictions were performed using the TPRpred server (https://toolkit.tuebingen.mpg.de/#/tools/tprpred) (Karpenahalli et al., 2007).
All alignments in the manuscript were performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) (Sievers et al., 2011) and visualized using ESPript 3.0 (http://espript.ibcp.fr/ESPript/ESPript/) (Robert and Gouet, 2014).
hNatA/HYPK Crystallization and Structure Determination
Ternary hNatA/HYPK (4.2 mg/ml) was incubated with acetyl-CoA at a 1:3 molar ratio (although density for acetyl-CoA was not resolved in the crystal structure). Initial crystals were obtained after 1–2 days with hanging-drop vapor diffusion in a drop containing a 1:1 mixture of protein to a 500 uL well solution containing 21% PEG 3350, 10% tascimate, pH 6.5 at 20°C, and yielded poor diffraction. These crystals were used as seeds (1:100,000) for further optimization where large crystals were obtained in a 1:1:0.2 mixture of protein, well solution (19.5% PEG 3350, 11% tascimate, pH 6.5), and seed solution. All crystals were cryoprotected by transferring them to their respective well solutions supplemented with 20% glycerol before being flash-frozen in liquid nitrogen. Data for the hNatA/HYPK complex were collected at beamline 24ID-C at the Advanced Photon Source.
For phasing, a total of 360 degrees (1° per frame) were collected on a single crystal to reach desirable completeness and redundancy in the P63 space group, and the data set was processed to 3.15 Å resolution HYPK using 80 frames. The hNatA structure was used as a search model in order to generate a solution for the ternary hNatA/HYPK molecular data set and HYPK was initially built as a polyalanine chain. All data processing and refinement, was performed using the same software packages detailed above for the hNatA structure.
Acetyltransferase Assays
hNatA acetyltransferase assays were carried out in 100 mM HEPES, pH 8.0, 50 mM NaCl 2 mg/ml BSA, where reactions were incubated with 10 nM of 6xHis-tagged hNatA alone or mixed with 5 μM MBP-HYPK (wild-type or mutant) in a 30 μl reaction volume containing 50 μM substrate peptide and [14C]acetyl-CoA (4 mCi/mmol; PerkinElmer Life Sciences) for 12 min at 25°C. The substrate peptide used in the assay corresponds to the first 19 residues of human H4 (Genscript), which was selected because it did not generate a substrate inhibition kinetic profile. However, we did observe comparable HYPK-mediated inhibition of hNatA towards the peptide corresponding to the in vivo substrate, SASEA.
SpNatA acetyltransferase assays were carried out under conditions previously reported using 50 μM radiolabeled [14C]acetyl-CoA and 400 μM H4 peptide, which was used for consistency with the hNatA assays.
hNatB (25 nM) acetyltransferase assays were carried out in 25 mM HEPES 7.5 200 mM NaCl using 400 μM each of actin peptide and [14C]acetyl-CoA in a 50 μl reaction volume for 20 min at 25°C.
hNatD (225 nM) acetyltransferase assays were carried out in 25 mM HEPES 7 100 mM NaCl using 60 μM H4 peptide and 100 μM [14C]acetyl-CoA in a 50 μl reaction volume for 15 min at 25°C.
hNaa50 (300 nM) acetyltransferase assays were carried out in 100 mM HEPES, pH 8.0, 50 mM NaCl 2 mg/ml BSA using 800 μM MLG peptide and 100 μM [14C]acetyl-CoA in a 50 μl reaction volume for 25 min at 25°C.
hNatF (1 μM) acetyltransferase assays were carried out in 50 mM HEPES 7.5 100 mM NaCl using 60 μM MLG-peptide and 100 μM [14C]acetyl-CoA in a 50 μl reaction volume for 15 min at 50°C.
hMOF (50 nM) acetyltransferase assays were carried out in 40 mM Tris 8.0 100 mM NaCl 800 μM cysteine 0.25 mg/ml using 400 μM H4 peptide and 50 μM [14C]acetyl-CoA in a 50 μl reaction volume for 60 min at 25°C.
hPCAF (500 nM) acetyltransferase assays were carried out in 100 mM HEPES, pH 8.0, 50 mM NaCl 2 mg/ml BSA using 400 μM H4 peptide and 50 μM [14C]acetyl-CoA in a 50 μl reaction volume for 45 min at 25°C.
All HAT and NAT reactions above were performed in the presence and absence 5 μM WT MBP-HYPK and followed the same quenching method described below.
To quench the reaction, 20 μl of the reaction mixture was added to negatively charged P81 phosphocellulose squares (EMD Millipore), and the paper disks were immediately placed in wash buffer (10 mM HEPES, pH 7.5). The paper disks were washed three times, at 5 min per wash, to remove unreacted acetyl-CoA. The papers were then dried with acetone, added to 4 ml of scintillation fluid, and the signal was measured with a PerkinElmer Life Sciences Tri-Carb 2810 TR liquid scintillation analyzer. Background control reactions were performed in the absence of enzyme or in the absence of substrate peptide to ensure that any possible signal due to chemical acetylation was negligible. Each reaction was performed in triplicate. The counts per minute were converted to molar units using a standard curve of known [14C]acetyl-CoA concentrations in scintillation fluid.
In order to determine steady-state catalytic parameters of hNatA, a saturating concentration of radiolabeled [14C]acetyl-CoA (100 μM) was incubated at seven different concentrations of the substrate peptide (ranging from 3.9–250 μM). Additionally, the acetyl-CoA Km values were determined by titration of the acetyl-CoA at eight different concentrations (ranging from 0.73 to 94 μM) in the presence of 350 μM substrate peptide. Assays were performed in the presence and absence of MBP-HYPK (10 nM). GraphPad Prism, version 5.01, was used for all data fitting to the Michaelis–Menten equation.
IC50 Assays
hNatA IC50 assays were performed under the same reaction conditions described above, except substrates H4 peptide and radiolabeled [14C]acetyl-CoA were both set at a constant sub-saturating concentration of 50 μM. To evaluate protein-induced inhibition, 6xHis-hNatA was incubated with various concentrations of MBP-HYPK (660 pM-38nM for ~15 minutes at room temperature). To evaluate the ability of a peptide of HYPK35–48 to inhibit hNatA, hNatA was incubated with concentrations of HYPK peptide or a non-specific gag peptide (16 μM-2 mM) for 1 hr. at room temperature. The reactions were initiated by addition of [14C]acetyl-CoA and proceeded as detailed above. Reactions were performed in triplicate and converted to molar units using a standard curve. The results were normalized using the reaction lacking MBP-HYPK. An IC50 was calculated using a log (inhibitor) vs. response fit on Prism 5.0. Error bars correspond to the SEM of each point.
Differential scanning calorimetry (DSC)
Wild-type and mutant hNatA, and ternary 6xHis-tagged hNatA/HYPK construct samples were purified as described above and prepared in the same sizing buffer by overnight dialysis at 4°C. Samples were recovered from dialysis and spin filtered to remove particulates (0.22 μm; Millipore). The final dialysis buffer was also filtered (0.22 μm aPES; Thermo Fisher) and used for all sample dilutions and as a reference solution for DSC studies. DSC data were collected with a MicroCal VP-Capillary DSC instrument (Malvern Instruments, Malvern, United Kingdom). Prior to sample scans, buffer scans were performed until baseline reproducibility was achieved. DSC scans of binary hNatA and ternary hNatA/HYPK samples were performed at concentrations of 7–8 μM (WT hNaa15), 8 μM (hNaa15 Y834F) and 9 μM (hNaa15 Y834A) and 6–8 μM (hNatA/HYPK), respectively. DSC scans were recorded from 10°C to 90°C at a scan rate of 1 °C/min with a pre-scan thermostat of 10 minutes, mid feedback mode and a filtering period of 30 seconds. DSC data were analyzed using Origin 7.0 (OriginLab Corporation, Northampton, MA). Raw DSC data were corrected for the instrumental baseline by subtraction of a suitable buffer reference scan. DSC scans of samples were performed in triplicate to ensure reproducibility. Fig 1F and 4A are representative curves from these experiments. Traces were plotted using MagicPlot Student using the processed data exported from Origin (https://www.magicplot.com).
MBP and Glutathione Pull-Down Assays
6xHis-tagged proteins as well as MBP-tagged HYPK proteins were prepared as described above. Free MBP was prepared as described for MBP-HYPK above. SpNatA, hNaa10, as well as untagged and GST-tagged hNaa50 were prepared as previously described (Liszczak et al., 2011; Liszczak et al., 2013; Magin et al., 2016). The pull-down experiments represented in Fig 2B, 4B, and 6C were conducted by incubating 2 μM MBP-tagged protein (or free MBP) with 6 μM bait (6xHis-tagged hNatA or hNaa10FL or SpNatA; or for the competition pull-down: 6xHis-tagged hNatA with or without equimolar untagged hNaa50) in sizing buffer at 4°C for 30 min. Proteins were then subjected to pull-down by incubation with amylose agarose resin (70μL slurry, New England BioLabs) for 30 min. Resin was washed with 80 CV of sizing buffer before elution of bound proteins by boiling resin in SDS gel-loading buffer.
The glutathione competition pull-down was performed by incubation of 2 μM GST-tagged protein with 6 μM 6xHis-tagged hNatA alone or in complex with an equimolar concentration of HYPKΔ1–33 in the same pull-down buffer as above for 30 min. Proteins were then subjected to pull-down by incubation with glutathione agarose resin (70μL slurry, Gold Biotechnology) for 30 min. Resin was washed with 80 CV of pull-down buffer before elution of bound proteins by boiling resin in SDS gel-loading buffer.
Selection of the HYPKΔ1–33 crystallization construct for the competition pull-down was predicated on the need to decipher between HYPK from hNaa101–160; at full-length, the HYPK migration distance is equal to that of hNaa101–160 by SDS-PAGE. This would otherwise significantly impede visual inspection by SDS-PAGE gel.
Results of all pull-down assays were analyzed though visual inspection of input and pull-down samples with 15% SDS-PAGE. Gels were stained using Coomassie Brilliant Blue G-250.
Analytical Ultracentrifugation (AUC)
Sedimentation equilibrium AUC experiments were performed at 4°C with absorbance optics at 280 nm using a Beckman Optima XL-I. We employed the use of a four-hole rotor containing six-channel centerpieces with quartz windows, spinning at 6,000, 9,000, 12,000 rpm. Samples of 6xHis-tagged hNatA was analyzed at A280 = 0.8, 0.5, and 0.2 in sizing buffer. The most representative runs were included to calculate a theoretical molecular weight using the program HeteroAnalysis (http://www.biotech.uconn.edu/auf/?i=aufftp).
Sedimentation velocity AUC experiments were performed at 20°C with absorbance optics at 280 nm using the same Beckman Optima XL-I. We employed a rotor containing two-channel centerpieces with quartz, spinning at 42,000 rpm. Data were obtained over ~8 h of centrifugation. Concentrations of Naa15 variants were achieved by dilution to an A280=0.6 in sizing buffer. Data were analyzed using SEDFIT to calculate a continuous c(s) distribution (Schuck et al., 2002), and data were graphed using GraphPad.
Quantification and Statistical Analysis
In order to calculate the inhibition constant (Ki) of the hNatA-HYPK interaction (in Figure 5E), we fit the data generated from the IC50 assays using a noncompetitive tight-binding (Morrison Fit) model represented by the following equation,
Where,
[E]tot = Total active enzyme concentration
[I]tot = Concentration of HYPK
KM = Peptide Michaelis-Menten Constant
[S] = Concentration of peptide
The Ki was calculated using GraphPad Prism 5.0, where the following parameters were fixed, β =0.081, KM = 30×103 nM, and [S]= 50 ×103 nM.
Statistical details of experiments can be found in the figure legends and in the methods details.
Data and Software Availability
Coordinates of the two structures described in this article have been deposited in the PDB with accession numbers PDB: 6C9M (human NatA) and PDB: 6C95 (human NatA/HYPK).
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli Rosetta (DE3)pLysS | EMD Millipore | Cat#70956-3 |
| Spodoptera frugiperda (Sf9) cells | Invitrogen | Cat#11496015 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 6xHis-hNaa15/Naa101–160 (i.e. hNatA) | This paper | N/A |
| 6xHis-hNaa15Δ672–866/Naa101–160 | This paper | N/A |
| 6xHis-hNaa15Y834F/Naa101–160 | This paper | N/A |
| 6xHis-hNaa15Y834A/Naa101–160 | This paper | N/A |
| MBP-HYPK | This paper | N/A |
| MBP-HYPKΔ1–33 | This paper | N/A |
| MBP-HYPKΔ1–59 | This paper | N/A |
| MBP-HYPKΔ60–129 | This paper | N/A |
| MBP-HYPKΔ1–39 | This paper | N/A |
| MBP-HYPKΔ1–89 | This paper | N/A |
| MBP-HYPKH36A | This paper | N/A |
| MBP-HYPKE74A | This paper | N/A |
| MBP-HYPKL43A/L46A | This paper | N/A |
| MBP-HYPKE101A/E103A | This paper | N/A |
| MBP-HYPKV121A/I25A | This paper | N/A |
| hNaa15/Naa101–160 (i.e. hNatA) | This paper | N/A |
| hNatA/HYPK | This paper | N/A |
| hNatA/HYPK34–129 | This paper | N/A |
| HNatB | This paper | N/A |
| cOmplete™ Protease Inhibitor Cocktail | Roche | 4693116001 |
| Amylose Resin | New England Biolabs | Cat#E8021L |
| Nickel Resin | Thermo Scientific | Cat#88223 |
| Glutathione Agarose Resin | Gold Biotechnology | Cat#G-250-100 |
| HiTrap Q HP, 5 mL | GE Healthcare | Cat#17115401 |
| HiTrap SP HP, 5 mL | GE Healthcare | Cat#17115201 |
| Superdex 200 10/300 GL | GE Healthcare | Cat#17517501 |
| Additive screen | Hampton Research | Cat#HR2-138 |
| Acetyl-CoA | Sigma-Aldrich | Cat#A2181 |
| Glycerol | EMD Millipore | Cat#4760-4L |
| PEG 3350 | Hampton Research | Cat#HR2-527 |
| Tacsimate, pH 6.5 and 7.5 | Hampton Research | N/A |
| [14C]Acetyl-CoA (4 mCi/mmol) | PerkinElmer Life Sciences | Cat#NEC313050UC |
| P81 Phosphocellulose squares | EMD Millipore | Cat#20-134 |
| Human H4 Peptide (NH2-SGRGKGGKGLGKGGAKRHR-COOH) | Genscript | N/A |
| Actin Peptide (NH2-MEEEIAARWGRPVGRRRRP-COOH) | Genscript | N/A |
| Gag Peptide (NH2-MLRFVTKRWGRPVGRRRRP-COOH) | Genscript | N/A |
| HYPK35–48 Peptide (NH2-KHDSGAADLERVTD-COOH) | Genscript | N/A |
| MLG Peptide (NH2-MLGPEGGRWGRPVGRRRRP-COOH) | Genscript | N/A |
| SpNatA | Liszczak et al., 2013 | N/A |
| HMOF | Yuan et al., 2012 | N/A |
| HPCAF | Rojas et al, 1999 | N/A |
| HNatD | Magin et al., 2015 | N/A |
| Untagged and GST-tagged hNatE | Liszczak et al., 2011 | N/A |
| HNatF | Støve et al., 2016 | N/A |
| Deposited Data | ||
| Atomic coordinates and structure factors | This paper | PDB: 6C9M |
| Atomic coordinates and structure factors | This paper | PDB: 6C95 |
| Recombinant DNA | ||
| pFB Dual | This paper | N/A |
| pFB 1 | This paper | N/A |
| pET-m41 | Arnesen et al., 2010 | N/A |
| Software and Algorithms | ||
| XDS | Kabsch, W., 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org/documentation/reference/refinement.html |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Schrodinger LLC | http://www.pymol.org |
| PDBePISA | PDBe | http://www.ebi.ac.uk/pdbe/pisa/ |
| TPRpred | Karpenahalli et al., 2007 | https://toolkit.tuebingen.mpg.de/#/tools/tprpred |
| Clustal Omega | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| ESPript 3.0 | Robert and Gouet, 2014 | http://espript.ibcp.fr/ESPript/ESPript/ |
| Prism 5.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MagicPlot Student | MagicPlot Systems, LLC | https://www.magicplot.com |
| HeteroAnalysis | Cole and Lary, University of Connecticut | http://www.biotech.uconn.edu/auf/?i=aufftp |
| SEDFIT | Schuck et al., 2002 | https://sedfitsedphat.nibib.nih.gov/software/default.aspx |
| Origin 7.0 | OriginLab | https://www.originlab.com/ |
Supplementary Material
Highlights.
The Naa15 metazoan-specific domain contributes to NatA activity and stability.
Human NatA structure reveals bound stabilizing inositol hexaphosphate.
HYPK bipartite structure facilitates intrinsic NatA-specific inhibitory activity.
HYPK-induced conformation changes blocks Naa50 in vitro localization to NatA.
Acknowledgments
This work was supported by NIH grants R01 GM060293 and R35 GM118090 awarded to R.M. and T32 GM071339 grant awarded to L.G. We acknowledge the support of the Proteomics core facility at the Wistar Institute as well as the DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania. We thank T. Arnesen (University of Bergen) for sharing the HYPK plasmid with us; E. Dean for providing the Sf9 cells for this study; and we thank E. Skordalakes as well as A. Vogt for helpful discussions.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Author Contributions
Conceptualization, LG and R.M.; Methodology, L.G. And R.M.; Investigation, L.G.; Formal Analysis, L.G.; Writing – Original Draft, L.G.; Visualization, L.G.; Writing – Review and Editing, L.G. and R.M.; Funding Acquisition; R.M.; Resources, R.M.; Supervision, R.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksnes H, Drazic A, Marie M, Arnesen T. First Things First: Vital Protein Marks by N-Terminal Acetyltransferases. Trends Biochem Sci. 2016;41:746–760. doi: 10.1016/j.tibs.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Aksnes H, Van Damme P, Goris M, Starheim KK, Marie M, Støve SI, Hoel C, Kalvik TV, Hole K, Glomnes N, et al. An organellar nα-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell Reports. 2015;10:1362–1374. doi: 10.1016/j.celrep.2015.01.053. [DOI] [PubMed] [Google Scholar]
- Andersen KM, Semple CA, Hartmann-Petersen R. Characterisation of the nascent polypeptide-associated complex in fission yeast. Mol Biol Rep. 2007;34:275–281. doi: 10.1007/s11033-006-9043-5. [DOI] [PubMed] [Google Scholar]
- Arnesen T, Starheim KK, Van Damme P, Evjenth R, Dinh H, Betts MJ, Ryningen A, Vandekerckhove J, Gevaert K, Anderson D. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol Cell Biol. 2010;30:1898–1909. doi: 10.1128/MCB.01199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A. 2009;106:8157–8162. doi: 10.1073/pnas.0901931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. BioEssays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Dinh TV, Bienvenut WV, Linster E, Feldman-Salit A, Jung VA, Meinnel T, Hell R, Giglione C, Wirtz M. Molecular identification and functional characterization of the first Nalpha-acetyltransferase in plastids by global acetylome profiling. Proteomics. 2015;15:2426–2435. doi: 10.1002/pmic.201500025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazic A, Aksnes H, Marie M, Boczkowska M, Varland S, Timmerman E, Foyn H, Glomnes N, Rebowski G, Impens F, et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc Natl Acad Sci U S A. 2018 doi: 10.1073/pnas.1718336115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth R, Hole K, Karlsen OA, Ziegler M, Arnesen T, Lillehaug JR. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J Biol Chem. 2009;284:31122–31129. doi: 10.1074/jbc.M109.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998;7:1463–1474. doi: 10.1093/hmg/7.9.1463. [DOI] [PubMed] [Google Scholar]
- Fluge O, Bruland O, Akslen LA, Varhaug JE, Lillehaug JR. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene. 2002;21:5056–5068. doi: 10.1038/sj.onc.1205687. [DOI] [PubMed] [Google Scholar]
- Gautschi M, Just S, Mun A, Ross S, Rucknagel P, Dubaquie Y, Ehrenhofer-Murray A, Rospert S. The Yeast N -Acetyltransferase NatA Is Quantitatively Anchored to the Ribosome and Interacts with Nascent Polypeptides. Mol Cell Biol. 2003;23:7403–7414. doi: 10.1128/MCB.23.20.7403-7414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AO, Rosati S, Pijnappel PW, van Breukelen B, Timmers MH, Mohammed S, Slijper M, Heck AJ. Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC Genomics. 2010;11:685. doi: 10.1186/1471-2164-11-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P. The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem Sci. 1996;21:172–173. [PubMed] [Google Scholar]
- Hole K, Van Damme P, Dalva M, Aksnes H, Glomnes N, Varhaug JE, Lillehaug JR, Gevaert K, Arnesen T. The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS One. 2011;6:e24713. doi: 10.1371/journal.pone.0024713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Chu CW, Kong X, Yokomori K, Zou H. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J Cell Biol. 2007;177:587–597. doi: 10.1083/jcb.200701043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvik TV, Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene. 2013;32:269–276. doi: 10.1038/onc.2012.82. [DOI] [PubMed] [Google Scholar]
- Karpenahalli MR, Lupas AN, Soding J. TPRpred: a tool for prediction of TPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Kim RR, Oh JH, Cho H, Varshavsky A, Hwang CS. The N-terminal methionine of cellular proteins as a degradation signal. Cell. 2014;156:158–169. doi: 10.1016/j.cell.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplin A, Preissler S, Ilina Y, Koch M, Scior A, Erhardt M, Deuerling E. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J Cell Biol. 2010;189:57–68. doi: 10.1083/jcb.200910074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Arnesen T, Marmorstein R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J Biol Chem. 2011;286:37002–37010. doi: 10.1074/jbc.M111.282863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Goldberg JM, Foyn H, Petersson EJ, Arnesen T, Marmorstein R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol. 2013;20:1098–1105. doi: 10.1038/nsmb.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hu Y, Li X, Niu L, Teng M. The crystal structure of the human nascent polypeptide-associated complex domain reveals a nucleic acid-binding region on the NACA subunit. Biochemistry. 2010;49:2890–2896. doi: 10.1021/bi902050p. [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, VanDemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin RS, Deng S, Zhang H, Cooperman B, Marmorstein R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS One. 2017;12:e0186278. doi: 10.1371/journal.pone.0186278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin RS, Liszczak GP, Marmorstein R. The molecular basis for histone H4- and H2A-specific amino-terminal acetylation by NatD. Structure. 2015;23:332–341. doi: 10.1016/j.str.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin RS, March ZM, Marmorstein R. The N-terminal Acetyltransferase Naa10/ARD1 Does Not Acetylate Lysine Residues. J Biol Chem. 2016;291:5270–5277. doi: 10.1074/jbc.M115.709428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough CE, Song S, Shin MH, Johnson FB, Marmorstein R. Structural and Functional Role of Acetyltransferase hMOF K274 Autoacetylation. J Biol Chem. 2016;291:18190–18198. doi: 10.1074/jbc.M116.736264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittl PR, Schneider-Brachert W. Sel1-like repeat proteins in signal transduction. Cell Signal. 2007;19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Mueller TD, Feigon J. Solution Structures of UBA Domains Reveal a Conserved Hydrophobic Surface for Protein–Protein Interactions. J Mol Biol. 2002;319:1243–1255. doi: 10.1016/S0022-2836(02)00302-9. [DOI] [PubMed] [Google Scholar]
- Neri L, Lasa M, Elosegui-Artola A, D’Avola D, Carte B, Gazque C, Alve S, Roca-Cusachs P, Inarrairaegui M, Herrero J, et al. NatB-mediated protein N-α-terminal acetylation is a potential therapeutic target in hepatocellular carcinoma. Oncotarget. 2017;8:40967–40981. doi: 10.18632/oncotarget.17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer JL. Pharmacology and Cancer Biology. Ann Arbor, MI: Duke University; 2012. Structural Analysis of the N-Terminal Acetyltransferase A Complex; p. 136. [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Rathore OS, Faustino A, Prudencio P, Van Damme P, Cox CJ, Martinho RG. Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci Rep. 2016;6:21304. doi: 10.1038/srep21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Banerjee R, Mukhopadhyay S, Bhattacharyya NP. Conserved C-terminal nascent peptide binding domain of HYPK facilitates its chaperone-like activity. J Biosci. 2014;39:659–672. doi: 10.1007/s12038-014-9442-z. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Majumder P, Sarkar S, Giri K, Mukhopadhyay D, Bhattacharyya NP. Huntingtin interacting protein HYPK is intrinsically unstructured. Proteins. 2008a;71:1686–1698. doi: 10.1002/prot.21856. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Sinha M, Mukhopadhyay D, Bhattacharyya NP. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum Mol Genet. 2008b;17:240–255. doi: 10.1093/hmg/ddm301. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Z, Ouyang Z, Magin RS, Marmorstein R, Yu H. Opposing Functions of the N-terminal Acetyltransferases Naa50 and NatA in Sister-chromatid Cohesion. J Biol Chem. 2016;291:19079–19091. doi: 10.1074/jbc.M116.737585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.1. 2015. [Google Scholar]
- Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsuddin AK, Bose S. IP6 (Inositol Hexaphosphate) as a Signaling Molecule. Curr Signal Transduction Ther. 2012;7:289–304. [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. In Mol Syst Biol. 2011:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starheim KK, Arnesen T, Gromyko D, Ryningen A, Varhaug JE, Lillehaug JR. Identification of the human N(alpha)-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem J. 2008;415:325–331. doi: 10.1042/BJ20080658. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gromyko D, Evjenth R, Ryningen A, Varhaug JE, Lillehaug JR, Arnesen T. Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol Cell Biol. 2009;29:3569–3581. doi: 10.1128/MCB.01909-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Støve SI, Magin RS, Foyn H, Haug BE, Marmorstein R, Arnesen T. Crystal Structure of the Golgi-Associated Human Nalpha-Acetyltransferase 60 Reveals the Molecular Determinants for Substrate-Specific Acetylation. Structure. 2016;24:1044–1056. doi: 10.1016/j.str.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolajska E, Chroboczek J. Faithful chaperones. Cell Mol Life Sci. 2011;68:3307–3322. doi: 10.1007/s00018-011-0740-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Evjenth R, Foyn H, Demeyer K, De Bock PJ, Lillehaug JR, Vandekerckhove J, Arnesen T, Gevaert K. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(alpha)-acetyltransferases and point to hNaa10p as the post-translational actin N(alpha)-acetyltransferase. Mol Cell Proteomics. 2011a;10:M110-004580–004581–M004110.004580–004512. doi: 10.1074/mcp.M110.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Hole K, Pimenta-Marques A, Helsens K, Vandekerckhove J, Martinho RG, Gevaert K, Arnesen T. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 2011b;7:e1002169. doi: 10.1371/journal.pgen.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Lasa M, Polevoda B, Gazquez C, Elosegui-Artola A, Kim DS, De Juan-Pardo E, Demeyer K, Hole K, Larrea E, et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc Natl Acad Sci U S A. 2012;109:12449–12454. doi: 10.1073/pnas.1210303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Støve SI, Glomnes N, Gevaert K, Arnesen T. A Saccharomyces Cerevisiae Model Reveals in vivo Functional Impairment of the Ogden Syndrome N-Terminal Acetyltransferase Naa10 S37P Mutant. Mol Cell Proteomics. 2014;13:2031–2041. doi: 10.1074/mcp.M113.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Yao T, Robinson H, Hill CP. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J Biol Chem. 2017;292:9493–9504. doi: 10.1074/jbc.M117.785287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011;20:1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer FA, Gumiero A, Lapouge K, Bange G, Kopp J, Sinning I. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat Commun. 2017;8:15726. doi: 10.1038/ncomms15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson CR, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, Gordon C. Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol. 2001;3:939–943. doi: 10.1038/ncb1001-939. [DOI] [PubMed] [Google Scholar]
- Yu M, Gong J, Ma M, Yang H, Lai J, Wu H, Li L, Li L, Tan D. Immunohistochemical analysis of human arrest-defective-1 expressed in cancers in vivo. Oncol Rep. 2009;21:909–915. doi: 10.3892/or_00000303. [DOI] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, Gabler F, Soding J, Lupas AN, Alva V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol. 2017 doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.