

Abstract
We propose that the normal adrenarche-related rise in DHEA secretion is ultimately caused by the rise in cortisol production occurring during childhood and adolescent growth, by the following mechanisms. (1) The onset of childhood growth leads to a slight fall in serum cortisol concentration due to growth-induced dilution, and a decrease in the negative feedback of cortisol upon ACTH secretion. (2) In response, ACTH rises and stimulates increased cortisol synthesis and secretion in the growing body, to restore the serum cortisol concentration to normal. (3) The cortisol concentration produced within and taken up by adrenocortical steroidogenic cells may rise during this time. (4) Cortisol competitively inhibits 3-beta-hydroxysteroid dehydrogenase II (3βHSD2)-mediated conversion of 17αOH-pregenenolone to cortisol, causing a further fall in serum cortisol, a further decrease in the negative feedback of cortisol upon ACTH, a further rise in ACTH, and further stimulation of adrenal steroidogenesis. (5) The cortisol-mediated inhibition of 3βHSD2 also blocks the conversion of DHEA to androstenedione, causing a rise in adrenal DHEA and DHEA sulfate relative to androstenedione secretion. Thus, the combination of normal body growth plus inhibition of 3βHSD2 by intra-adrenal cortisol may cause normal adrenarche. Childhood obesity may hasten this process by causing a pathologic increase in body size that triggers these same processes at an earlier age, resulting in the premature onset of adrenarche.
Introduction
In 1947 Fuller Albright introduced the term “adrenarche” [1] to describe the initiation of dehydroepiandrosterone (DHEA) and DHEA-sulfate secretion from the zona reticularis of the adrenal gland during childhood, first documented by Talbot in 1943 as a rise in urinary 17-ketosteroid secretion [2]. Adrenarche is a gradual process beginning during early childhood [3, 4]. Premature adrenarche is a common pediatric disorder, usually benign but occasionally the harbinger of an adrenal tumor or enzyme deficiency. In some children, premature adrenarche may precede the polycystic ovarian syndrome or the metabolic syndrome [5].
Rationale for intra-adrenal cortisol regulation of adrenarche
Despite decades of research, the trigger for adrenarche remains unknown. A pituitary factor, analogous to ACTH, that initiates adrenal secretion of DHEA has been sought [6] but not found [7]. As blood concentrations of cortisol remain constant during adrenarche while DHEA levels rise 5- to 10-fold, cortisol has not been thought to play a role in initiating adrenarche [8]. The rise in adrenal DHEA secretion could be due to its increased synthesis, controlled by the enzyme, 17, 20 lyase, or decreased metabolism, controlled by the enzymes, 3-beta hydroxysteroid dehydrogenase Type II (3βHSD2), and DHEA sulfotransferase (SULT2A1). The activity of 17, 20 lyase is enhanced by both its phosphorylation [9], as well as by the presence in the zona reticularis of cytochrome b5 [10], an allosteric cofactor [11], and P450 oxidoreductase, an electron donor [12], all of which facilitate electron transfer [13]. However, none of these factors has been shown to initiate adrenarche [14]. Both older children [15] and adults [16] have decreased expression of 3βHSD2 in the zona reticularis, but it is not known whether this is an initiating or later event. Loss of function mutations in the gene encoding PAPS synthase, a cofactor for SULT2A1, cause premature adrenarche [17], although changes in the activity of neither PAPSS2 nor SULT2A has been shown to initiate adrenarche.
Although the average serum cortisol concentration remains constant throughout life, its daily production rate increases several-fold during the transition into adulthood [18]. As the relative increase in adrenal weight is approximately one third that of body weight during childhood [19–21], we reasoned that the concentration of intra-adrenal cortisol likely rises as children grow, due to the increase in hormone production without a proportionate increase in adrenal gland size to match the increase in body size. A rise in intra-adrenal cortisol has been previously hypothesized on theoretical grounds to trigger adrenarche [22]. Winter and his colleagues had considered that inhibition of 3βHSD2 by intra-adrenal steroids might contribute to the initiation of adrenarche [23], but showed no effect with 1 μM cortisol, the highest dose they tried despite their previously finding a fifty-fold higher cortisol concentration in adrenal homogenates [24]. During the same period, Rabe et al. [25] showed that cortisol inhibited human placental 3βHSD1 activity by 50%, and found cortisone’s inhibitory effect to be much more potent. Further, treatment of primary cultures of human fetal adrenal cells with high concentrations of glucocorticoid leads to increased DHEA secretion [26]. These observations led us to ask whether cortisol regulates the synthesis and secretion of DHEA in human adrenocortical cells and might play a role in initiating human adrenarche.
Cortisol competitively inhibits adrenal 3-beta-hydroxysteroid dehydrogenase II
We performed cell-based studies [27] by growing either NCI-H295R human adrenal cells or COS-7 monkey kidney cells in tissue culture. Full length complementary DNAs encoding human CYP17A1 or HSD3β2 were synthesized, cloned into expression vectors, and transiently transfected into COS-7 cells. To investigate the effect of cortisol on steroidogenic enzyme activity, these COS-7 cells or NCI-H295R cells were treated with the radiolabeled steroid precursors, [3H]-pregnenolone or [3H]-DHEA. DHEA in media was measured by ELISA and radioimmunoassay. DHEA and androstenedione in media were also measured by liquid chromatography-tandem mass spectroscopy (LC/MS/MS). Tritiated steroids in media were separated by thin layer chromatography (TLC) and detected and quantified using a phosphorImager or x-ray film. We also studied the regulation of purified human 3βHSD2 by adrenal steroids under cell-free conditions [28].
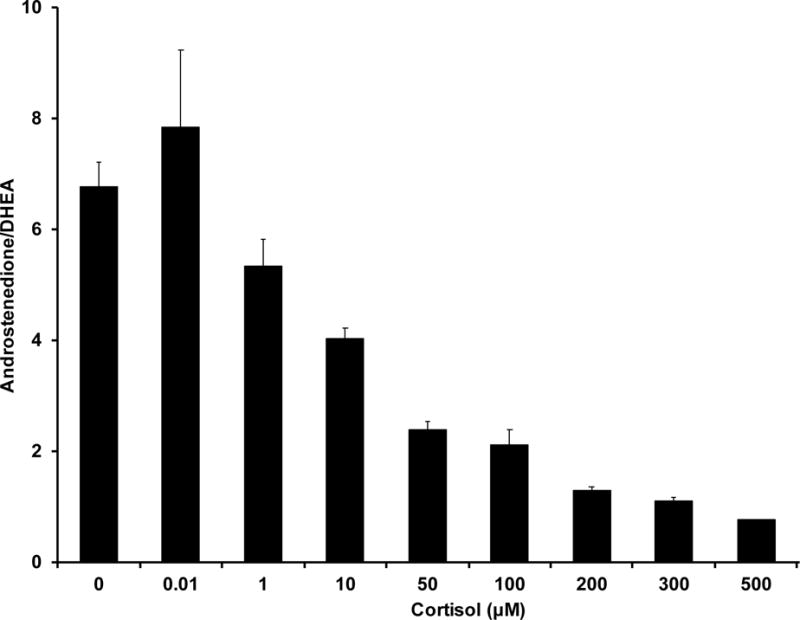
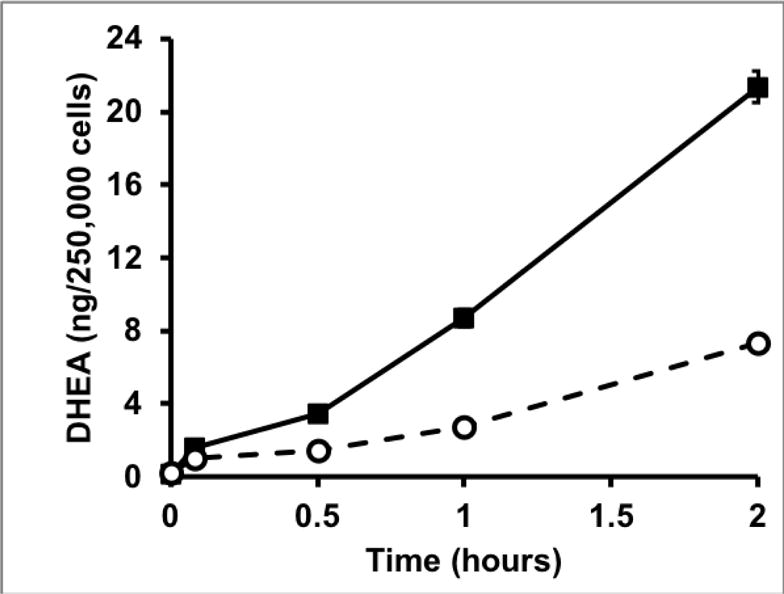
NCI-H295R cells contain all of the enzymes and steroids of the human adrenal gland [29, 30] to determine the effect of cortisol on adrenal steroidogenesis. We found that cortisol stimulated DHEA secretion in a dose-dependent manner at concentrations ≥ 50 μM but had no effect at lower concentrations (Fig. 1A). The increase in DHEA secretion was accompanied by a fall in androstenedione secretion, indicating that cortisol was acting to inhibit 3βHSD2 activity (Fig. 1B). Surprisingly, the onset of DHEA secretion occurred within 30 minutes after the addition of cortisol, more rapidly than expected for a transcriptional mechanism (Fig. 1C). We reasoned that if cortisol was working through a glucocorticoid receptor-dependent transcriptional mechanism, glucocorticoid receptor agonists would have a similar effect. However, neither dexamethasone nor prednisolone, both potent mediators of glucocorticoid-dependent transcription, stimulated DHEA secretion, whereas cortisone and 11-deoxycortisol, transcriptionally inactive and weakly active glucocorticoids, respectively, were even more potent than cortisol [27]. Finally, measurement of mRNAs encoding the two key regulators of adrenal androgen steroidogenesis, CYP17 and 3βHSD2, revealed no significant differences in either mRNA following cortisol treatment of NCI-H295R cells [27].
Figure 1.
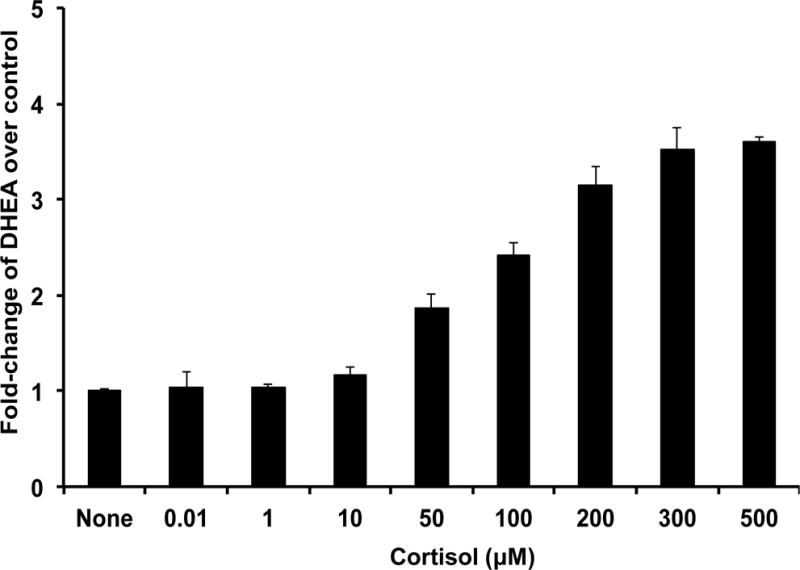
A. Cortisol increases secretion of DHEA from NCI-H295R adrenal cells. ([27] with permission of the Endocrine Society)
B. Cortisol decreases 3βHSD2 activity in NCI-H295R adrenal cells. ([27] with permission of the Endocrine Society)
C. Rapid time course effect of cortisol on adrenal secretion of DHEA in NCI-H295R adrenal cells. Solid line, cortisol (500 μM), dashed line, vehicle. ([27] with permission of the Endocrine Society)
These somewhat surprising results indicated that stimulation of DHEA secretion is not mediated by a transcriptional mechanism: its onset is rapid; the required concentration of cortisol is much greater than that needed to activate the glucocorticoid receptor (~1 nM); dexamethasone and prednisolone are inactive, whereas cortisone and 11-deoxycortisol are more active than cortisol; it occurs with no changes in CYP17A1 or HSD3B2 mRNA levels; and, as discussed subsequently, cortisol stimulates DHEA secretion from COS-7 cells, which lack glucocorticoid receptors. Interestingly, the steroids which increase DHEA secretion from NCI-H295R cells uniquely share a single A ring (Δ4) double bond with androstenedione, the end product of DHEA metabolism by 3βHSD2, whereas those steroids which do not increase DHEA secretion possess two A ring double bonds (Δ1 and Δ4) (Figure 2). The high degree of structural similarity among cortisol, cortisone and 11-deoxycortisol despite their divergent transcriptional activities suggests that they may stimulate DHEA secretion via direct interaction with an enzyme in the adrenal androgen biosynthetic pathway, rather than through a glucocorticoid-receptor-mediated mechanism. This, as well as the decreased androstenedione/DHEA ratio following cortisol treatment of NCI-H295R cells, is consistent with the possibility that cortisol may inhibit 3βHSD2 activity via competition with end-product androstenedione.
Figure 2.
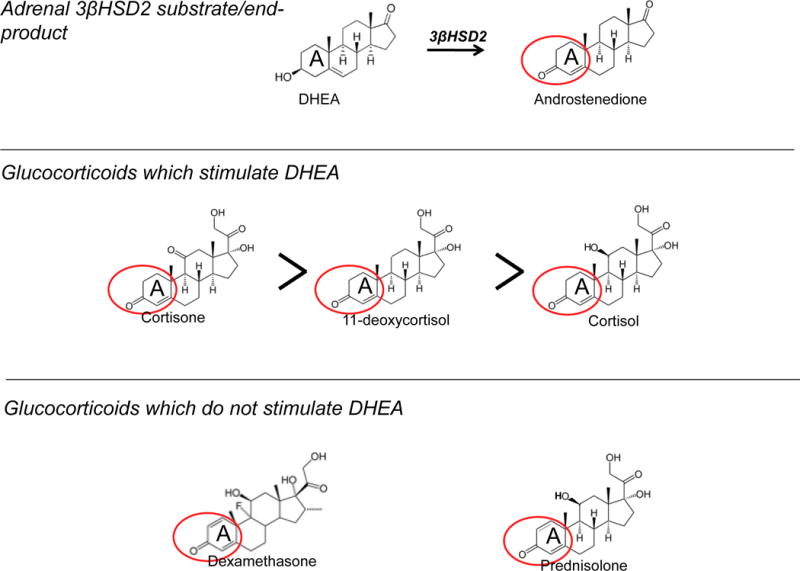
Steroids which stimulate secretion of DHEA in NCI-H295R adrenal cells share A ring structural similarities with 3βHSD2 end-product androstenedione.
We next sought more direct evidence for the enzyme(s) whose activity is affected by cortisol to mediate the rise in DHEA secretion from human adrenal cells using pathway analysis with tritiated steroid precursors. We considered two possible mechanisms to explain cortisol’s effect on DHEA secretion from NCIH295R cells: stimulation of 17, 20 lyase or inhibition of 3βHSD2. We first analyzed cortisol’s effect on 17, 20 lyase function. Using [3H]-pregnenolone, we found only a modest (1.3-fold) stimulation of 17, 20 lyase conversion of 17αOH-pregnenolone to DHEA [27], which is unlikely to account for the 4- to 10-fold stimulation of DHEA secretion by cortisol. In contrast, using [3H]-DHEA as the substrate, we found a sevenfold reduction in enzymatic activity of 3βHSD2-catalyzed conversion of DHEA to androstenedione (Fig. 3), and an eight-fold inhibition of 17αOH-pregnenolone conversion to 17αOH-progesterone [27] following cortisol treatment.
Figure 3.
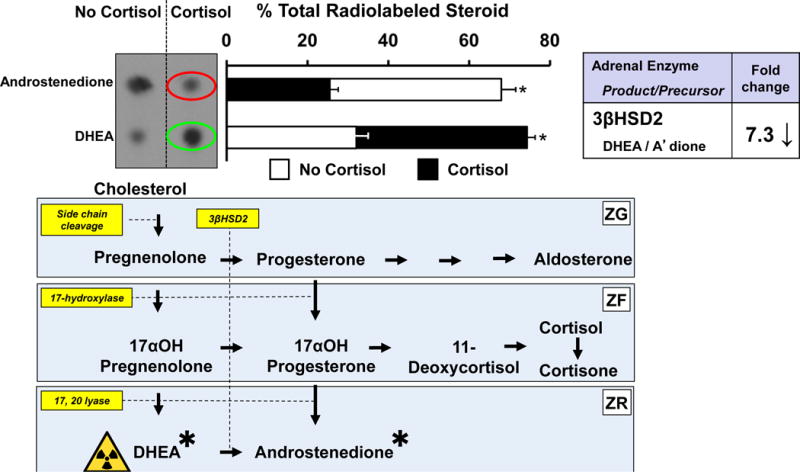
Cortisol decreases 3βHSD2 activity in NCI-H295R cells, causing a rise in labeled DHEA (green oval) and a fall in androstenedione (red oval). ([27] with permission of the Endocrine Society)
To more specifically determine if the nature of cortisol’s inhibitory effect on 3βHSD2 was competitive, we expressed HSD3B2 in COS-7 monkey kidney cells, which express neither steroidogenic enzymes nor glucocorticoid receptor (GR) [31–33], and examined Michaelis-Menten kinetics. Untransfected cells which lack 3βHSD2 cannot convert [3H]-DHEA to androstenedione (Fig. 4). Following transient transfection with HSD3B2, DHEA was converted to androstenedione, and cortisol competitively inhibited this activity, revealed by Dixon plot analysis, which indicated a Ki for cortisol of ~36 μM (Fig. 4). This is more than twenty times greater than the Km for DHEA (1.6 μM) [34]. Because glucocorticoid receptor is absent from COS-7 cells, the inhibitory effect of cortisol upon 3βHSD2 is clearly mediated by a GR-independent mechanism. We found no stimulatory effect of cortisol upon the 17α-hydroxylase and 17,20 lyase activities of CYP17 using COS-7 cells transfected with CYP171A [27].
Figure 4.
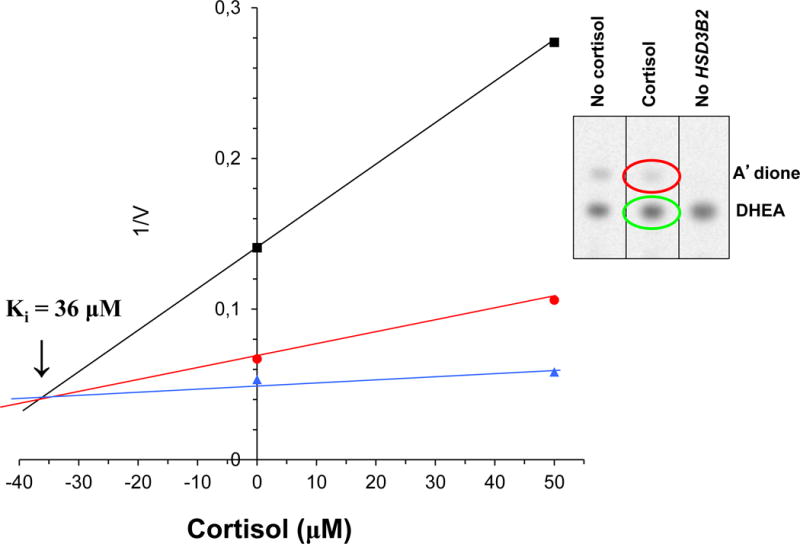
Cortisol competitively inhibits human 3βHSD2 in COS-7 cells. Dixon plot of Cortisol vs. 1/Velocity at DHEA substrate concentrations (μM) of 0.5 (black line), 2 (red line) and 4 (blue line) to obtain Cortisol Ki of 36 μM. Inset: Cortisol (50 μM) addition to DHEA (0.5 μM) increases DHEA substrate (green oval) and decreases androstenedione product (red oval).
When we evaluated the ability of cortisol to inhibit purified human 3βHSD2 conversion of DHEA to androstenedione, we observed uncompetitive inhibition only at very high, nonphysiological concentrations (>500 μM), whereas androstenedione showed the expected competitive end-product inhibition upon its substrate at physiological concentrations (6 μM) [28].
Implications of cortisol inhibition of adrenal 3 beta hydroxysteroid dehydrogenase II
Cortisol, in physiologically relevant concentrations present in the human adrenal, is capable of marked competitive inhibition of adrenal 3βHSD2. We hypothesize that by this mechanism it may participate in the initiation of adrenarche.
Others have wondered whether cortisol might have such a role in adrenarche, including by inhibition of 3βHSD2. Anderson suggested almost forty years ago that adrenarche was triggered by a rise in intra-adrenal cortisol, which might occur due to a hypothetical age-dependent change in intra-adrenal blood flow leading to induction of 17,20-lyase and perhaps partial loss of 3βHSD2 activity [22]. He believed it was not due to any differences between cortisol production in children and adults, although it was known [18] and later confirmed [35, 36] that cortisol production increases several-fold with growth in body size. Winter and his colleagues had considered that inhibition of 3βHSD2 by intra-adrenal steroids might contribute to the initiation of adrenarche [23], but showed no effect on enzyme activity with 1 μM cortisol, the highest dose they tried despite their previously finding a fifty-fold higher cortisol concentration in adrenal homogenates [24]. During the same period, others [25] showed that cortisol, in amounts used in our study, inhibited human placental 3βHSD1 activity by 50%, and as we demonstrated for adrenal 3βHSD2, found cortisone’s inhibitory effect to be much more potent.
How a rise in intra-adrenal cortisol might initiate adrenarche is depicted in Figure 5. Diurnal variation in blood cortisol levels are similar throughout childhood and adulthood, but (1) in response to body growth, blood cortisol falls slightly due to growth-induced dilution, leading to (2) a rise in ACTH and (3) an increase total cortisol production in the growing body. Increased ACTH would also (4) stimulate 17,20 lyase and zona reticularis growth. Before the onset of adrenarche, we suggest that the intra-adrenal cortisol is insufficient to inhibit 3βHSD2 (blue boxed arrows), directing steroidogenesis along the cortisol pathway, resulting in little DHEA synthesis. The increase in cortisol production would raise the intra-adrenal cortisol concentration. Cortisol in high concentrations is a competitive inhibitor of 3βHSD2, causing (5) decreased conversion of DHEA to androstenedione and of 17αOH-pregnenolone to cortisol. The net result would be a rise in serum DHEA, a fall in androstenedione, and a transient fall in serum cortisol. Decreased negative feedback of cortisol upon ACTH would result in (6) a further increase in ACTH leading to more adrenal stimulation, restoration of cortisol levels to normal, as well as further rise in DHEA secretion and the physiological manifestations of adrenarche. Childhood obesity may hasten this process by causing a pathologic increase in body size that triggers these same processes at an earlier age, resulting in the premature onset of adrenarche.
Figure 5.

A new model for adrenarche: inhibition of adrenal 3βHSD2 by intra-adrenal cortisol. A steroidogenic adrenal cell is outlined by the blue dashed line. Red pulses indicate cortisol secretion. 17OH preg, 17-hydroxypregnenolone.
In other words, we propose that normal adrenarche is a state of mild, physiological 3βHSD2 deficiency, including secondary activation of the pituitary-adrenal axis. Many investigators have observed an increase in adrenal 17, 20 lyase activity, determined by enzyme substrate/product ratios during adrenarche [3, 4, 37, 38], and much effort has therefore been focused on the regulation of this enzyme [14]. On the other hand, several investigators have documented decreased activity of 3βHSD2 determined by substrate product ratios during adrenarche [4, 38–41]. We found that cortisol caused a marked stimulation of adrenal DHEA secretion by competitively inhibiting 3βHSD2, without significant effect on 17, 20 lyase. However, we hypothesize that in vivo, in response to decreased negative feedback on ACTH caused by both growth-induced dilution of blood cortisol and by intra-adrenal inhibition of cortisol synthesis, ACTH would rise. This postulated increase in ACTH would stimulate 17, 20 lyase activity due to increased delivery of its substrate, 17αOH-pregnenolone, and possibly due to increased CYP17A1 transcription [42]. This rise in ACTH might also be responsible for the growth in the size of the zona reticularis during adrenarche [21, 43]. Although ACTH dynamics during childhood growth have not been studied, there is a strong positive correlation between ACTH secretion and body size in adults [44].
This hypothesis of initiation of adrenarche is consistent with many observations:
adrenarche is a gradual process [3, 4], dependent on childhood growth [45] rather than the converse [46];
adrenarche requires ACTH action [47];
“benign” premature adrenarche is associated with steroid profiles resembling mild 3βHSD2 deficiency [48];
“benign” premature adrenarche is common among obese children and adolescents, who have increased cortisol production rates [49–51];
patients with untreated [22] and glucocorticoid-treated [52, 53] 21 hydroxylase deficiency have diminished or absent adrenarche (due to low intra-adrenal cortisol);
patients with 11β hydroxysteroid dehydrogenase type 1 (cortisone reductase) deficiency may have premature adrenarche not only because of decreased conversion of cortisone to cortisol causing increased ACTH stimulation of DHEA secretion [54], but also because high intra-adrenal levels of cortisone in this disease might contribute by inhibiting 3βHSD2 activity, further stimulating ACTH and blocking conversion of DHEA to androstenedione.
Unexplained by our hypothesis is the profound reduction in 3βHSD2 protein [15, 16] and mRNA [55] during the adrenarchal process.
In summary, cortisol stimulates DHEA production in adrenal cells through inhibition of 3βHSD2 activity and may be a major regulator of adrenarche. Further studies in children are needed to determine if intra-adrenal cortisol actually increases during adrenarche, whether an early or more marked rise in intra-adrenal cortisol is responsible for benign premature adrenarche, and to provide a way to distinguish these patients from those with more serious adrenal pathologies.
Acknowledgments
Funding. This work was supported in part by NIHT32DK007699, by the Clinical Investigator Training Program, Harvard/MIT Health Sciences and Technology - Beth Israel Deaconess Medical Center, in collaboration with Pfizer Inc. and Merck & Co., as well as by funding from Boston Children’s Hospital and Harvard Medical School.
Footnotes
Conflicts of Interest. The authors have no conflicts of interest to disclose.
Ethics. The article does not include any original research. Of that carried out by the authors, no human or animal studies were performed.
Contributor Information
Joseph A. Majzoub, Email: joseph.majzoub@childrens.harvard.edu, Division of Endocrinology, Department of Medicine, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115.
Lisa Swartz Topor, Email: lisa_swartz_topor@brown.edu, Division of Pediatric Endocrinology, Hasbro Children’s Hospital, Warren Alpert Medical School of Brown University, Providence, RI 02903.
References
- 1.Albright F. Osteoporosis. Ann Intern Med. 1947;27:861–882. doi: 10.7326/0003-4819-27-6-861. [DOI] [PubMed] [Google Scholar]
- 2.Talbot NR, Butler AM, Berman RA, Rodriguez PM, MacLachlan EA. Excretion of 17-ketosteroids by normal and by abnormal childrenAm J Diseases of Children Am J Diseases of Children. 1943;65:364–375. [Google Scholar]
- 3.Palmert MR, Hayden DL, Mansfield MJ, Crigler JF, Jr, Crowley WF, Jr, Chandler DW, Boepple PA. The longitudinal study of adrenal maturation during gonadal suppression: evidence that adrenarche is a gradual process. Journal of Clinical Endocrinology and Metabolism. 2001;86:4536–4542. doi: 10.1210/jcem.86.9.7863. [DOI] [PubMed] [Google Scholar]
- 4.Remer T, Boye KR, Hartmann MF, Wudy SA. Urinary markers of adrenarche: reference values in healthy subjects, aged 3-18 years. Journal of Clinical Endocrinology and Metabolism. 2005;90:2015–2021. doi: 10.1210/jc.2004-1571. [DOI] [PubMed] [Google Scholar]
- 5.Ibanez L, Diaz R, Lopez-Bermejo A, Marcos MV. Clinical spectrum of premature pubarche: links to metabolic syndrome and ovarian hyperandrogenism. Rev Endocr Metab Disord. 2009;10:63–76. doi: 10.1007/s11154-008-9096-y. [DOI] [PubMed] [Google Scholar]
- 6.Parker LN, Odell WD. Evidence for existence of cortical androgen-stimulating hormone. American Journal of Physiology. 1979;236:E616–620. doi: 10.1152/ajpendo.1979.236.6.E616. [DOI] [PubMed] [Google Scholar]
- 7.Mellon SH, Shively JE, Miller WL. Human proopiomelanocortin-(79-96), a proposed androgen stimulatory hormone, does not affect steroidogenesis in cultured human fetal adrenal cells. Journal of Clinical Endocrinology and Metabolism. 1991;72:19–22. doi: 10.1210/jcem-72-1-19. [DOI] [PubMed] [Google Scholar]
- 8.Parker LN. Adrenarche. Endocrinology and Metabolism Clinics of North America. 1991;20:71–83. [PubMed] [Google Scholar]
- 9.Zhang LH, Rodriguez H, Ohno S, Miller WL. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: implications for adrenarche and the polycystic ovary syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:10619–10623. doi: 10.1073/pnas.92.23.10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mapes S, Corbin CJ, Tarantal A, Conley A. The primate adrenal zona reticularis is defined by expression of cytochrome b5, 17alpha-hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3beta-hydroxysteroid dehydrogenase/delta5-4 isomerase (3beta-HSD) Journal of Clinical Endocrinology and Metabolism. 1999;84:3382–3385. doi: 10.1210/jcem.84.9.6105. [DOI] [PubMed] [Google Scholar]
- 11.Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. Journal of Biological Chemistry. 1998;273:3158–3165. doi: 10.1074/jbc.273.6.3158. [DOI] [PubMed] [Google Scholar]
- 12.Yanagibashi K, Hall PF. Role of electron transport in the regulation of the lyase activity of C21 side-chain cleavage P-450 from porcine adrenal and testicular microsomes. J Biol Chem. 1986;261:8429–8433. [PubMed] [Google Scholar]
- 13.Miller WL. Minireview: regulation of steroidogenesis by electron transfer. Endocrinology. 2005;146:2544–2550. doi: 10.1210/en.2005-0096. [DOI] [PubMed] [Google Scholar]
- 14.Miller WL. Androgen synthesis in adrenarche. Rev Endocr Metab Disord. 2009;10:3–17. doi: 10.1007/s11154-008-9102-4. [DOI] [PubMed] [Google Scholar]
- 15.Gell JS, Carr BR, Sasano H, Atkins B, Margraf L, Mason JI, Rainey WE. Adrenarche results from development of a 3beta-hydroxysteroid dehydrogenase-deficient adrenal reticularis. Journal of Clinical Endocrinology and Metabolism. 1998;83:3695–3701. doi: 10.1210/jcem.83.10.5070. [DOI] [PubMed] [Google Scholar]
- 16.Endoh A, Kristiansen SB, Casson PR, Buster JE, Hornsby PJ. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3 beta-hydroxysteroid dehydrogenase. Journal of Clinical Endocrinology and Metabolism. 1996;81:3558–3565. doi: 10.1210/jcem.81.10.8855801. [DOI] [PubMed] [Google Scholar]
- 17.Noordam C, Dhir V, McNelis JC, Schlereth F, Hanley NA, Krone N, Smeitink JA, Smeets R, Sweep FC, Claahsen-van der Grinten HL, Arlt W. Inactivating PAPSS2 mutations in a patient with premature pubarche. The New England journal of medicine. 2009;360:2310–2318. doi: 10.1056/NEJMoa0810489. [DOI] [PubMed] [Google Scholar]
- 18.Kenny FM, Preeyasombat C, Migeon CJ. Cortisol production rate. II. Normal infants, children, and adults. Pediatrics. 1966;37:34–42. [PubMed] [Google Scholar]
- 19.Harris JA, Jackson CM, Paterson DG, Scammon RE. The measurement of man [Minneapolis] The University of Minnesota press; 1930. [Google Scholar]
- 20.Stowens D. Pediatric pathology. 2nd. Baltimore: Williams & Wilkins Co; 1966. [Google Scholar]
- 21.Dhom G. The prepuberal and puberal growth of the adrenal (adrenarche) Beitrage zur Pathologie. 1973;150:357–377. doi: 10.1016/s0005-8165(73)80086-1. [DOI] [PubMed] [Google Scholar]
- 22.Anderson DC. The adrenal androgen-stimulating hormone does not exist. Lancet. 1980;2:454–456. doi: 10.1016/s0140-6736(80)91889-9. [DOI] [PubMed] [Google Scholar]
- 23.Byrne GC, Perry YS, Winter JS. Steroid inhibitory effects upon human adrenal 3 beta-hydroxysteroid dehydrogenase activity. The Journal of clinical endocrinology and metabolism. 1986;62:413–418. doi: 10.1210/jcem-62-2-413. [DOI] [PubMed] [Google Scholar]
- 24.Dickerman Z, Grant DR, Faiman C, Winter JS. Intraadrenal steroid concentrations in man: zonal differences and developmental changes. Journal of Clinical Endocrinology and Metabolism. 1984;59:1031–1036. doi: 10.1210/jcem-59-6-1031. [DOI] [PubMed] [Google Scholar]
- 25.Rabe T, Kiesel L, Runnebaum B. Regulation of human placental progesterone synthesis in vitro by naturally occurring steroids. J Steroid Biochem. 1985;22:657–664. doi: 10.1016/0022-4731(85)90220-1. [DOI] [PubMed] [Google Scholar]
- 26.Kahri AI, Voutilainen R, Salmenpera M. Different biological action of corticosteroids, corticosterone and cortisol, as a base of zonal function of adrenal cortex. Acta endocrinologica. 1979;91:329–337. doi: 10.1530/acta.0.0910329. [DOI] [PubMed] [Google Scholar]
- 27.Topor LS, Asai M, Dunn J, Majzoub JA. Cortisol stimulates secretion of dehydroepiandrosterone in human adrenocortical cells through inhibition of 3betaHSD2. The Journal of clinical endocrinology and metabolism. 2011;96:E31–39. doi: 10.1210/jc.2010-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas JL, Rajapaksha M, Mack VL, DeMars GA, Majzoub JA, Bose HS. Regulation of human 3beta-hydroxysteroid dehydrogenase type 2 by adrenal corticosteroids and product-feedback by androstenedione in human adrenarche. The Journal of pharmacology and experimental therapeutics. 2015;352:67–76. doi: 10.1124/jpet.114.219550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rainey WE, Bird IM, Sawetawan C, Hanley NA, McCarthy JL, McGee EA, Wester R, Mason JI. Regulation of human adrenal carcinoma cell (NCI-H295) production of C19 steroids. Journal of Clinical Endocrinology and Metabolism. 1993;77:731–737. doi: 10.1210/jcem.77.3.8396576. [DOI] [PubMed] [Google Scholar]
- 30.Rainey WE, Saner K, Schimmer BP. Adrenocortical cell lines. Molecular and Cellular Endocrinology. 2004;228:23–38. doi: 10.1016/j.mce.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 31.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell. 1986;46:645–652. doi: 10.1016/0092-8674(86)90339-9. [DOI] [PubMed] [Google Scholar]
- 32.Codner E, Okuma C, Iniguez G, Boric MA, Avila A, Johnson MC, Cassorla FG. Molecular study of the 3 beta-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. The Journal of clinical endocrinology and metabolism. 2004;89:957–964. doi: 10.1210/jc.2002-020873. [DOI] [PubMed] [Google Scholar]
- 33.Low SC, Chapman KE, Edwards CR, Seckl JR. ‘Liver-type’ 11 beta-hydroxysteroid dehydrogenase cDNA encodes reductase but not dehydrogenase activity in intact mammalian COS-7 cells. J Mol Endocrinol. 1994;13:167–174. doi: 10.1677/jme.0.0130167. [DOI] [PubMed] [Google Scholar]
- 34.Rheaume E, Lachance Y, Zhao HF, Breton N, Dumont M, de Launoit Y, Trudel C, Luu-The V, Simard J, Labrie F. Structure and expression of a new complementary DNA encoding the almost exclusive 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4-isomerase in human adrenals and gonads. Mol Endocrinol. 1991;5:1147–1157. doi: 10.1210/mend-5-8-1147. [DOI] [PubMed] [Google Scholar]
- 35.Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. The Journal of pediatrics. 1990;117:892–896. doi: 10.1016/s0022-3476(05)80128-3. [DOI] [PubMed] [Google Scholar]
- 36.Esteban NV, Loughlin T, Yergey AL, Zawadzki JK, Booth JD, Winterer JC, Loriaux DL. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. The Journal of clinical endocrinology and metabolism. 1991;72:39–45. doi: 10.1210/jcem-72-1-39. [DOI] [PubMed] [Google Scholar]
- 37.Kelnar CJ, Brook CG. A mixed longitudinal study of adrenal steroid excretion in childhood and the mechanism of adrenarche. Clinical Endocrinology. 1983;19:117–129. doi: 10.1111/j.1365-2265.1983.tb00750.x. [DOI] [PubMed] [Google Scholar]
- 38.Lashansky G, Saenger P, Fishman K, Gautier T, Mayes D, Berg G, Di Martino-Nardi J, Reiter E. Normative data for adrenal steroidogenesis in a healthy pediatric population: age- and sex-related changes after adrenocorticotropin stimulation. The Journal of clinical endocrinology and metabolism. 1991;73:674–686. doi: 10.1210/jcem-73-3-674. [DOI] [PubMed] [Google Scholar]
- 39.Rich BH, Rosenfield RL, Lucky AW, Helke JC, Otto P. Adrenarche: changing adrenal response to adrenocorticotropin. Journal of Clinical Endocrinology and Metabolism. 1981;52:1129–1136. doi: 10.1210/jcem-52-6-1129. [DOI] [PubMed] [Google Scholar]
- 40.Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. Journal of Clinical Endocrinology and Metabolism. 2001;86:2364–2368. doi: 10.1210/jcem.86.6.7603. [DOI] [PubMed] [Google Scholar]
- 41.Shi L, Wudy SA, Buyken AE, Hartmann MF, Remer T. Body fat and animal protein intakes are associated with adrenal androgen secretion in children. Am J Clin Nutr. 2009;90:1321–1328. doi: 10.3945/ajcn.2009.27964. [DOI] [PubMed] [Google Scholar]
- 42.Brentano ST, Picado-Leonard J, Mellon SH, Moore CC, Miller WL. Tissue-specific, cyclic adenosine 3′,5′-monophosphate-induced, and phorbol ester-repressed transcription from the human P450c17 promoter in mouse cells. Molecular Endocrinology. 1990;4:1972–1979. doi: 10.1210/mend-4-12-1972. [DOI] [PubMed] [Google Scholar]
- 43.Dhom G. Morphology of the human adrenarche. Acta Endocrinologica Supplementum. 1973;173:27. doi: 10.1530/acta.0.072s027. [DOI] [PubMed] [Google Scholar]
- 44.Veldhuis JD, Roelfsema F, Iranmanesh A, Carroll BJ, Keenan DM, Pincus SM. Basal, pulsatile, entropic (patterned), and spiky (staccato-like) properties of ACTH secretion: impact of age, gender, and body mass index. The Journal of clinical endocrinology and metabolism. 2009;94:4045–4052. doi: 10.1210/jc.2009-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Remer T, Manz F. The midgrowth spurt in healthy children is not caused by adrenarche. Journal of Clinical Endocrinology and Metabolism. 2001;86:4183–4186. doi: 10.1210/jcem.86.9.7862. [DOI] [PubMed] [Google Scholar]
- 46.Kaplowitz P. Clinical characteristics of 104 children referred for evaluation of precocious puberty. The Journal of clinical endocrinology and metabolism. 2004;89:3644–3650. doi: 10.1210/jc.2003-031532. [DOI] [PubMed] [Google Scholar]
- 47.Weber A, Clark AJ, Perry LA, Honour JW, Savage MO. Diminished adrenal androgen secretion in familial glucocorticoid deficiency implicates a significant role for ACTH in the induction of adrenarche. Clinical Endocrinology. 1997;46:431–437. doi: 10.1046/j.1365-2265.1997.1580969.x. [DOI] [PubMed] [Google Scholar]
- 48.Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, Castro-Magana M, Copeland KC, David R, Pang S. Newly proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid dehydrogenase deficiency. The Journal of clinical endocrinology and metabolism. 2002;87:2611–2622. doi: 10.1210/jcem.87.6.8615. [DOI] [PubMed] [Google Scholar]
- 49.Wirix AJ, Finken MJ, von Rosenstiel-Jadoul IA, Heijboer AC, Nauta J, Groothoff JW, Chinapaw MJ, Kist-van Holthe JE. Is There an Association Between Cortisol and Hypertension in Overweight or Obese Children? Journal of clinical research in pediatric endocrinology. 2017;9:344–349. doi: 10.4274/jcrpe.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandon DD, Isabelle LM, Samuels MH, Kendall JW, Loriaux DL. Cortisol production rate measurement by stable isotope dilution using gas chromatography-negative ion chemical ionization mass spectrometry. Steroids. 1999;64:372–378. doi: 10.1016/s0039-128x(98)00112-3. [DOI] [PubMed] [Google Scholar]
- 51.Vierhapper H, Nowotny P, Waldhausl W. Production rates of cortisol in obesity. Obesity research. 2004;12:1421–1425. doi: 10.1038/oby.2004.178. [DOI] [PubMed] [Google Scholar]
- 52.Brunelli VL, Chiumello G, David M, Forest MG. Adrenarche does not occur in treated patients with congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. Clinical Endocrinology. 1995;42:461–466. doi: 10.1111/j.1365-2265.1995.tb02663.x. [DOI] [PubMed] [Google Scholar]
- 53.Sellers EP, MacGillivray MH. Blunted adrenarche in patients with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Research. 1995;21:537–544. doi: 10.1080/07435809509030471. [DOI] [PubMed] [Google Scholar]
- 54.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- 55.Dardis A, Saraco N, Rivarola MA, Belgorosky A. Decrease in the expression of the 3beta-hydroxysteroid dehydrogenase gene in human adrenal tissue during prepuberty and early puberty: implications for the mechanism of adrenarche. Pediatric Research. 1999;45:384–388. doi: 10.1203/00006450-199903000-00016. [DOI] [PubMed] [Google Scholar]