

Abstract
Recent evidence suggests that metformin exerts glucose‐lowering effects via its action in the gut. The accumulation in the colon of 18F‐labeled fluorodeoxyglucose (FDG), a nonmetabolizable derivative of glucose, is markedly augmented after metformin administration, indicating that metformin affects the handling of glucose in colon whereas the underlying mechanism and the significance to glucose metabolism of the finding remain to be determined.
One hundred years have passed since guanidine, a precursor compound of biguanides and thus of metformin, was found to have a glucose‐lowering effect in live animals1. In this historical study, Watanabe1 did not set out to identify glucose‐lowering agents, but rather to understand the mechanism underlying the development of tetany in animals that have undergone parathyroidectomy. Watanabe1 concluded that guanidine is a pathogenic factor responsible for tetany, because its administration in rabbits resulted in ‘tetany‐like’ muscle cramps associated with low blood glucose levels. Since this serendipitous discovery of a glucose‐lowering agent, which occurred 3 years before the discovery of insulin, various mechanisms for how biguanides exert their effects have been proposed. Despite the time elapsed, however, a complete picture of the pharmacological action of metformin, the most widely administered antidiabetic drug, has yet to be realized.
Metformin is thought to exert its primary antidiabetic action through suppression of gluconeogenesis in the liver2, 3. To date, two key molecular targets of metformin have been identified, both of which are localized to mitochondria (Figure 1)2, 4. Metformin thus inhibits the function of mitochondrial respiratory complex I, which leads to an increase in the cellular ratio of adenosine monophosphate (AMP) to adenosine triphosphate (ATP) as a result of a reduction in the efficiency of ATP production. This increase in the AMP:ATP ratio triggers the activation of AMP‐activated protein kinase (AMPK), which has a variety of effects on energy metabolism, including downregulation of the expression of gluconeogenic genes2. The increase in AMP concentration is also thought to inhibit the activity of adenylate cyclase5, an important mediator of glucagon action, and thus in turn also results in the inhibition of gluconeogenesis. The second target of metformin in mitochondria is mitochondrial glycerol‐3‐phosphate dehydrogenase4, which plays a key role in the glycerophosphate shuttle. This oxidoreduction shuttle between the cytosol and mitochondria is necessary for production of the oxidized form of coenzymes required for biochemical reactions, such as nicotinamide adenine dinucleotide in gluconeogenic reactions. Metformin inhibits the activity of mitochondrial glycerol‐3‐phosphate dehydrogenase, which thus results in the suppression of gluconeogenic reactions including the conversion of lactate to pyruvate4. These various biochemical findings, together with the results of glucose‐clamp analyses3, indicate that the liver is the principal target organ of metformin in the circulation.
Figure 1.
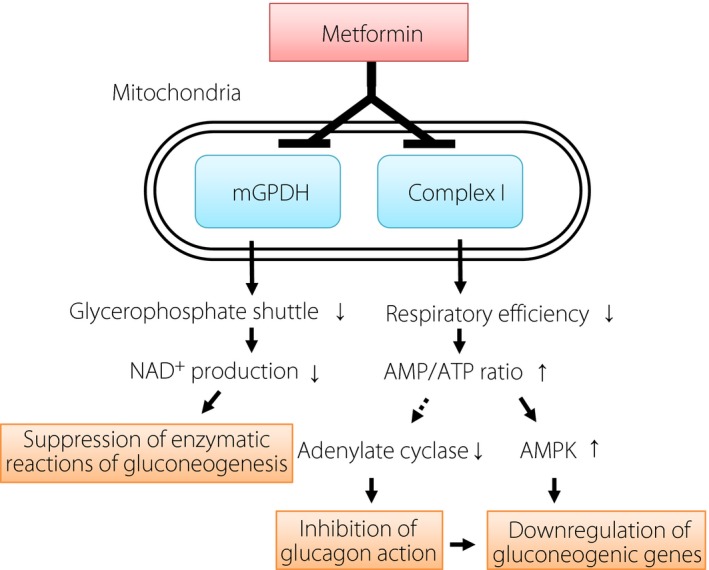
Intracellular targets of metformin and the mechanisms underlying its inhibition of gluconeogenesis. Metformin attenuates mitochondrial respiration through inhibition of respiratory complex I, which results sequentially in an increase in the cellular adenosine monophosphate (AMP) to adenosine triphosphate (ATP) ratio, activation of AMP‐activated protein kinase (AMPK) and suppression of gluconeogenic gene expression. The increase in the cellular AMP concentration likely also inhibits adenylate cyclase activity and thereby suppresses glucagon action. Metformin also inhibits mitochondrial glycerol‐3‐phosphate dehydrogenase (mGPDH) and thereby impairs the production of nicotinamide adenine dinucleotide (NAD +) required for gluconeogenic reactions.
The half‐life of orally administered metformin in blood is as short as 3–4 h. This rapid disappearance of metformin from the circulation appears inconsistent with the duration of the glucose‐lowering effect generally recognized by clinicians. In contrast, orally administered metformin remains in the gastrointestinal tract for a substantially longer time, a phenomenon thought by some investigators to account for the difference between the half‐life of metformin in blood and the apparent time‐course of its clinical action. In 1968, Czyzyk et al.6 showed that administration of butyl‐biguanide (buformin) in dogs both attenuated the increase in blood glucose levels during glucose infusion from a catheter placed in the duodenum and increased the amount of unabsorbed glucose recovered in the distal small intestine. They thus concluded that suppression of glucose absorption from the intestine might explain not only the blood glucose‐lowering effect, but also the anti‐obesity effect and gastrointestinal adverse effects of this class of drugs. Although inhibition of glucose absorption from the intestine is no longer recognized as a principal mechanism underlying the glucose‐lowering effect of metformin, advances in imaging technology have shown that metformin influences glucose handling in the human intestine. It was thus found, again by chance, that the accumulation in the colon of 18F‐labeled fluorodeoxyglucose, a non‐metabolizable derivative of glucose, is markedly augmented after metformin administration (Figure 2)7. Whereas both the underlying mechanism and the significance to glucose metabolism of this observation remain to be determined, this finding indeed suggests that metformin affects the handling of glucose in the colon.
Figure 2.
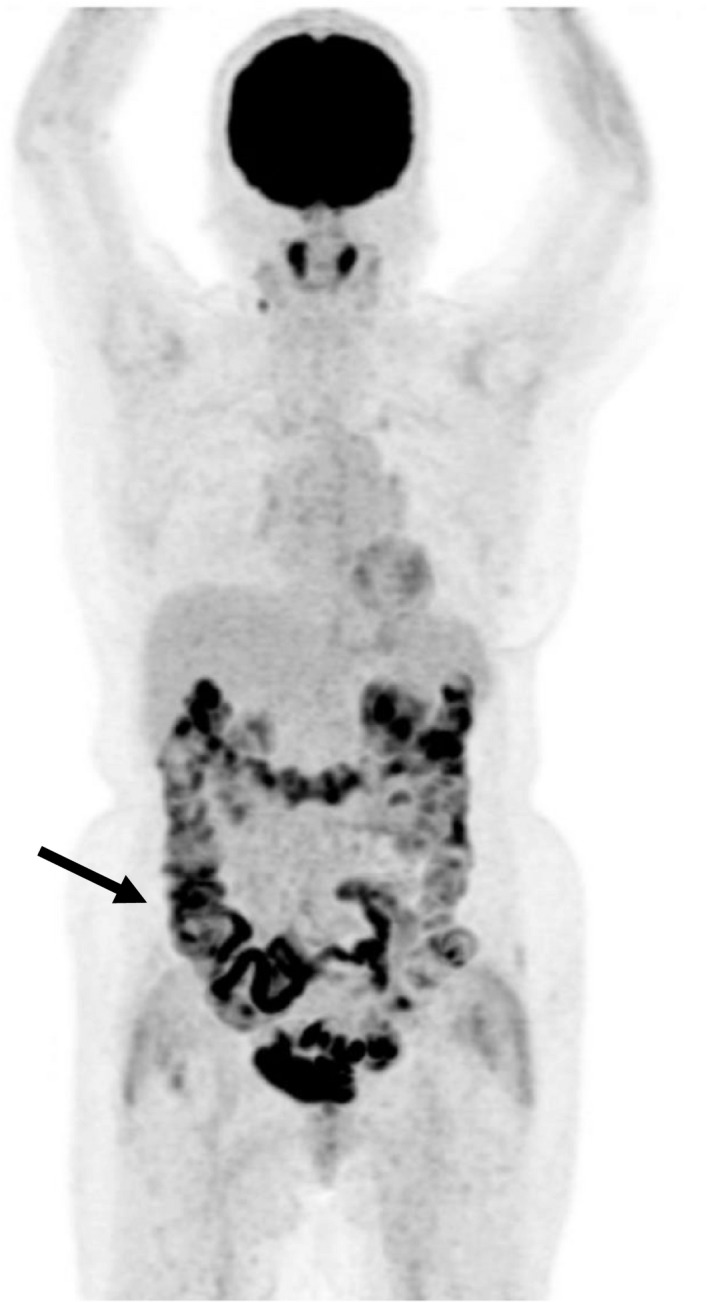
Representative image of 18F‐labeled fluorodeoxyglucose positron emission tomography of an individual taking metformin. The image was obtained from a 70‐year‐old woman with type 2 diabetes and paraganglioma who was taking 1,000 mg of metformin daily. The arrow indicates accumulation of 18F‐labeled fluorodeoxyglucose in the colon.
A recent clinical trial has provided strong evidence that metformin that remains in the intestine exerts a glucose‐lowering effect. This phase 2 trial measured the blood concentrations and therapeutic effects both of extended‐release metformin – which is on the market in several countries including the USA and some European nations – and of the newly developed delayed‐release metformin (Met DR)8. Met DR is formulated to be released slowly in the intestine and to be absorbed from the region of the gut comprising the distal jejunum to the colon. The bioavailability of Met DR is thus low, and its absorption is ~50% of that of extended‐release metformin. The glucose‐lowering effect of Met DR, however, was found to be similar to that of the same dose of extended‐release metformin, supporting the notion that metformin lowers blood glucose levels not only through an action in the circulation, but also through one in the intestine.
How might metformin achieve such an intraintestinal effect? Duca et al.9 demonstrated an action of preabsorptive metformin in the intestine mediated by the central nervous system. Administration of metformin through a catheter placed in the duodenal lumen of rats resulted in marked suppression of hepatic glucose production in association with the activation of AMPK in duodenal cells. Expression of a dominant negative form of AMPK in duodenal cells or administration either of an antagonist of the glucagon‐like peptide‐1 receptor or of an inhibitor of protein kinase A prevented this effect of metformin on glucose production, suggesting that the effect is mediated by an AMPK–glucagon‐like peptide‐1–protein kinase A pathway. The attenuation of hepatic glucose production by the intraduodenal administration of metformin was also prevented by inhibition of signaling by afferent or efferent branches of the vagus nerve, or by suppression of the activity of the nucleus of the solitary tract, a relay nucleus of the vagus, suggesting that this effect of metformin is mediated by a duodenum–central nervous system–liver axis and relayed by the vagus nerve. Whereas some missing links remain to be filled in, such as the mechanistic link from duodenal AMPK to glucagon‐like peptide‐1 signaling, this study uncovered a previously unrecognized role of the duodenum in metformin action. The finding that little Met DR is released in the duodenum, however, also suggests that metformin is able to exert its antidiabetic action in a more distal portion of the intestine.
The application of next‐generation deoxyribonucleic acid sequencing has shown that the gut microbiota plays a key role in a variety of physiological, pathological and pharmacological phenomena. Whereas several studies have shown a correlation between metformin action and changes in the gut microbiota, a recent study by Wu et al.10 provided evidence for a causal relationship between the two. In this double‐blind, randomized, controlled clinical trial, in which treatment‐naïve individuals who had been recently diagnosed with type 2 diabetes mellitus took either metformin or a placebo for 4 months, metformin administration was found to alter the gut microbiota. The changes included an increase in the abundance of Akkermansia muciniphila – which was previously shown to undergo expansion in the gut of humans or rodents treated with metformin – as well as stimulation of the growth of Bifidobacterium adolescentis, the extent of which was correlated with the improvement in glycosylated hemoglobin level. Short‐chain fatty acids produced by the gut microbiota are thought to mediate biological interactions with the host. Metformin treatment altered the expression of genes related to short‐chain fatty acid metabolism and increased short‐chain fatty acid levels in feces. Furthermore, Wu et al.10 transferred feces from mice before and 4 months after treatment to germ‐free mice, and found that feces of the metformin‐treated mice improved glucose tolerance in the recipient mice. Although it remains unclear which functions of which bacterial species are important for metformin action, the results of this study indicate the existence of a strong causal link between changes in the gut microbiota and the glucose‐lowering effect of metformin. To confirm that the observed changes to the microbiota were attributable to a direct action of metformin, and not to a secondary influence resulting from the improvement in systemic metabolism, Wu et al.10 cultured fecal samples from non‐treated subjects in ‘a gut simulator’ in the presence of metformin. This ex vivo experiment confirmed that metformin directly modulated the function and growth of the microbiota in a gut‐like environment.
The hypothesis that the glucose‐lowering effect of metformin is mediated in part through the microbiota is attractive. The observation that the effect of metformin on the accumulation of 18F‐labeled fluorodeoxyglucose in the colon had not completely disappeared 3 days after cessation of metformin administration7 suggests that this long‐lasting effect might also be influenced by the gut microbiota. Although the efficacy and safety of metformin have been established by its ≥60 years of clinical administration, the possibility of the development of lactic acidosis, a rare adverse event associated with the drug, sometimes limits its use. Guidelines and labels in many countries warn healthcare providers against the prescription of metformin for individuals at risk for this serious side‐effect, including those with dysfunction of major organs, such as the lungs, heart, liver and kidneys, as well as elderly patients. Given that the inhibition of mitochondrial function might increase the production of lactate, lactic acidosis is likely related to metformin action outside of the gut (Figure 1). Further knowledge of metformin action in the gut might provide the basis for the development of novel antidiabetic drugs that exert their effects only in the intestine. As suggested by Buse et al.8, such drugs might possess good clinical efficacy without the risk for lactic acidosis.
Disclosure
WO has received lecture fees and research support from Sumitomo Dainippon Pharma, Novartis, Takeda Pharmaceutical and Sanwa Kagaku Kenkyusho. The other authors declare no conflict of interest.
References
- 1. Watanabe CK. Studies in the metabolic changes induced by administration of guanidine bases. I. Influence of injected guanidine hydrochloride upon blood sugar content. J Biol Chem 1918; 33: 253–265. [Google Scholar]
- 2. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017; 60: 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hundal RS, Krssak M, Dufour S, et al Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000; 49: 2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Madiraju AK, Erion DM, Rahimi Y, et al Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014; 510: 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller RA, Chu Q, Xie J, et al Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013; 494: 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Czyzyk A, Tawecki J, Sadowski J, et al Effect of biguanides on intestinal absorption of glucose. Diabetes 1968; 17: 492–498. [DOI] [PubMed] [Google Scholar]
- 7. Lee SH, Jin S, Lee HS, et al Metformin discontinuation for less than 72 h is suboptimal for F‐18 FDG PET/CT interpretation of the bowel. Ann Nucl Med 2016; 30: 629–636. [DOI] [PubMed] [Google Scholar]
- 8. Buse JB, DeFronzo RA, Rosenstock J, et al The primary glucose‐lowering effect of metformin resides in the gut, not the circulation: results from short‐term pharmacokinetic and 12‐week dose‐ranging studies. Diabetes Care 2016; 39: 198–205. [DOI] [PubMed] [Google Scholar]
- 9. Duca FA, Cote CD, Rasmussen BA, et al Metformin activates a duodenal AMPK‐dependent pathway to lower hepatic glucose production in rats. Nat Med 2015; 21: 506–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu H, Esteve E, Tremaroli V, et al Metformin alters the gut microbiome of individuals with treatment‐naïve type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 2017; 23: 850–858. [DOI] [PubMed] [Google Scholar]