

Abstract
The ability to adapt behavior to environmental fluctuations is critical for survival of organisms ranging from invertebrates to mammals. Caenorhabditis elegans can learn to avoid sodium chloride when it is paired with starvation. This behavior may help animals avoid areas without food. Although some genes have been implicated in this salt-aversive learning behavior, critical genetic components, and the neural circuit in which they act, remain elusive. Here, we show that the sole worm ortholog of mammalian CaMKI/IV, CMK-1, is essential for salt-aversive learning behavior in C. elegans hermaphrodites. We find that CMK-1 acts in the primary salt-sensing ASE neurons to regulate this behavior. By characterizing the intracellular calcium dynamics in ASE neurons using microfluidics, we find that loss of cmk-1 has subtle effects on sensory-evoked calcium responses in ASE axons and their modulation by salt conditioning. Our study implicates the expression of the conserved CaMKI/CMK-1 in chemosensory neurons as a regulator of behavioral plasticity to environmental salt in C. elegans.
SIGNIFICANCE STATEMENT Like other animals, the nematode Caenorhabditis elegans depends on salt for survival and navigates toward high concentrations of this essential mineral. In addition to its role as an essential nutrient, salt also causes osmotic stress at high concentrations. A growing body of evidence indicates that C. elegans balances the requirement for salt with the danger it presents through a process called salt-aversive learning. We show that this behavior depends on expression of a calcium/calmodulin-dependent kinase, CMK-1, in the ASE salt-sensing neurons. Our study identifies CMK-1 and salt-sensitive chemosensory neurons as key factors in this form of behavioral plasticity.
Keywords: calcium imaging, CaM kinase, chemotaxis, learning, salt-seeking
Introduction
Behavioral plasticity allows animals to survive a constantly changing environment. This trait enables organisms to seek environments rich in essential resources like food and potential mates, and to avoid barren or perilous ones. Commensurate with its importance, there have been widespread efforts to identify the genes and neural circuits involved in behavioral plasticity. The nematode Caenorhabditis elegans exhibits behavioral plasticity in response to a variety of external stimuli, including temperature, touch, smell, and taste (Ardiel and Rankin, 2010). For this reason and because of its well-characterized nervous system and ease of genetic manipulation, C. elegans is an informative model system for investigating behavioral plasticity.
All animals require salt (sodium chloride) for survival and, as a result, salt-seeking is a highly conserved behavior (Geerling and Loewy, 2008; Hurley and Johnson, 2015). However, high salt can also be a threat especially to small, terrestrial animals like C. elegans because it generates osmotic stress (Choe, 2013; Kim and Jin, 2015). Thus, the worm experiences an intense pressure to balance the need for salt with the need to avoid toxic concentrations. A considerable body of evidence establishes that C. elegans nematodes seek salt by performing positive chemotaxis (Ward, 1973; Bargmann and Horvitz, 1991) and this attraction can be transformed into aversion (negative chemotaxis) following conditioning in solutions rich in salt and poor in bacterial food (Saeki et al., 2001; Tomioka et al., 2006). Although several signaling pathways (Table 1), including those linked to insulin signaling (Tomioka et al., 2006; Vellai et al., 2006; Adachi et al., 2010; Oda et al., 2011; Kunitomo et al., 2013), modulate this salt-aversive learning behavior, the majority of mutants analyzed thus far exhibit modest deficits in learning ability. Thus, genes essential for salt-aversive learning behavior and their cellular locus of action remain under-studied.
Table 1.
Pathways implicated in salt-aversive learning
| Pathway | Citations |
|---|---|
| G-protein signaling | (Jansen et al., 2002; Hukema et al., 2006; Matsuki et al., 2006; Sakashita et al., 2008; Adachi et al., 2010; Kunitomo et al., 2013) |
| Insulin signaling | (Tomioka et al., 2006; Vellai et al., 2006; Adachi et al., 2010; Oda et al., 2011; Kunitomo et al., 2013) |
| Synaptic proteins | (Ishihara et al., 2002; Ikeda et al., 2008; Stetak et al., 2009; Iwata et al., 2011) |
| Neurotransmitters | (Hukema et al., 2008; Kano et al., 2008; Stetak et al., 2009) |
| Other | (Saeki et al., 2001; Iino and Yoshida, 2009; Sakai et al., 2013) |
Calcium and calmodulin (CaM)-dependent kinases play evolutionarily conserved roles in the nervous system in signal transduction, synaptic development, and behavioral plasticity. Among other functions, CaM kinases respond to changes in cytosolic calcium levels and regulate gene transcription in the nucleus. Although CaMKII is the most intensively studied, CaMKI and CaMKIV have been found to be important for specific functions such as developmental and activity-dependent growth (Hook and Means, 2001; Wayman et al., 2008; Cohen et al., 2015). Recently, the sole worm ortholog of mammalian CaMKI/IV, CMK-1, has emerged as a versatile regulator of neuronal plasticity in C. elegans. CMK-1 is expressed broadly throughout the worm's nervous system (Kimura et al., 2002; Satterlee et al., 2004), and regulates behavioral plasticity in responses to thermal stimuli (Satterlee et al., 2004; Schild et al., 2014; Yu et al., 2014; Kobayashi et al., 2016). It also regulates developmental plasticity by affecting entry into the “dauer” diapause state in response to food deprivation (Neal et al., 2015), and synaptic plasticity by regulating expression of an AMPA-type glutamate receptor (Moss et al., 2016). However, whether CMK-1 is implicated in salt attraction or its transformation to salt aversion following conditioning is unknown.
Here, we show that CMK-1 is absolutely required for salt-aversive learning behavior yet dispensable for salt attraction. In contrast, disrupting the only other CaM kinase encoded by the C. elegans genome, UNC-43 CaMKII, or other kinases such as AMP kinase and protein kinase C, has no detectable effect on either positive chemotaxis or its transformation to negative chemotaxis following conditioning in high-salt and the absence of bacterial food. By expressing wild-type CMK-1 in a neuron-specific manner, we find that cmk-1 functions in the ASEL and ASER chemosensory neurons known to be required for salt chemotaxis (Bargmann and Horvitz, 1991). Having identified the ASE neurons as a critical cellular locus for cmk-1 action, we analyze salt-evoked calcium signals in wild-type and cmk-1 mutant ASE neurons. Consistent with prior studies, we show that conditioning sufficient to induce salt-aversive learning blunts salt-evoked calcium signals in ASE cell bodies. Loss of cmk-1 has subtle effects on salt-evoked calcium responses in ASE dendrites and axons, affecting ASEL to a greater extent than ASER. Collectively, our results expand the forms of behavioral plasticity that depend upon CMK-1 function in primary sensory neurons.
Materials and Methods
Worm strains and husbandry.
C. elegans strains were cultured using standard conditions at 20°C on NGM plates seeded with OP50-1 bacteria. All experiments were performed using adult hermaphrodites. Transgenic worms were generated using microinjection, resulting in formation of extra-chromosomal arrays. A subset of the C. elegans strains we used were generated specifically for this study, the remainder were obtained from our in-house collections, as gifts from colleagues, or from the Caenorhabditis Genetics Center. Table 2 lists all strains, their RRIDs (if available) or origin, and the figures showing data from each strain.
Table 2.
List of C. elegans strains used in this study
| Strain name | Genotype | RRID or source | Figure |
|---|---|---|---|
| N2 | Wild-type, Bristol | WB-STRAIN:N2_(ancestral) | Throughout |
| RB754 | aak-2(ok524) X | WB-STRAIN:RB754 | Fig. 1C |
| RB781 | pkc-1(ok563) V | WB-STRAIN:RB781 | Fig. 1C |
| VC127 | pkc-2(ok328) X | WB-STRAIN:VC127 | Fig. 1C |
| VC1052 | unc-43(gk452) IV | WB-STRAIN:VC1052 | Fig. 1C |
| JT200 | unc-43(sa200) IV | WB-STRAIN:JT200 | Fig. 1C |
| VC220 | cmk-1(ok287) IV | WB-STRAIN:VC220 | Throughout |
| PY1589 | cmk-1(oy21) IV | WB-STRAIN:PY1589 | Figs. 1C, 3B |
| VC691 | ckk-1(ok1033) III | WB-STRAIN:VC691 | Fig. 2A |
| YT17 | crh-1(tz2) III | WB-STRAIN:YT17 | Fig. 2D |
| MT9973 | crh-1(n3315) III | (Kauffman et al., 2010) | Fig. 2D |
| PS3551 | hsf-1(sy441) I | WB-STRAIN:PS3551 | Fig. 2D |
| GN190 | cmk-1(pg58) IV | (Schild et al., 2014) | Fig. 4D |
| GN634 | cmk-1(ok287) IV; oyIs17[gcy-8p::GFP] V; pgEx167[cmk-1p::cmk-1(WT)::GFP; myo-3p::mCherry] | This study | Figs. 1D, 4A |
| DAG307 | cmk-1(ok287) IV; domEx307[cmk-1p::cmk-1(1–348)T179A; unc-122p::RFP] | (Schild et al., 2014) | Fig. 2B |
| GN720 | cmk-1(ok287) IV; domEx328[cmk-1p::cmk-1(K52A)::GFP; unc-122p::RFP] | This study | Fig. 2C |
| DAG610-3 | domEx610–3[flp-6p::cmk-1::GFP; cmk-1p::QF;QUAS::cmk-1::mCherry; unc-122p::RFP] | This study | Fig. 3A |
| GN636 | cmk-1(ok287) IV; domEx122[mec-3p::cmk-1(WT)::GFP; unc-122::RFP] | This study | Fig. 3B |
| GN639-40 | cmk-1(ok287) IV; oyIs17[gcy-8p::GFP] V; pgEx170–1[flp-6p::cmk-1(WT)::GFP; myo-3p::mCherry] | This study | Fig. 3B |
| PY8395 | cmk-1(oy21) IV; Ex[ceh-36Dp::cmk-1(WT); unc-122p::dsRed] line 1 | (Neal et al., 2015) | Fig. 3B |
| DAG176 | cmk-1(ok287) IV; domEx176[cmk-1p::cmk-1(WT)::NES::GFP; unc-122p::RFP] | (Schild et al., 2014) | Fig. 4B,C |
| DAG178 | cmk-1(ok287) IV; domEx178[cmk-1p::cmk-1(WT)::GFP::NLS; unc-122p::RFP] | (Schild et al., 2014) | Fig. 4B,C |
| ZC1600 | yxEx738 [flp-6p::GCaMP3; unc-122p::GFP] | (Luo et al., 2014) | Figs. 5, 6A,B,E, 7A,C, 7-1A |
| GN637, GN670 | cmk-1(ok287) IV; yxEx738 [flp-6p::GCaMP3; unc-122p::GFP] | This study | Figs. 6C–E, 7B,C, 7-1B |
| DAG142 | cmk-1(ok287) IV | This study | Fig. 3C |
| DAG720 | cmk-1(ok287) IV; domEx720[gcy-5p::cmk-1::SL2::mCherry; gcy-6p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3-1 |
| DAG721 | cmk-1(ok287) IV; domIs721[gcy-5p::cmk-1::SL2::mCherry; gcy-6p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3C |
| DAG716 | cmk-1(ok287) IV; domEx716[gcy-6p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3-1 |
| DAG717 | cmk-1(ok287) IV; domEx717[gcy-6p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3C |
| DAG712 | cmk-1(ok287) IV; domEx712[gcy-5p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3-1 |
| DAG713 | cmk-1(ok287) IV; domEx713[gcy-5p::cmk-1::SL2::mCherry; coel::GFP] | This study | Fig. 3C |
Transgenic strains expressing full-length wild-type CMK-1 in one or both ASE neurons were created for this study using standard transgenesis methods (Evans, 2006). Rescue plasmids encoding wild-type CMK-1 were generated with the three-fragment Multisite Gateway system (Invitrogen), as previously described (Schild et al., 2014). A 2027bp gcy-5 promoter fragment, a 710 bp gcy-6 promoter fragment, and a 2682 bp flp-6 promoter fragment were amplified by PCR and cloned into pDONR P4P1r through BP recombination reactions to produce slot1 Entry clones. The primers for amplification of the gcy-5, gcy-6, and flp-6 promoter fragments were as follows:
gcy-5pF, ggggacaactttgtatagaaaagttgGCTCGTTAACTCTTCACATG;
gcy-5pR, ggggactgcttttttgtacaaacttgTTTTCATCAGAATAAGTAATTTTTCGAAAAC;
gcy-6pF, ggggacaactttgtatagaaaagttgACGTAATTGTGTCATTTTTGAAGTC;
gcy-6pR, ggggactgcttttttgtacaaacttgTCACTGGAAAGGAGTTATGAAAATG)
flp-6pF, ggggacaactttgtatagaaaagttgTCGCTTGACTTCTGATG
flp-6pR, ggggactgcttttttgtacaaacttgTTCTGGAATAATCATATTG
For the gcy-5 and gcy-6 strains, expression clones were made through LR reactions between each slot1 Entry clone, a slot2 Entry clone containing cmk-1 coding sequence (mg271), a slot3 Entry clone containing SL2::mCherry (mg277) and a pDEST R4R3 destination vector. The resulting expression plasmids, dg616[gcy-5p::cmk-1::SL2::mCherry] and dg617[gcy-6p::cmk-1::SL2::mCherry], were microinjected together with a [coel::GFP] marker (Addgene #8937, gift from Piali Sengupta; Miyabayashi et al., 1999) in the gonad of DAG142 cmk-1(ok287) animals according to standard procedures (Evans et al., 2006). The final concentration of each plasmid was 20 ng/μl. Stable lines (Table 2) were recovered and epifluorescence microscopy was used to confirm the expression of the trans-spliced mCherry reporter in the expected ASE neurons. The extrachromosomal array in one of the dual rescue lines (DAG721) spontaneously integrated into the genome.
For the flp-6 strains, expression clones were made through LR reactions between each slot1 Entry clone, a slot2 Entry clone containing cmk-1 coding sequence (mg273), a slot3 Entry clone containing GFP::unc-54 (mg208), and a pDEST R4R3 destination vector. The resulting expression plasmid, pJPL3[flp-6p::cmk-1::GFP], was microinjected together with a [myo-3p::mCherry] marker (Addgene #19328) in the gonad of VC220 cmk-1(ok287) animals according to standard procedures (Evans et al., 2006). The final concentration of the plasmid was 50 ng/μl. Stable lines (Table 2) were recovered and epifluorescence microscopy was used to confirm the expression of the GFP reporter in the expected ASE neurons.
Salt conditioning.
Following a previously reported method (Tomioka et al., 2006), we transferred adult hermaphrodites from culture plates to 1.5 ml microcentrifuge tubes containing one of two conditioning buffers before each assay. The first buffer contained no added NaCl (0 mm) and the second buffer contained 20 mm NaCl. Both buffers also contained the following (in mm): 5 KPO4, pH 6.0, 1 MgSO4, and 1 CaCl2. We removed residual bacteria by washing animals once in fresh buffer and conditioned worms for the indicated time periods (between 5 min and 10 h).
Chemotaxis assays.
We adapted methods for assessing salt chemotaxis and salt-aversive learning from Tomioka et al. (2006) and diagram the process in Figure 1A. We generated radial salt gradients on assay plates by placing a 5-mm-thick high-salt agar plug 5 mm away from the edge of the 3.5 cm assay plate (with a 5-mm-thick agar plate) and leaving it in place for 3 h at 23°C. The high-salt 2% agar plug contained the following (in mm): 5 KPO4, pH 6.0, 1 MgSO4, 1 CaCl2, and 100 NaCl. The assay 2% agar assay plate contained the following (in mm): 5 KPO4, pH 6.01, MgSO4, and 1 CaCl2. Immediately before the start of the chemotaxis assay, the plug was removed and a drop (1 μl) of sodium azide (0.1 m) was spotted on opposite sides of the assay plate to immobilize animals near their final positions.
Figure 1.

CMK-1 is required for salt chemotaxis learning. A, Procedure for testing salt-aversive learning. Left to right: Young adult (Day 1) worms are collected from culture plates, conditioned in food-free buffer containing 0 or 20 mm NaCl for 180 min, unless otherwise specified, transferred to the center of an assay plate pretreated to create a radial salt gradient, allowed to migrate for 30 min. Results were quantified by computing a Chemotaxis Index (CI; see Materials and Methods). B, Salt-aversion development following several hours of conditioning in wild-type (N2) adults. Symbols are the mean ± SD of three to nine assays for animals conditioned in 0 mm NaCl (open circles) or 20 mm NaCl (filled circles) for the indicated times. Data were pooled from four independent experiments consisting of three to six assays/condition. ****p < 0.0001. C, Chemotaxis behavior of kinase loss-of-function mutants conditioned without food and either 0 or 20 mm NaCl for 180 min (180′). Each circle is the result of a single assay; horizontal bars indicate the mean ± SD of all the data. Data were pooled from at least two independent experiments consisting of three to six replicates/condition. All assays were performed blind to genotype. A two-way ANOVA revealed a significant interaction between genotype and condition (F(7,237) = 62.34, p < 0.0001). D, Transgenic expression of wild-type CMK-1::GFP under its own promoter restores wild-type salt-aversive learning to cmk-1(ok287) mutants. Each symbol represents the results of a single assay; horizontal bars indicate mean ± SD of all assays. Data were pooled from at least two independent experiments consisting of three to six assays/condition. A two-way ANOVA revealed a significant interaction between genotype and condition (F(2,114) = 44.2, p < 0.0001). Sidak's posthoc tests were used to assess the effect of conditioning in panels C and D. The results are indicated by asterisks: *, **, and **** denote p < 0.1, p < 0.01, and p < 0.0001 and ns denotes not significant.
Conditioned worms were deposited in the middle of a 3.5 cm diameter radial salt gradient assay plate. Residual buffer was wicked off with a Kimwipe, and worms were allowed to crawl for 30 min at 20°C. The number of worms in each region (origin, low salt, high salt) was counted and used to compute a chemotaxis index:
 |
where CI is the chemotaxis and N is the number of worms in the indicated category. For each assay, we collected well-fed worms from a single growth plate (6 cm) into one 1.5 ml microcentrifuge tube for conditioning and then divided this sample into three equal aliquots to perform chemotaxis assays in triplicate.
Locomotion assay.
We collected adult (age: 1 d) worms from growth plates and conditioned them for 180 min, as described above for chemotaxis assays. Locomotion speed was assessed in the absence of food on standard NGM plates and we allowed animals to acclimate for 5 min before imaging. We collected short (1 min) movies at 6× magnification at 20 frames/s (50 ms/frame) with a CCD camera (Mightex, M/N SME-B050-U) mounted on zoom lens (Navitar) with a C-mount adaptor and 0.5× lens and an illumination ring positioned in the same plane as the agar plate and providing radial illumination around it. Tracks showing locomotion were generated using the automated Parallel Worm Tracker software (Ramot et al., 2008) and were used to compute the average speed/track. The average speed of each sample was determined by computing the ensemble average of speed/track.
Colocalization imaging.
Colabeling imaging for flp-6p and cmk-1p fluorescent reporters was performed on an Axioplan 2 Zeiss epifluorescence microscope equipped with an Axiocam camera and a 40× objective (air, NA = 0.95), as previously described (Hostettler et al., 2017).
Subcellular localization imaging.
We collected adult (age: 1 d) worms from growth plates and conditioned them for 180 min. In buffer containing the following (in mm): 5 KPO4, pH 6.0, 1 MgSO4, 1 CaCl2, and 0 NaCl, 20 NaCl, or 0 NaCl with OP50-1 bacteria for 0, 10, 30, and 180 min. After conditioning we put the animals into a microfluidic chip (Nekimken et al., 2017) to restrict the worm's motility during imaging. We took images using a Leica DMI 4000 B microscopy system, consisting of a cyan LED (Spectra X light engine, Lumencor), a fluorescence cube with a beam splitter (Chroma, Q495lp) and an emission filter (Chroma, HQ500lp), a 63×/1.32 NA oil objective (Leica), and a Hamamatsu Orca-Flash 4.0LT digital CMOS camera. The subcellular localization was quantified by comparing the relative average fluorescence intensity of a hand-drawn ROI covering the nucleus versus the average fluorescence intensity of an ROI covering the cytoplasmic compartments each subtracted by the average fluorescence of an ROI outside the cell.
In vivo calcium imaging.
We collected worms (age: 1 d) expressing the flp-6p::GCaMP3 transgene and conditioned them for at least 180 min, using the same procedures and buffers as described for chemotaxis assays. Following conditioning, worms were maintained in the conditioning buffer and positioned in a polydimethylsiloxane microfluidic device (MicroKosmos) that immobilizes the worm's body while leaving the nose exposed to laminar buffer flow. The chip design is based on a device reported by Chronis et al. (2007). Under baseline conditions, the worm's nose was exposed to a NaCl-free buffer for <10 s before the start of image acquisition. Chemosensory stimulation and imaging occurred during 90 s time blocks consisting of a control period (10 s), a 40 s NaCl pulse (20 mm), and a recovery period (40 s). The solutions were switched as described by Chalasani et al. (2007) by a three-way valve (778360, The Lee Company) that was controlled by a power supply (6212B, Hewlett Packard). We added fluorescein (~20 mm) to the solution coming from the two side channels. The stream from these two channels is used to redirect the flow of the NaCl-free and the 20 mm NaCl buffer and to visualize solution switches.
We performed calcium imaging of ASE neurons of worms held in the chip, which was mounted on a Leica DMI 4000 B equipped for epifluorescence imaging based on a cyan LED light source (Spectra X light engine, Lumencor), image splitting optics (W-view Gemini, Hamamatsu), and an sCMOS camera (Orca-Flash 4.0LT, Hamamatsu). We mounted a long-pass filter (Chroma, Q495lp) and an emission filter (Chroma, HQ500lp) in the beam splitter to collect GCaMP3 fluorescence. Fluorescence signals were collected using a 20×/0.4 NA objective (Leica) and recorded at 10 Hz for the entire 90 s of the stimulus protocol. This setup was also used to perform calcium imaging of indentation-evoked calcium responses in C. elegans touch receptors neurons (Nekimken et al., 2017; Fehlauer et al., 2018).
All image sequences were analyzed using custom code running inside Fiji software (Schindelin et al., 2012). Briefly, a desired region of the cell, the sensory ending (dendrite), cell body, or axons was selected manually in the first image of each sequence. In the first image the program identifies the brightest (b) and the dimmest (d) pixel in an area (5 × 5 μm for cell bodies and 1 × 1 μm for dendrites and axons) around this selection. To track the cell region in the following images the brightest pixel of the previous image is used as the midpoint to search for the new brightest and dimmest pixel as described before. Based on the fluorescence of the brightest (Fb) and the dimmest (Fd) pixel a threshold background. For background correction, the intensity of the background (Fbg) was subtracted from the intensity of the region of cell (F). The difference was divided by the mean prestimulus intensity of the region of cell (F0, corrected by Fbg). The result is the background subtracted, relative change of the intensity of the cell region, which is reported as a percentage: F/F0 = 100 × [(F − Fbg)/(F0 − Fbg)].
The signals we report in Figures 5, 6, 7 are normalized to fluorescence levels measured before application of a salt pulse in all conditions and genotypes and recordings were normalized individually before being averaged. Although this normalization strategy differs from those used previously in which stimulus-evoked calcium signals are normalized to values immediately before an up-step or a down-step (Suzuki et al., 2008; Ortiz et al., 2009; Thiele et al., 2009; Oda et al., 2011; Rabinowitch et al., 2014), it enables a simple experimental design in which a single pulse can be used to interrogate responses to both up-steps and down-steps. We also normalized somatic ASER responses to values immediately before a down-step and include the pooled, average responses as insets in Figure 6B and D.
Figure 5.
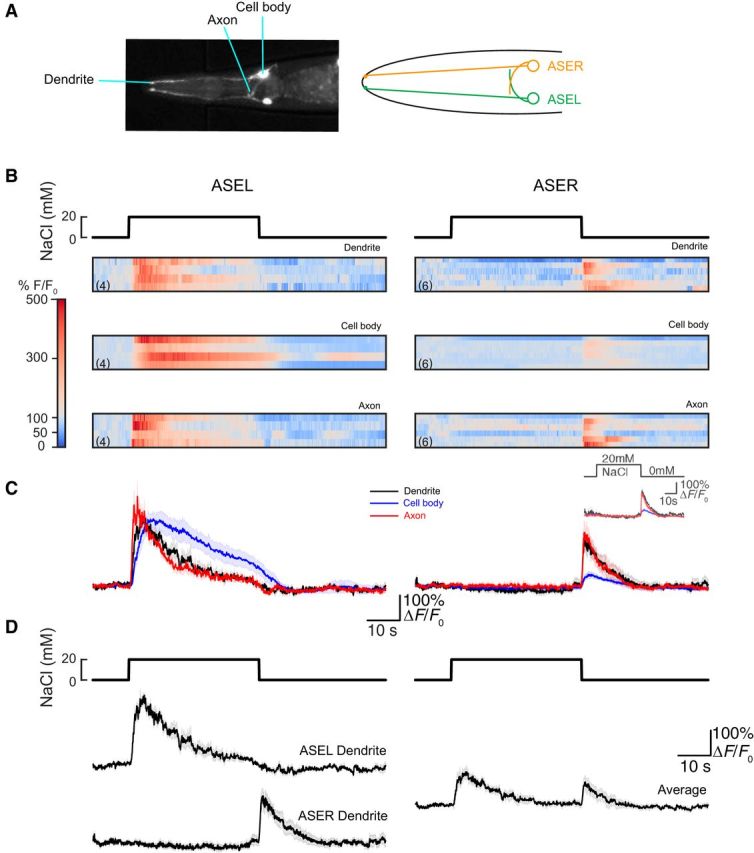
Calcium signals in wild-type ASEL and ASER dendrites, cell bodies, and axons. A, Image and schematic of ASE neurons. Left, Wild-type worms expressing yxEx738 [flp-6p::GCaMP3 + unc-122p::GFP]. The dendrite tips, cell bodies, and axons are easily visualized, allowing for calcium imaging of different regions of these cells. Right, Schematic of ASE neurons. Neurite tips and cell bodies of ASEL and ASER are readily distinguishable from each other, but axons overlap in the part of the neurons that synapse onto the nerve ring of the worm. B, Raster plots of GCaMP3 fluorescence as a function of time in the dendrite, cell body, and axon of the ASEL (left) and ASER (right) neurons of wild-type worms conditioned in 0 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Color indicates fluorescence relative to prestimulus control values, ΔF/F0: white indicates values similar to F0, red and blue indicate increases and decreases, respectively. All recordings were obtained in animals expressing yxEx738 in ASEL or ASER, but not in both neurons and the number of recordings is indicated in parentheses. C, Average calcium signals in the dendrite, cell body, and axon of the ASEL (left) and ASER (right) neurons of wild-type worms derived from the data shown in B, normalized to prestimulus values. Lines indicate mean of corresponding traces and light shading indicates ±SEM. Inset, The average (±SEM) responses recorded in ASER and normalized to values recorded before the down-step. D, Normalized fluorescence of calcium traces in dendrites of the ASE neurons of wild-type worms conditioned in 0 mm NaCl and no food and exposed to up-step (0–20 mm) and down-step (20–0 mm) of NaCl (left; same data as in C). Average of calcium traces in dendrites of ASEL and ASER (right). Lines in normalized fluorescence plots indicate mean of corresponding traces and light shading indicates ±SEM.
Figure 6.
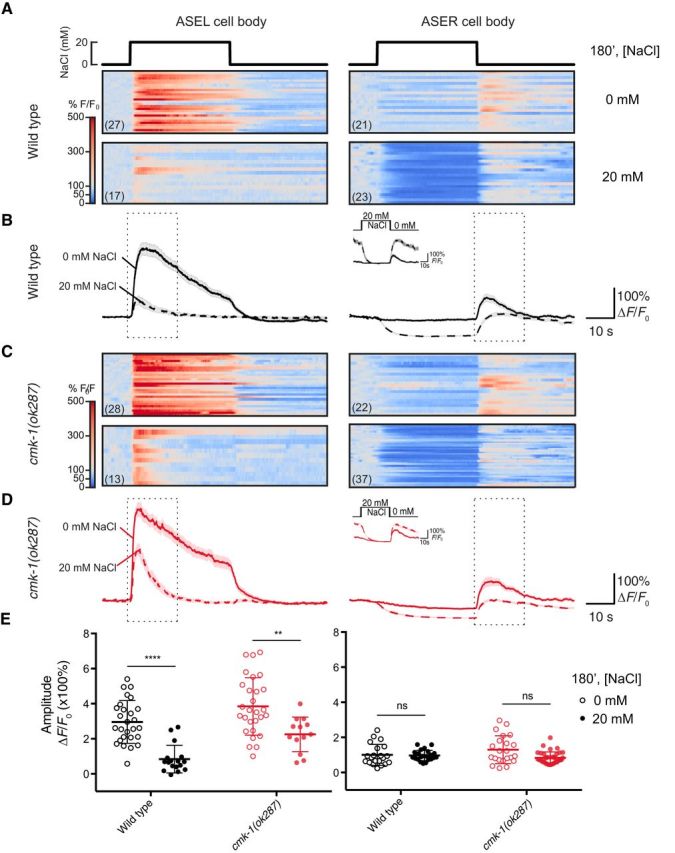
Salt conditioning modifies salt-evoked calcium signals in wild-type and cmk-1 mutant ASER and ASEL cell bodies. A, Raster plots of GCaMP3 fluorescence as a function of time in the cell bodies of the ASEL (left) and ASER (right) neurons of wild-type worms conditioned in 0 or 20 mm NaCl and no food. Animals were exposed to 0 mm NaCl saline and stimulated by a 40 s pulse of 20 mm NaCl. Each horizontal line corresponds to a recording from one cell body; the number of recordings is indicated in parentheses. Color indicates fluorescence relative to prestimulus control values, ΔF/F0: white indicates control values, red and blue indicate increases and decreases, respectively. B, Average calcium signals in the cell bodies of the ASEL (left) and ASER (right) neurons of wild-type worms. The traces are derived from the data shown in A. Inset, The waveform generated when signals generated by ASER cell body are normalized to values measured before the down-step. Lines indicate the mean and light shading indicates ±SEM; solid and dashed lines indicate data collected from animals conditioned in 0 and 20 mm NaCl, respectively. Figure 6-1 shows the response of ASEL and ASER cell bodies to transient decreases in external salt (from 20 to 0 mm) recorded from animals conditioned in 0 and 20 mm NaCl. C, Raster plots of GCaMP3 fluorescence as a function of time in the cell bodies of the ASEL (left) and ASER (right) of cmk-1(ok287) worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Each horizontal line corresponds to a recording of a single cell body; the number of recordings is indicated in parentheses. Color indicates fluorescence relative to prestimulus control values, ΔF/F0: white indicates control values, red and blue indicate increases and decreases, respectively. D, Average fluorescence of calcium traces in cell bodies of the ASEL (left) and ASER (right) neurons of cmk-1(ok287) worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. The traces are derived from the data shown in C. Lines indicate the mean and light shading indicates ±SEM. E, Maximum amplitude of the change in calcium signals recorded in cell bodies of the ASEL (left) and ASER (right) neurons of wild-type and cmk-1(ok287) worms conditioned in 0 or 20 mm NaCl and no food. The amplitude was extracted from calcium traces by subtracting the average signal of 10 s before the outlined region from the maximum signal in the region outlined by dotted line in the normalized fluorescence plots in B and D. Data were analyzed using the Wilcoxon rank sum test for nonparametric data, where p values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. There was a significant effect of salt conditioning upon ON responses in the ASEL body in both wild-type and cmk-1(ok287) animals (wild-type p < 0.0001, cmk-1(ok287) p < 0.01). A significant effect of salt conditioning upon OFF responses in the ASER cell bodies was detected only in cmk-1(ok287) mutants: wild-type p = 0.5728, cmk-1(ok287) p = 0.0691). ** and **** indicate p < 0.01 and p < 0.0001, respectively and ns denotes not significant.
Figure 7.

CMK-1 is required for the effect of salt conditioning on chemosensory calcium signals in dendrites and axons of ASE sensory neurons. A, Average chemosensory calcium signals recorded from ASEL (left) and ASER dendrites (right) in wild-type worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Lines indicate the mean and light shading indicates ±SEM of at least 12 recordings. Figure 7-1A shows the individual replicates for ASEL (left) and ASER (right) dendrites. B, Average chemosensory calcium signals recorded from ASEL (left) and ASER dendrites (right) in cmk-1(ok287) worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Lines indicate the mean and light shading indicates ±SEM of at least 10 recordings. Figure 7-1B shows the individual replicates for ASEL (left) and ASER (right) dendrites. C, Maximum amplitude of ON (left) and OFF (right) responses in dendrites of ASE neurons of wild-type and cmk-1(ok287) worms conditioned in 0 mm NaCl (open circles) or 20 mm NaCl (closed circles). Maximum amplitude was extracted from calcium traces by subtracting the average signal measured 10 s before the outlined region from the maximum signal in the region outlined by dotted line in the normalized fluorescence plots in A and B. For the ASEL dendrite, a two-way ANOVA revealed a significant interaction between genotype and condition (F(1,40) = 4.546, p = 0.0392). For the ASER dendrite, a two-way ANOVA failed to detect an interaction between genotype and condition (F(1,73) = 0.303, p = 0.5837). D, Average chemosensory calcium signals recorded from ASE axons in wild-type worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Lines indicate the mean and light shading indicates ±SEM of at least 11 recordings. Figure 7-1A (right) shows the individual replicates of wild-type axon recordings. The solid cyan lines are a weighted average of the mean traces of the ASEL and ASER dendrites, which were fitted individually [dASEL(t), dASER(t), as described in Materials and Methods]. The weighting factor, dAxon(t) = A * dASEL(t) + (1 − A) * dASER(t), was estimated by minimizing the r2 of the difference between d Axon(t) and the mean fluorescence signal measured from the axon (black line). The weighting factors were 0.4 and 0.29 for 0 and 20 mm NaCl conditioned animals. E, Average chemosensory calcium signals in the axons of ASE neurons in cmk-1(ok287) worms conditioned in 0 or 20 mm NaCl and no food and exposed to a 40 s pulse of 20 mm NaCl. Lines indicate the mean and light shading indicates ±SEM of at least 11 recordings. Figure 7-1B (right) shows the individual replicates for cmk-1 mutant axon recordings. The solid magenta lines are a weighted average of fits to the mean traces of the ASEL and ASER dendrites [dASEL(t), dASER(t); as described in Materials and Methods]. The weighting factor, dAxon (t) = A * dASEL(t) + (1 − A) * dASER(t), was estimated by minimizing the r2 of the difference between dAxon (t) and the mean fluorescence signal measured from the axon. The weighting factors were 0.47 and 0.67 for 0 and 20 mm NaCl conditioned animals, respectively. F, Maximum amplitude of ON (left) and OFF (right) responses in the ASE axons recorded from wild-type and cmk-1(ok287) worms conditioned in 0 mm NaCl (open circles) or 20 mm NaCl (closed circles). Maximum amplitude was extracted from calcium traces by subtracting the average signal of 10 s before the outlined region from the maximum signal in the region outlined by dotted line in the normalized fluorescence plots in D and E. A two-way ANOVA failed to reveal an interaction between genotype and condition (F(3,116) = 1.203, p = 0.3119). Sidak's posthoc tests were used to assess the effect of conditioning and to compare genotypes in panels C and F. The results are indicated by asterisks: ** and **** denote p < 0.01 and p < 0.0001, respectively and ns denotes not significant.
The mean traces of the ASEL and the ASER dendrites were fitted individually with functions describing the ON response in ASEL and the OFF response in ASER as the difference of two exponential functions and the OFF response in ASEL and the ON response in ASER as an exponential function.
 |
 |
Experimental design and statistical analyses.
This study used adult C. elegans hermaphrodites (age: day 1, first day of egg-laying) for all experiments, which included chemotaxis assays, analysis of gene and protein expression by fluorescence microscopy, and neuronal activity by imaging a genetically-encoded calcium indicator. For each chemotaxis assay, we placed between 30 and 450 worms on an assay plate and analyzed at least three assay plates for each condition. We pooled data from at least two independent replicates, which were performed blind to genotype, except in assays involving strains carrying transgenic arrays. In the latter case, genotype was determined post hoc by visual inspection for the presence or absence of the transgene and behavioral performance was scored for both genotypes in parallel. To achieve this goal, we used sodium azide (0.1 m) to immobilize animals after the 30 min chemotaxis assay period and counted animals of both genotypes on the stage of stereomicroscope equipped for epifluorescence.
We built and analyzed four independent transgenic lines coexpressing CMK-1::GFP from a flp-6 promoter active in the ASE neurons and mCherry from the cmk-1 promoter (DAG610, DAG611, DAG612, DAG613). We analyzed the subcellular position of CMK-1 using a transgenic line expressing CMK-1::GFP from a flp-6 promoter (10–18 neurons; see Fig. 4A) and verified subcellular localization of strains where CMK-1 was targeted to the nucleus or cytoplasm (14–37 neurons; see Fig. 4C). We analyzed salt-evoked calcium transients in animals held in a microfluidic trap. In transgenic animals with the yxEx738 [flp-6p::GCaMP3 + unc-122p::GFP] in both ASE neurons, we analyzed signals in 10–27 individual ASER and ASEL neurons across conditions and genotypes. Except in rare instances (4–6 animals), we analyzed a single ASE neuron in each animal. Animals expressing yxEx738 [flp-6p::GCaMP3 + unc-122p::GFP] in either ASER or ASEL, but not both, were rare. As a result, we analyzed signals in four ASEL neurons and six ASER neurons. These data were collected during three independent imaging sessions. We generated two strains expressing ykEx738 in a cmk-1(ok287)-null mutant background, GN637 and GN670, and pooled the results after determining that calcium signals were indistinguishable between the two strains.
Figure 4.
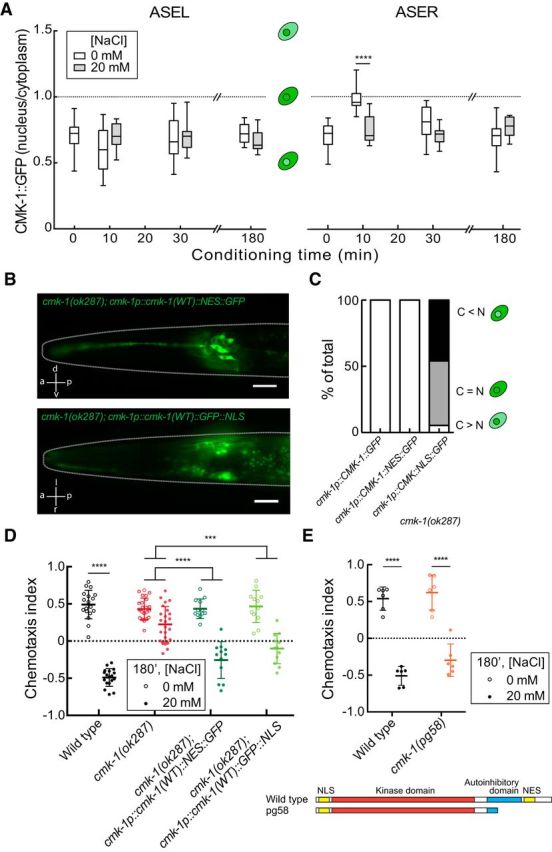
Salt conditioning does not depend on the exact subcellular localization of the CMK-1 protein. A, Subcellular localization of CMK-1 in the ASE neurons. The ratio of fluorescence in the nuclear compartment versus the cytoplasmic compartment is plotted separately for ASEL (left) and ASER (right). Each box-and-whisker bar is the result of the analysis of between 10 and 18 neurons; data were pooled from seven independent experiments. Boxes extend from the 25th to 75th percentiles of the data, and whiskers extend from the minimum value to the maximum value. For ASEL, a two-way ANOVA revealed no interaction between conditioning time and condition (F(2,70) = 2.346, p = 0.1033). For ASER, a two-way ANOVA revealed a significant interaction between conditioning time and condition (F(2,62) = 12.94, p < 0.0001). B, Photomicrographs of transgenic animals expressing GFP-tagged CMK-1fused to an exogenous NES (top) or NLS (bottom). The genotypes analyzed were as follows: cmk-1(ok287);cmk-1p::cmk-1(WT)::GFP, cmk-1(ok287);cmk-1p::cmk-1(WT)::NES::GFP, and cmk-1(ok287);cmk-1p::cmk-1(WT)::GFP::NLS. Scale bar, 20 μm. C, Proportion of neurons expressing more CMK-1 in the cytoplasm (C > N), the nucleus (C < N), or similar levels in both compartments (C = N). The genotypes analyzed were as follows: cmk-1(ok287);cmk-1p::cmk-1(WT)::GFP, cmk-1(ok287);cmk-1p::cmk-1(WT)::NES::GFP, and cmk-1(ok287);cmk-1p::cmk-1(WT)::GFP::NLS. Each bar represents between 14 and 37 neurons. D, Behavior of cmk-1(ok287) mutants expressing CMK-1 fused to an NES or an NLS. The genotypes analyzed were as follows: cmk-1(ok287), cmk-1(ok287);cmk-1p::cmk-1(WT)::NES::GFP, and cmk-1(ok287);cmk-1p::cmk-1(WT)::GFP::NLS. Each circle is the result of a single assay and bars indicate mean ± SD. Data were pooled from at least two independent experiments, where each experiment consisted of three to six plates per condition. A two-way ANOVA across all genotypes and conditions revealed a significant interaction between genotype and condition (F(3,124) = 29.21, p < 0.0001). E, A gain-of-function allele of cmk-1 behaves like wild-type. Each circle represents the results of a single assay and bars indicate mean ± SD. Data were pooled from least two independent experiments, where each experiment consisted of three to six plates per condition and was blinded to genotype. A two-way ANOVA revealed a nonsignificant interaction between genotype and condition (F(1,20) = 0.6954, p = 0.4142). The schematic (bottom) shows the protein predicted to be encoded by cmk-1(pg58) and is adapted from (Schild et al., 2014). Sidak's posthoc tests were used to assess the effect of conditioning and to compare genotypes in panels A, D and E. The results are indicated by asterisks: *** and **** denotes p < 0.001 and p < 0.0001 and ns denotes not significant.
Data were tested for normality using the D'Agostino and Pearson omnibus normality test. Data passing this test were compared for differences using two-way ANOVA followed by Sidak's post hoc test. These analyses were performed using Prism 6/7 (GraphPad). Data that did not pass normality were compared using the Wilcoxon rank sum test and adjusted for multiple hypothesis testing using the Benjamini–Hochberg method using R Studio. The outcomes of these statistical analyses are given in each figure legend.
Results
As a first step in our investigation of salt-aversive learning in C. elegans, we determined the relationship between conditioning time and the conversion of positive salt chemotaxis (attraction) to negative salt chemotaxis (aversion). We conditioned animals in buffer with or without salt in the absence of bacterial food and tested their ability to perform chemotaxis (Fig. 1A). Under initial conditions, the chemotaxis index was close to +1, which would indicate perfect attraction (see Materials and Methods). As shown in Figure 1B, animals converted attraction to aversion over a few hours, and the conversion followed an approximately exponential time course. This time course was fit by a single exponential function (time constant, τ = 21.7 min). Control animals, conditioned without salt and in the absence of bacterial food, continued to perform positive chemotaxis even after 10 h of conditioning. This analysis reveals that conditioning animals for 180 min (3 h) was sufficient to saturate the aversion response. Thus, we conditioned animals for 180 min in the absence of bacterial food without salt (0 mm) or with 20 mm salt for all subsequent experiments, unless otherwise specified.
CMK-1 is essential for salt-learning behavior in C. elegans
Protein kinases often transduce extracellular signals into cellular responses. To understand whether and how kinases contribute to salt-aversive learning, we compared the learning ability of wild-type worms to that of worms carrying mutations in several protein kinases known to be involved in relaying key environmental stimuli: AMP kinase (AMPK), protein kinase C (PKC), and CaM kinases (CaMKI and II). Loss-of-function mutants in the worm AMPK [aak-2(ok524)], PKC [pkc-1(ok563), pkc-2(ok328)], and CaMKII genes [unc-43(gk452) and unc-43(sa200)], behaved like wild-type worms, suggesting that these genes are dispensable for both salt seeking and for salt-aversive learning (Fig. 1C). By contrast, cmk-1 loss-of-function mutants, cmk-1(ok287) and cmk-1(oy21), performed positive chemotaxis regardless of whether they were conditioned with or without salt. The defect in salt-aversive learning is due to the loss of cmk-1 function because re-expressing wild-type CMK-1 protein under its endogenous promoter was sufficient to restore salt-aversive learning to cmk-1(ok287) mutants (Fig. 1D). The defect does not appear to be due to a change in the conditioning time required for learning, however, as cmk-1 mutants failed to convert salt attraction into aversion even after 10 h of conditioning. Indeed, the chemotaxis index of cmk-1 worms conditioned in the presence and absence of 20 mm NaCl for 10 h was 0.59 ± 0.21 (mean ± SD, n = 6) and 0.51 ± 0.07 (mean ± SD, n = 6), respectively. The failure of cmk-1 mutants to convert salt attraction into salt avoidance was not due to general defects in salt-sensing or in locomotion since naive cmk-1 mutants performed like wild-type animals (Fig. 1B) and had average locomotion speeds similar to wild-type when assayed on a sterile agar surface: 90 ± 48 μm/s (mean ± SD, n = 37) and 115 ± 55 μm/s (mean ± SD, n = 37) for cmk-1 and wild-type, respectively. These speeds fall within the range of previously reported values (Ramot et al., 2008). Collectively, these results indicate that CaMKI, but not CaMKII or other kinases tested in this study, is essential for salt-aversive learning but not for salt chemotaxis per se.
Activation of CaMKI by its upstream kinase and CaMKI kinase activity are necessary for salt-aversive learning
CMK-1 is a substrate of CaM kinase kinase (CaMKK; CKK-1 in worms) via phosphorylation of threonine 179 (Eto et al., 1999; Wayman et al., 2008) and ckk-1 is coexpressed with cmk-1 in many neurons (Kimura et al., 2002). To determine whether or not phosphorylation of CMK-1 was needed for salt-aversive learning, we analyzed ckk-1 mutants and found that these mutants also exhibit defects in salt-aversive learning (Fig. 2A). We then tested the learning behavior of cmk-1(ok287) mutants overexpressing a form of CMK-1 harboring a mutation ablating the CKK-1 phosphorylation site (T179A; Schild et al., 2014). As expected if phosphorylation of CMK-1 by CKK-1 were critical for salt-aversive learning, transgenic animals expressing CMK-1(T179A) were unable to convert salt attraction into aversion (Fig. 2B). These results are consistent with a model in which ckk-1 regulates salt-aversive learning upstream of cmk-1 through phosphorylation of the CMK-1 protein, but do not exclude alternative models in which CKK-1 and CMK-1 act in separate pathways.
Figure 2.
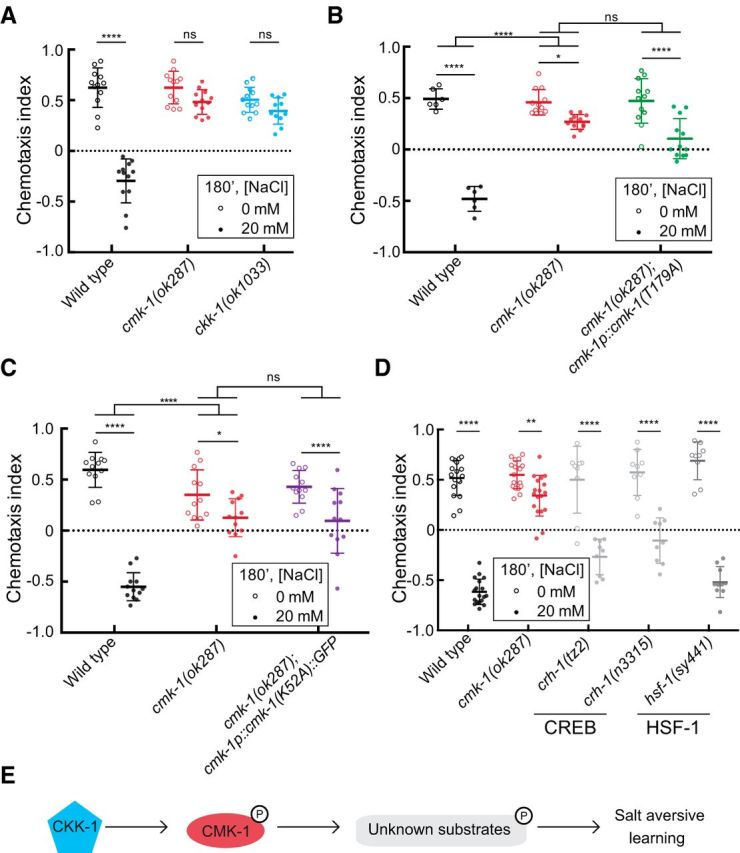
Salt-aversive learning requires CaMKK, CKK-1, and CMK-1 kinase activity. A, CKK-1 is needed for salt-aversive learning. A two-way ANOVA revealed a significant interaction between genotype and condition (F(2,65) = 47.37, p < 0.0001) and Sidak's post hoc test was used to test the effect of conditioning. B, A CMK-1 mutant that cannot be phosphorylated fails to restore salt-aversive learning to cmk-1 mutants. A two-way ANOVA across all genotypes and conditions revealed a significant interaction between genotype and condition (F(2,54) = 26.17, p < 0.0001) and Sidak's post hoc test revealed a significant effect of conditioning for all genotypes. A two-way ANOVA between wild-type and cmk-1(ok287) revealed a significant interaction between genotype and condition (F(1,32) = 112, p < 0.0001) and a two-way ANOVA between cmk-1(ok287) and cmk-1(ok287);cmk-1p::cmk-1(T179A) revealed a nonsignificant interaction between genotype and condition (F(1,44) = 3.521, p = 0.0672). C, A kinase-defective CMK-1(K52A) mutant protein cannot restore salt-aversive learning to cmk-1(ok287) mutants. A two-way ANOVA across all genotypes and conditions revealed a significant interaction between genotype and condition (F(2,66) = 33.96, p < 0.0001) and a Sidak's post hoc test revealed a significant effect of conditioning for all genotypes. A two-way ANOVA between wild-type and cmk-1(ok287) revealed a significant interaction between genotype and condition (F(1,44) = 71.16, p < 0.0001) and a two-way ANOVA between cmk-1(ok287) and cmk-1(ok287);cmk-1p::cmk-1(K52A)::GFP revealed a nonsignificant interaction between genotype and condition (F(1,44) = 0.6384, p = 0.4286). D, Behavior of known CMK-1 substrates. A two-way ANOVA revealed a significant interaction between genotype and condition (F(4,117) = 34.05, p < 0.0001). Sidak's post hoc test was used to assess the effect of conditioning for each genotype. For each panel in this figure, data were pooled from at least two independent experiments, consisting of three to six assays/condition performed blind to genotype. Animals of the indicated genotypes were conditioned for 180 min in either 0 or 20 mm NaCl and tested in a salt chemotaxis assay. Each symbol represents the results of a single assay; open and closed symbols indicate animals conditioned in 0 and 20 mm NaCl, respectively, and bars indicate mean ± SD of all assays conducted under the indicated condition and genotype. E, A schematic model, where CMK-1 is activated via phosphorylation by its upstream kinase CKK-1, and subsequently activates unknown downstream substrates to regulate salt-aversive learning. Sidak's posthoc tests were used to assess the effect of conditioning in panels A–D. The results are indicated by asterisks: *, **, and **** denote p < 0.1, p < 0.01, and p < 0.0001 and ns denotes not significant.
To determine whether the kinase activity of CMK-1 is needed for salt-aversive learning, we tested a strain overexpressing CMK-1(K52A), a mutant isoform that cannot function as a kinase, but lacking endogenous CMK-1 (Schild et al., 2014). We found that transgenic worms expressing the kinase-dead CMK-1 isoform had intact salt attraction behavior, but had defects in salt-aversive learning like cmk-1(ok287) mutants (Fig. 2C). This result suggests that CMK-1 regulates salt-aversive learning behavior through its function as a kinase.
CaM kinases are known to phosphorylate transcription factors such as CREB and HSF-1, thereby regulating activity-dependent changes in gene expression (Sheng et al., 1991; Eto et al., 1999; Holmberg et al., 2001; Wayman et al., 2008). However, loss-of-function mutants in CREB, crh-1, or HSF-1, hsf-1, responded to salt conditioning like wild-type animals (Fig. 2D). Thus, salt-aversive learning is independent of CREB and HSF-1. Similar results were observed in some (Satterlee et al., 2004; Yu et al., 2014; Kobayashi et al., 2016), but not all (Moss et al., 2016), forms of cmk-1-dependent plasticity studied to date. Collectively, these data implicate a pathway whereby CMK-1 is activated via phosphorylation by CKK-1 and subsequently phosphorylates unknown substrates responsible for salt-aversive learning (Fig. 2E).
CMK-1 acts in ASE neurons to regulate salt learning
Because CMK-1 is expressed broadly throughout the worm nervous system (Kimura et al., 2002; Satterlee et al., 2004) and proper chemotaxis behavior depends not only on salt-sensing, but also on neural circuits linking sensory events to behavior, we sought to determine the neuronal locus of cmk-1 action vis-à-vis salt-aversive learning. Given that prior work has shown that CMK-1 acts in primary sensory neurons to regulate behavioral (Schild et al., 2014; Yu et al., 2014) and developmental (Neal et al., 2015) plasticity, we focused on the primary salt-sensing neurons, ASER and ASEL. First, we verified that CMK-1 is expressed in the ASE neurons by coexpressing CMK-1::GFP with a known ASE marker (Fig. 3A). Next, we generated and tested transgenic lines that could restore wild-type CMK-1 protein to selected neurons in a cmk-1-null mutant background. We found that expressing wild-type CMK-1 protein under the control of the ASE-specific flp-6 promoter was sufficient to restore salt-aversive learning to cmk-1(ok287) mutants. The ability to rescue salt-aversive learning appears to be specific to flp-6p, because expressing CMK-1 under the control of the mechanosensory neuron-specific mec-3p or the AWC chemosensory-neuron specific ceh-36Dp promoters failed to rescue wild-type behavior (Fig. 3B). These findings indicate that not only is CMK-1 expressed in the ASE neurons, but also that CMK-1 functions in a cell-autonomous manner in these sensory neurons to control salt-aversive learning behavior.
Figure 3.
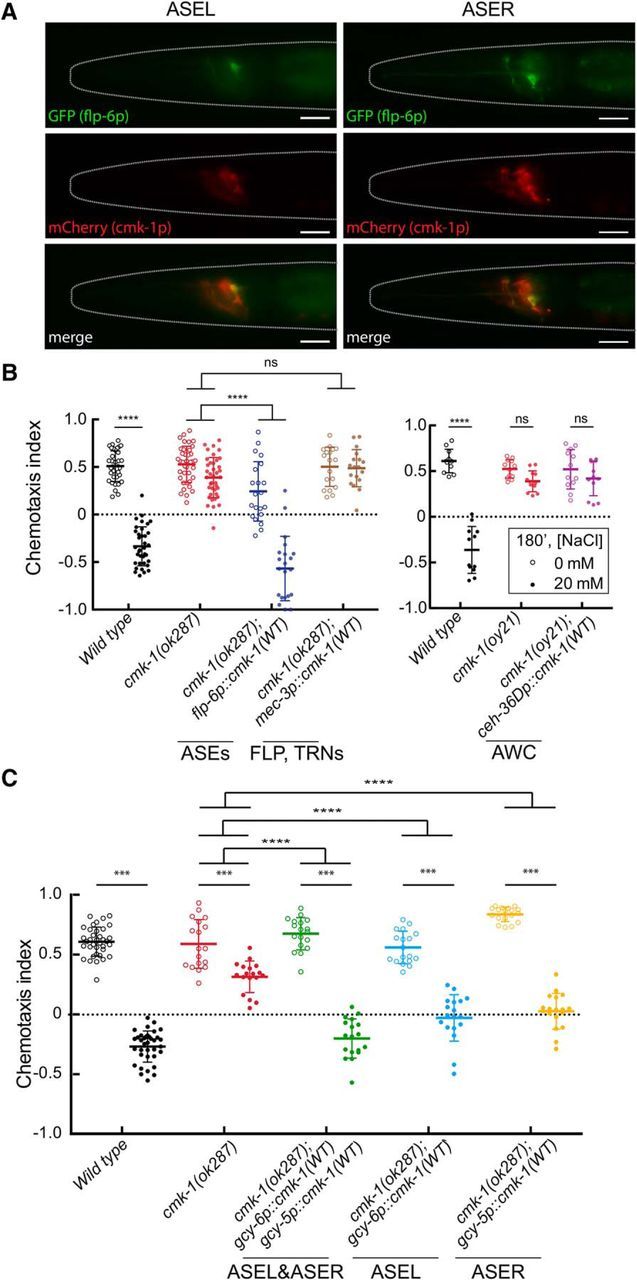
CMK-1 acts in ASE neurons to regulate learning. A, CMK-1 is expressed in ASE sensory neurons. GFP expressed under the flp-6 promoter (known to drive expression in ASE neurons) colocalizes with mCherry expressed under the cmk-1 promoter in both ASE neurons of wild-type worms. Shown are the ASEL (left) and ASER (right) neurons from the same worm imaged using epifluorescence microscopy. Worms are oriented anterior side to the left and dorsal side to top of the image. Scale bar, 20 μm. Similar results were observed in four (of 4) independent transgenic lines. B, Behavior of cmk-1(ok287) (left) and cmk-1(oy21) (right) mutants expressing wild-type CMK-1 under neuron-specific promoters. Each circle represents the results of a single assay and bars indicate mean ± SD; open and closed circles indicate conditioning in the absence and presence of 20 mm NaCl, respectively. Data were pooled from two independent experiments, where each experiment consisted of three to six plates per condition. Left, A two-way ANOVA across all genotypes and conditions revealed a significant interaction between genotype and condition (F(3,220) = 51, p < 0.0001). A two-way ANOVA between cmk-1(ok287) and cmk-1(ok287);flp-6p::cmk-1(WT)::GFP revealed a significant interaction between genotype and condition (F(1,116) = 48.67, p < 0.0001). Similar results were observed in animals carrying a second, independent cmk-1(ok287); flp-6p::cmk-1(WT)::GFP transgene, which had an average chemotaxis index of −0.64 ± 0.12 (mean ± SD, n = 6) and 0.28 ± 0.45 (mean ± SD, n = 6) with and without conditioning in 20 mm NaCl, respectively. A two-way ANOVA between cmk-1(ok287) and cmk-1(ok287);mec-3p::cmk-1(WT)::GFP revealed a nonsignificant interaction between genotype and condition (F(1,110) = 2.318, p = 0.1308). Right, A two-way ANOVA revealed a significant interaction between genotype and condition (F(2,65) = 45.12, p < 0.0001). C, Behavior of cmk-1(ok287) mutants expressing wild-type CMK-1 under ASEL- and ASER-specific promoters. Each circle represents the results of a single assay and bars indicate mean ± SD; open and closed circles indicate conditioning in the absence and presence of 20 mm NaCl, respectively. For clarity, data were pooled from three independent experiments with the following strains: N2, DAG142, DAG712, DAG716, and DAG721, where each experiment consisted of 6 plates per condition. A two-way ANOVA across all genotypes and conditions revealed a significant interaction between genotype and condition (F(4,170) = 29.5, p < 0.0001). Data for independent ASEL, ASER, and ASEL&ASER rescue lines are shown in Figure 3-1. Sidak's posthoc tests were used to assess the effect of conditioning and to compare genotypes in panels B and C. The results are indicated by asterisks: *** and **** denote p < 0.001 and p < 0.0001, respectively and ns denotes not significant.
Behavior of cmk-1(ok287) mutants expressing wild-type CMK-1 under ASEL- and ASER-specific promoters (biological replicate of assays shown in Figure 3C). Each circle represents the results of a single assay and bars indicate mean ± SD; open and closed circles indicate conditioning in the absence and presence of 20mM NaCl, respectively. Data were pooled from 3 independent experiments with the following strains: N2, DAG142, DAG713, DAG717, DAG720, where each experiment consisted of 6 plates per condition. A two-way ANOVA revealed a significant interaction between genotype and conditioning [F(4, 170) = 29.94, p < 0.0001] and a Sidak’s posthoc test revealed a significant effect of conditioning for all genotypes. Download Figure 3-1, EPS file (708.9KB, eps)
Having determined that CMK-1 acts in the two ASE neurons, we next tested whether or not expressing CMK-1 in only the ASEL or ASER neuron might be sufficient to restore salt-avoidance behavior to cmk-1(ok287)-null mutants. To achieve this goal, we used the ASEL-specific gcy-6 promoter and the ASER-specific gcy-5 promoter (Yu et al., 1997) to drive expression of wild-type CMK-1. In two independent transgenic lines, we found that expressing wild-type CMK-1 in either the ASEL or the ASER neuron was sufficient to partially restore salt-avoidance behavior (Fig. 3C and Fig. 3-1). Expressing CMK-1 from both the gcy-6 and gcy-5 promoters had a similar effect. These findings reinforce the idea that CMK-1 functions in the ASE neurons to enable salt-aversive learning, but do not implicate either of the two ASE neurons as a dominant factor in this process.
We then examined how the CMK-1 protein responded to salt stimulation in these neurons. Previous studies reported nuclear-cytoplasmic shuttling of CMK-1 in primary sensory neurons (Schild et al., 2014; Yu et al., 2014; Neal et al., 2015) and in concert with modulation of glutamate receptor expression (Moss et al., 2016). We sought to observe the subcellular localization of CMK-1 in the cell bodies of the two ASE neurons by imaging a fusion between CMK-1 and GFP. The fusion protein is sufficient to restore wild-type salt-aversive learning to cmk-1(ok287) mutants (Fig. 1D), indicating the fusion protein functions like the wild-type protein under these conditions. Before conditioning, the CMK-1 protein localizes primarily to the cytoplasm of both ASE neurons (Fig. 4A). We did not detect any changes in the nuclear-cytoplasmic ratio in the ASEL or ASER cell body during conditioning for between 10 and 180 min in 20 mm NaCl (Fig. 4A). Although conditioning in salt-free buffer transiently increased the localization of CMK-1 throughout cell body in ASER neurons, the significance of this finding for salt-aversive learning is unclear because learning does not occur under these conditions. We next expressed forms of CMK-1 targeted to the nucleus or the cytoplasm, thanks to the adjunction of ectopic nuclear localization signal (NLS) or nuclear export signal (NES), respectively (Fig. 4B,C). In both cases, the learning defect of cmk-1(ok287) mutants was rescued (Fig. 4D). Moreover, a mutant that encodes a form of CMK-1 with a deleted predicted NES (and a portion of the auto-inhibitory domain), cmk-1(pg58), exhibited normal salt learning (Fig. 4E). Collectively, these findings suggest that neither the precise localization of CMK-1 nor nuclear-cytoplasmic shuttling in the ASE cell bodies are essential for salt-aversive learning.
Calcium signals in sensory dendrites, cell bodies, and axons of ASE neurons
The two ASE neurons are bilaterally-symmetric, bipolar chemosensory neurons whose cell bodies and dendrites lie on the left and right sides of the animal and whose axons overlap in the nerve ring (Fig. 5A). Despite their similar appearance, the two ASE neurons are functionally distinct: ASEL detects salt increases and ASER is activated by salt decreases (Hobert, 2014). Ample evidence supports this general description of chemosensory responses in the ASE cell bodies (Tomioka et al., 2006; Suzuki et al., 2008; Ortiz et al., 2009; Thiele et al., 2009; Oda et al., 2011; Kunitomo et al., 2013; Luo et al., 2014; Rabinowitch et al., 2014), but less is known about how the calcium signals generated in ASE dendrites, cell bodies, and axons compared to one another. We sought to fill this gap in knowledge by recording salt-evoked calcium signals in the sensory endings (dendrites), cell bodies, and axons of the wild-type ASE neurons. To reach this goal, we took advantage of mosaic expression of the yxEx738 [flp-6p::GCaMP3; ttx-1p::GFP; unc-122p::GFP] transgene and recorded responses to pulses of sodium chloride salt in the ASEL dendrites, cell bodies, and axons independently of the ASER neuron and vice versa. Figure 5B shows raster plots of salt-evoked calcium signals measured in all three compartments (sensory dendrite, cell body, axon) of the ASEL and ASER neurons. Each line in the raster plot represents calcium signals as a function of time in the indicated compartment.
Salt pulses produced calcium transients synchronized to the up-step in ASEL and the down-step in ASER in all three subcellular domains (Fig. 5C). The inset shows ASER calcium transients normalized to the fluorescence recorded just before the down-step. Although dendritic endings and axons lie on opposite sides of the cell body and are at least 100 μm apart from one another, the time course of their calcium signals were indistinguishable from one another. Responses developed more slowly and lasted longer in cell bodies than in dendrites or axons, a result that could be explained by the cell body's larger volume acting as a low-pass filter for calcium signals. These observations are compatible with the electrically compact nature of the ASE neurons (Goodman et al., 1998) and a lack of distinct, subcellular calcium domains in these cells (Kuramochi and Doi, 2017).
We averaged the mean responses of ASEL and ASER axons to generate a prediction for the waveform expected in animals expressing a calcium indicator in both axons (Fig. 5D). These reconstructed traces recapitulate the phasic responses we detected in animals expressing in both ASE axons (see Fig. 7D). With this analysis, we confirm that ASEL functions as an ON cell that is sensitive primarily to salt up-steps whereas ASER functions as an OFF cell that responds primarily to salt down-steps. We establish that signals recorded in sensory dendrites, but not cell bodies, mirror the stimulus-induced calcium waveforms present in axons. We further propose that signals recorded from the nerve ring of animals expressing calcium indicator in both neurons can be interpreted as the average of the axonal signals generated independently by ASEL and ASER.
Salt conditioning alters salt-evoked calcium signals in wild-type and cmk-1 mutant ASEL and ASER cell bodies
Calcium signals recorded in the cell bodies of specific neurons are a common proxy for neuronal activity and have been used to understand how acclimation affects sensory signaling in several C. elegans behaviors (Clark et al., 2006; Biron et al., 2008; Yu et al., 2014). Following this approach, we analyzed chemosensory responses in ASE cell bodies of wild-type and cmk-1 mutants conditioned in the absence or presence of salt (Fig. 6). In wild-type worms, salt conditioning decreased ON responses in ASEL cell bodies (Fig. 6A,B,E, left). This effect of salt conditioning on wild-type ASEL chemosensory responses was also evident in recordings of the response to a 40 s decrease in salt from 20 to 0 mm (Fig. 6-1A). Consistent with its function as an OFF cell, salt application had little effect on calcium levels and down-steps evoked an increase in calcium in ASER cell bodies (Fig. 6A,B,E, right and Fig. 6-1B). This OFF response was also evident when we normalized to fluorescence levels recorded just before the down-step (Fig. 6B, inset). Following salt conditioning, however, calcium was suppressed during salt application and OFF responses were maintained in the ASER cell body (Fig. 6A,B). If we controlled for the salt-induced decrease in calcium signals by normalizing to fluorescence values at the end of the salt pulse, it appears that salt conditioning increases the amplitude of OFF responses (Fig. 6B, inset). These observations agree with prior studies (Oda et al., 2011) and indicate that salt conditioning modulates the chemosensory responses in both ASE cell bodies.
The effect of salt conditioning upon calcium signals evoked by a transient decrease in external salt in ASER and ASEL cell bodies. A. Average calcium signals recorded in ASER cell bodies exposed to a decrease in salt concentration from 20 mM to 0 mM. B. Average calcium signals recorded in ASER cell bodies exposed to a decrease in salt concentration from 20 mM to 0 mM. Solid and dashed lines show the average (±s.e.m.) recorded from animals conditioned in 0 mM and 20 mM NaCl. Download Figure 6-1, EPS file (1.4MB, eps)
Although cmk-1(ok287) mutants fail to develop salt avoidance (Fig. 1C) and this deficit is rescued by expression of wild-type CMK-1 in the ASE neurons (Fig. 3B,C), salt-evoked calcium signals in cmk-1(ok287) mutant ASEL and ASER cell bodies were similar to those observed in wild-type (Fig. 6E). As found in wild-type animals, salt conditioning decreased ON responses to salt pulses in the ASEL neurons in cmk-1 mutants (Fig. 6C,D, left) and had only minor effects on OFF responses in the ASER neurons in cmk-1 mutants (Fig. 6C,D, right). A subtle effect of loss of cmk-1 on ASER signals is apparent in our data: salt application decreases ASER GCaMP3 signals relative to prestimulus levels in animals conditioned in the absence and presence of 20 mm salt (Fig. 6C,D, right). The net result of this effect is a smaller effect of salt conditioning on ASER OFF responses and is evident in plots controlling for the salt-induced decrease (Fig. 6D, inset).
Peak ON responses in the ASEL neurons decrease in both wild-type and cmk-1(ok287) ASEL cell bodies upon conditioning (Fig. 6E, left). As noted above, salt conditioning had minor effects on peak OFF responses in wild-type and cmk-1(ok287) ASER cell bodies (Fig. 6E, right), unless the stimulus-induced decrease in GCaMP signaling was considered. In the latter situation, salt conditioning increases OFF responses in both wild-type and cmk-1 mutant ASER cell bodies and this effect is weaker in cmk-1 mutants. These findings indicate that ASEL and ASER neurons retain their function as ON and OFF cells, respectively, and that the modulation of ASE chemosensory responses following salt conditioning is weakened by loss of cmk-1 function.
The effect of salt conditioning on chemosensory calcium signals in ASEL and ASER dendrites and axons is regulated by cmk-1
Given that the response dynamics in ASE cell bodies are slower than signals observed at the tip of the ASE dendrites and along their axons (Fig. 5), we also determined the effect of salt conditioning and loss of cmk-1 function on calcium signals recorded in these subcellular domains. In particular, we reasoned that a closer look at the axons that harbor all of ASE's synaptic connections to the nervous system might provide additional insight into how loss of cmk-1 function in ASE neurons contributes to the deficit in salt-aversive learning. Consistent with current and prior findings in the cell bodies, ASEL sensory endings detect up-shifts in salt and ASER endings detect downshifts. In wild-type worms, the peak amplitude of the ON response in ASEL dendrites and the OFF response in ASER dendrites decreased after salt conditioning (Fig. 7A and Fig. 7-1B, left and center). This effect of conditioning was diminished in cmk-1 mutant ASEL dendrites (Fig. 7B and Fig. 7-1B, left and center). We observed a qualitatively similar, but less dramatic effect upon salt conditioning in cmk-1 mutant ASER dendrites. We summarized these results by comparing peak calcium values observed at the onset (ASEL) and offset (ASER) of the salt pulse (Fig. 7C). A two-way ANOVA revealed a significant effect of conditioning on the amplitude of ON responses in ASEL (F(1,40) = 22.67, p < 0.0001) and an interaction between genotype (wild-type vs cmk-1) and conditioning in ASEL (F(1,40) = 4.546, p = 0.0392). Although there was also a significant effect of conditioning on the amplitude of OFF responses in ASER(F(1,73) = 27.72, p < 0.0001), there was no detectable interaction between genotype (wild-type vs cmk-1) and conditioning. In summary, loss of cmk-1 function blunts the ability of salt conditioning to enhance ON responses in ASEL dendrites but has little or no effect on OFF responses in ASER dendrites. The subcellular locus of the defect in signaling remains ambiguous, however, because the ASE neurons are isopotential (Goodman et al., 1998) and appear to lack calcium subdomains (Kuramochi and Doi, 2017).
Individual traces shown as averages in Figure 7. A. Raster plots of chemosensory calcium signals in the ASEL and ASER dendrites in wild type worms conditioned in 0 mM or 20 mM NaCl and no food and exposed to a 40 second pulse of 20mM NaCl. Each horizontal line corresponds to a recording from a single dendrite tip; the number of recordings is shown in parentheses. Color indicates the change in fluorescence relative to pre-stimulus levels: red indicates an increase and blue indicates a decrease. B. Raster plots of chemosensory calcium signals in the ASEL and ASER dendrites in cmk-1(ok287) mutants conditioned in 0 mM or 20 mM NaCl and no food and exposed to a 40 second pulse of 20 mM NaCl. Each horizontal line corresponds to a recording from a single dendrite tip; the number of recordings is shown in parentheses. Color indicates the change in fluorescence relative to pre-stimulus levels: red indicates an increase and blue indicates a decrease. The recordings shown are the source data for average traces shown in Figure 7B. Download Figure 7-1, EPS file (1.5MB, eps)
Finally, we analyzed how conditioning in 20 mm NaCl and loss of cmk-1 function affected responses in the overlapping ASE axons (Fig. 7D–F), which are responsible for transmitting changes in sensory neuron activity to postsynaptic neurons. Conditioning wild-type animals in 20 mm NaCl had two main effects on axonal calcium signals in ASE neurons. First, responses to salt up-steps are transformed from a transient increase in axonal calcium to a sustained decrease in calcium (Fig. 7D and Fig. 7-1A, right). Second, OFF responses to salt down-steps decreased slightly (Fig. 7D and Fig. 7-1A, right). Interestingly, in cmk-1 mutant axons, conditioning in 20 mm NaCl failed to generate this sustained decrease in calcium in response to salt up-steps (Fig. 7E and Fig. 7-1B, right). In addition, cmk-1 mutant ASE axons failed to generate wild-type-like conditioned responses to salt down-steps (Fig. 7E,F and Fig. 7-1B, right). Thus, loss of cmk-1 does not abolish salt-evoked calcium responses in ASE axons upon salt conditioning, but alters the nature of the signal delivered to postsynaptic neurons.
We considered these observations regarding the influence regarding cmk-1 on axonal signals in the context of our earlier findings that: (1) chemosensory signals in overlapping axons are consistent with a summation of the signals generated by the ASEL and ASER axons (Fig. 5D) and (2) the chemosensory waveforms recorded in single ASE dendrites are virtually indistinguishable from those recorded in axons (Fig. 5C). Assuming these relationships hold for cmk-1 mutant ASE neurons, we fit chemosensory waveforms measured in wild-type and cmk-1 ASER and ASEL dendrites (see Materials and Methods), computed a weighted sum of these waveforms and compared the resulting waveforms to the measured axonal signals. As shown in Figure 7D (cyan curves), this simple strategy was sufficient to recapitulate the effects of conditioning by 20 mm NaCl on chemosensory signaling in wild-type animals, reaffirming that conditioning depresses calcium levels during salt pulses. This approach could reproduce the blunted responses to salt up-steps in cmk-1 mutants (though it was less successful at reproducing the responses to salt down-steps; Fig. 7E, purple curves).
The fact that cmk-1 mutants perform chemotaxis like wild-type animals when conditioned with 0 mm NaCl (Fig. 1C) is compatible with our finding that cmk-1(ok287) mutant ASE neurons retain stimulus-evoked calcium signals under these conditions (Figs. 6, 7). The response of cmk-1(ok287) mutant ASE neurons diverged from wild-type upon conditioning in 20 mm salt, however, and this effect was more pronounced in chemosensory responses recorded in dendrites and axons. The effects on ASE cell bodies were less pronounced, consistent with our inference that chemosensory signals in the ASE cell bodies are filtered compared with these detected in dendrites and axons. Collectively, our data show that although cmk-1 loss does not completely abolish the modulation of chemosensory responses by salt conditioning, but does blunt the effect of salt conditioning, especially in the ASEL dendrites and axons.
Discussion
CMK-1, the C. elegans ortholog of mammalian CaMKI/IV, enables behavioral plasticity related to thermal acclimation and developmental plasticity governed by chemosensation (Satterlee et al., 2004; Schild et al., 2014; Yu et al., 2014; Neal et al., 2015). Here, we demonstrate that CMK-1 is also critical for salt-aversive learning, a process that allows animals to balance the need for salt with the potentially damaging effects of this essential inorganic nutrient. We show that salt-aversive learning depends on CMK-1 expression in the ASE chemosensory neurons, uses an upstream activation pathway involving CKK-1, and requires the kinase activity of CMK-1.
In agreement with a number of prior studies (Suzuki et al., 2008; Ortiz et al., 2009; Thiele et al., 2009; Oda et al., 2011; Rabinowitch et al., 2014), we find that ASEL functions as an ON cell and ASER functions as an OFF cell and that salt conditioning appears to blunt salt-evoked signals. We analyzed stimulus-evoked calcium signals in the dendrites, cell bodies, and axons of both ASER and ASEL neurons, extending general knowledge of chemosensory signaling by these neurons. We found that signals in dendrites and axons were indistinguishable from one another and that somatic signals resembled a low-pass filtered version of these signals. These findings are consistent with isopotential electrical signaling by ASE (Goodman et al., 1998) and have implications for relying on somatic calcium signals as a proxy for global activity in the C. elegans nervous system.
Salt-evoked calcium signals in cmk-1 mutant ASE dendrites and axons are significantly less sensitive to salt conditioning than they are in their wild-type counterparts, although cmk-1 mutant ASE neurons continue to generate both ON and OFF chemosensory responses. Additional work will be needed to distinguish between the effects of cmk-1-dependent pathway on chemosensory transduction in dendrites or on presynaptic signaling in axons. This ambiguity follows from prior work demonstrating that the ASE neurons are isopotential (Goodman et al., 1998) and the finding that signaling dynamics are similar in wild-type dendrites and axons. Building on this finding, we hypothesized that signal dynamics in the overlapping axons could be predicted from a weighted sum of the signals in ASEL and ASER dendrites. This simple model recapitulated the principal features of the phasic responses to salt pulses we observed in wild-type ASE axons, but was not adequate to capture the effect of salt conditioning in cmk-1 mutant axons (Fig. 7E). This mismatch favors a model in which CMK-1 affects the sensitivity of transmitter release by ASE axons to salt conditioning, but does not exclude influences on chemosensory transduction or other pathways.
CMK-1 is expressed globally in the C. elegans nervous system (Kimura et al., 2002; Satterlee et al., 2004), but has been shown to act locally in sensory neurons to support plasticity (Schild et al., 2014; Yu et al., 2014; Neal et al., 2015). Its kinase activity is required for plasticity, but CREB, an established CaM kinase substrate, is not essential for the forms of behavioral and developmental plasticity analyzed thus far. At present, we cannot exclude the possibility that CREB acts redundantly with other CMK-1 substrates, including Raf/MAPK as shown for thermosensory adaptation (Kobayashi et al., 2016). Important next steps for understanding the influence of CMK-1 on learning will be to discover additional proteins phosphorylated by CMK-1 and to analyze their contribution to behavioral plasticity.
In conclusion, we have characterized the ability of cmk-1 to profoundly affect learning in C. elegans. This, along with the sufficiency of this gene in a single pair of neurons to rescue this behavior, illustrates a relatively simple circuit in this animal to adapt to the natural world. The fact that worm CMK-1 is thought to serve functions performed by two different proteins in mammals, CaMKII and CaMKI/CaMKIV (Cohen et al., 2015), speaks further to the efficiency of this system. Evolutionary advantages of such a model include the ability to perform relatively complex behaviors with very few neurons and molecular players. Potential risks, however, may include insufficient compensatory mechanisms to rescue the system when damaged. The ability to learn to avoid areas of high salt upon starvation likely evolved as a way for organisms to minimize energy exploring barren areas and instead seek food-rich habitats.
Footnotes
This work was supported by a National Science Foundation Graduate Research Fellowship (J.P.L.), NIH Grant NS047715 (M.B.G.), NIH Grant AG031198 (A.B.), and Swiss National Science Foundation Grants BSSGI0_155764 and PP00P3_150681 (D.A.G.). We thank C. Murphy (Princeton), Y. Zhang (Harvard), and the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), for worm strains; S. Chalasani (Scripps) for technical advice regarding microfluidics setup; A. Takeishi (Brandeis) for advice about analyzing CMK-1 subcellular localization; Z. Liao, V. Lauper, and L. Schild for superb technical support; J. Kubanek for help with worm tracking; and L. Booth, B. Dulken, S. Han, C.-K. Hu, K. Papsdorf, and P. Singh for critical reading of the paper.
The authors declare no competing financial interests.
References
- Adachi T, Kunitomo H, Tomioka M, Ohno H, Okochi Y, Mori I, Iino Y (2010) Reversal of salt preference is directed by the insulin/PI3K and Gq/PKC signaling in Caenorhabditis elegans. Genetics 186:1309–1319. 10.1534/genetics.110.119768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardiel EL, Rankin CH (2010) An elegant mind: learning and memory in Caenorhabditis elegans. Learn Mem 17:191–201. 10.1101/lm.960510 [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Horvitz HR (1991) Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7:729–742. 10.1016/0896-6273(91)90276-6 [DOI] [PubMed] [Google Scholar]
- Biron D, Wasserman S, Thomas JH, Samuel AD, Sengupta P (2008) An olfactory neuron responds stochastically to temperature and modulates Caenorhabditis elegans thermotactic behavior. Proc Natl Acad Sci U S A 105:11002–11007. 10.1073/pnas.0805004105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, Bargmann CI (2007) Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature 450:63–70. 10.1038/nature06292 [DOI] [PubMed] [Google Scholar]
- Choe KP. (2013) Physiological and molecular mechanisms of salt and water homeostasis in the nematode Caenorhabditis elegans. Am J Physiol Regul Integr Comp Physiol 305:R175–R186. 10.1152/ajpregu.00109.2013 [DOI] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI (2007) Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods 4:727–731. 10.1038/nmeth1075 [DOI] [PubMed] [Google Scholar]
- Clark DA, Biron D, Sengupta P, Samuel AD (2006) The AFD sensory neurons encode multiple functions underlying thermotactic behavior in Caenorhabditis elegans. J Neurosci 26:7444–7451. 10.1523/JNEUROSCI.1137-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Li B, Tsien RW, Ma H (2015) Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem Biophys Res Commun 460:88–99. 10.1016/j.bbrc.2015.02.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Takahashi N, Kimura Y, Masuho Y, Arai K, Muramatsu MA, Tokumitsu H (1999) Ca2+/calmodulin-dependent protein kinase cascade in Caenorhabditis elegans implication in transcriptional activation. J Biol Chem 274:22556–22562. 10.1074/jbc.274.32.22556 [DOI] [PubMed] [Google Scholar]
- Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME (2006) Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet 43:289–294. 10.1136/jmg.2005.036319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TC. (2006) Transformation and microinjection. In: The C. elegans Research Community (Ambros V, ed). Aurora, CO: WormBook; 10.1895/wormbook.1.108.1 [DOI] [Google Scholar]
- Fehlauer H, Nekimken AL, Kim AA, Pruitt BL, Goodman MB, Krieg M (2018) Using a microfluidics device for mechanical stimulation and high resolution imaging of C. elegans. J Vis Exp 132:e56530. 10.3791/56530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerling JC, Loewy AD (2008) Central regulation of sodium appetite. Exp Physiol 93:177–209. 10.1113/expphysiol.2007.039891 [DOI] [PubMed] [Google Scholar]
- Goodman MB, Hall DH, Avery L, Lockery SR (1998) Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20:763–772. 10.1016/S0896-6273(00)81014-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. (2014) Development of left/right asymmetry in the Caenorhabditis elegans nervous system: from zygote to postmitotic neuron. Genesis 52:528–543. 10.1002/dvg.22747 [DOI] [PubMed] [Google Scholar]
- Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M, Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson JE, Sistonen L (2001) Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J 20:3800–3810. 10.1093/emboj/20.14.3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Means AR (2001) Ca2+/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol 41:471–505. 10.1146/annurev.pharmtox.41.1.471 [DOI] [PubMed] [Google Scholar]
- Hostettler L, Grundy L, Käser-Pébernard S, Wicky C, Schafer WR, Glauser DA (2017) The bright fluorescent protein mNeonGreen facilitates protein expression analysis in vivo. G3 7:607–615. 10.1534/g3.116.038133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukema RK, Rademakers S, Dekkers MP, Burghoorn J, Jansen G (2006) Antagonistic sensory cues generate gustatory plasticity in Caenorhabditis elegans. EMBO J 25:312–322. 10.1038/sj.emboj.7600940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukema RK, Rademakers S, Jansen G (2008) Gustatory plasticity in C. elegans involves integration of negative cues and NaCl taste mediated by serotonin, dopamine, and glutamate. Learn Mem 15:829–836. 10.1101/lm.994408 [DOI] [PubMed] [Google Scholar]
- Hurley SW, Johnson AK (2015) The biopsychology of salt hunger and sodium deficiency. Pflugers Arch 467:445–456. 10.1007/s00424-014-1676-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino Y, Yoshida K (2009) Parallel use of two behavioral mechanisms for chemotaxis in Caenorhabditis elegans. J Neurosci 29:5370–5380. 10.1523/JNEUROSCI.3633-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda DD, Duan Y, Matsuki M, Kunitomo H, Hutter H, Hedgecock EM, Iino Y (2008) CASY-1, an ortholog of calsyntenins/alcadeins, is essential for learning in Caenorhabditis elegans. Proc Natl Acad Sci U S A 105:5260–5265. 10.1073/pnas.0711894105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Iino Y, Mohri A, Mori I, Gengyo-Ando K, Mitani S, Katsura I (2002) HEN-1, a secretory protein with an LDL receptor motif, regulates sensory integration and learning in Caenorhabditis elegans. Cell 109:639–649. 10.1016/S0092-8674(02)00748-1 [DOI] [PubMed] [Google Scholar]
- Iwata R, Oda S, Kunitomo H, Iino Y (2011) Roles for class IIA phosphatidylinositol transfer protein in neurotransmission and behavioral plasticity at the sensory neuron synapses of Caenorhabditis elegans. Proc Natl Acad Sci U S A 108:7589–7594. 10.1073/pnas.1016232108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Weinkove D, Plasterk RH (2002) The G-protein gamma subunit gpc-1 of the nematode C. elegans is involved in taste adaptation. EMBO J 21:986–994. 10.1093/emboj/21.5.986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano T, Brockie PJ, Sassa T, Fujimoto H, Kawahara Y, Iino Y, Mellem JE, Madsen DM, Hosono R, Maricq AV (2008) Memory in Caenorhabditis elegans is mediated by NMDA-type ionotropic glutamate receptors. Curr Biol 18:1010–1015. 10.1016/j.cub.2008.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AL, Ashraf JM, Corces-Zimmerman MR, Landis JN, Murphy CT (2010) Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol 8:e1000372. 10.1371/journal.pbio.1000372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Jin Y (2015) Neuronal responses to stress and injury in C. elegans. FEBS Lett 589:1644–1652. 10.1016/j.febslet.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Corcoran EE, Eto K, Gengyo-Ando K, Muramatsu MA, Kobayashi R, Freedman JH, Mitani S, Hagiwara M, Means AR, Tokumitsu H (2002) A CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep 3:962–966. 10.1093/embo-reports/kvf191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Nakano S, Amano M, Tsuboi D, Nishioka T, Ikeda S, Yokoyama G, Kaibuchi K, Mori I (2016) Single-cell memory regulates a neural circuit for sensory behavior. Cell Rep 14:11–21. 10.1016/j.celrep.2015.11.064 [DOI] [PubMed] [Google Scholar]
- Kunitomo H, Sato H, Iwata R, Satoh Y, Ohno H, Yamada K, Iino Y (2013) Concentration memory-dependent synaptic plasticity of a taste circuit regulates salt concentration chemotaxis in Caenorhabditis elegans. Nat Commun 4:2210. 10.1038/ncomms3210 [DOI] [PubMed] [Google Scholar]
- Kuramochi M, Doi M (2017) A computational model based on multi-regional calcium imaging represents the spatio-temporal dynamics in a Caenorhabditis elegans sensory neuron. PLoS One 12:e0168415. 10.1371/journal.pone.0168415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Wen Q, Ren J, Hendricks M, Gershow M, Qin Y, Greenwood J, Soucy ER, Klein M, Smith-Parker HK, Calvo AC, Colón-Ramos DA, Samuel AD, Zhang Y (2014) Dynamic encoding of perception, memory, and movement in a C. elegans chemotaxis circuit. Neuron 82:1115–1128. 10.1016/j.neuron.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki M, Kunitomo H, Iino Y (2006) Goalpha regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans. Proc Natl Acad Sci U S A 103:1112–1117. 10.1073/pnas.0506954103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P (1999) Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev Biol 215:314–331. 10.1006/dbio.1999.9470 [DOI] [PubMed] [Google Scholar]
- Moss BJ, Park L, Dahlberg CL, Juo P (2016) The CaM kinase CMK-1 mediates a negative feedback mechanism coupling the C. elegans glutamate receptor GLR-1 with its own transcription. PLoS Genet 12:e1006180. 10.1371/journal.pgen.1006180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal SJ, Takeishi A, O'Donnell MP, Park J, Hong M, Butcher RA, Kim K, Sengupta P (2015) Feeding state-dependent regulation of developmental plasticity via CaMKI and neuroendocrine signaling. eLife 4:e11547. 10.7554/eLife.11547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekimken AL, Fehlauer H, Kim AA, Manosalvas-Kjono SN, Ladpli P, Memon F, Gopisetty D, Sanchez V, Goodman MB, Pruitt BL, Krieg M (2017) Pneumatic stimulation of C. elegans mechanoreceptor neurons in a microfluidic trap. Lab Chip 17:1116–1127. 10.1039/C6LC01165A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda S, Tomioka M, Iino Y (2011) Neuronal plasticity regulated by the insulin-like signaling pathway underlies salt chemotaxis learning in Caenorhabditis elegans. J Neurophysiol 106:301–308. 10.1152/jn.01029.2010 [DOI] [PubMed] [Google Scholar]
- Ortiz CO, Faumont S, Takayama J, Ahmed HK, Goldsmith AD, Pocock R, McCormick KE, Kunimoto H, Iino Y, Lockery S, Hobert O (2009) Lateralized gustatory behavior of C. elegans is controlled by specific receptor-type guanylyl cyclases. Curr Biol 19:996–1004. 10.1016/j.cub.2009.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitch I, Chatzigeorgiou M, Zhao B, Treinin M, Schafer WR (2014) Rewiring neural circuits by the insertion of ectopic electrical synapses in transgenic C. elegans. Nat Commun 5:4442. 10.1038/ncomms5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramot D, Johnson BE, Berry TL Jr, Carnell L, Goodman MB (2008) The parallel worm tracker: a platform for measuring average speed and drug-induced paralysis in nematodes. PLoS One 3:e2208. 10.1371/journal.pone.0002208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki S, Yamamoto M, Iino Y (2001) Plasticity of chemotaxis revealed by paired presentation of a chemoattractant and starvation in the nematode Caenorhabditis elegans. J Exp Biol 204:1757–1764. [DOI] [PubMed] [Google Scholar]
- Sakai N, Iwata R, Yokoi S, Butcher RA, Clardy J, Tomioka M, Iino Y (2013) A sexually conditioned switch of chemosensory behavior in C. elegans. PLoS One 8:e68676. 10.1371/journal.pone.0068676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakashita T, Hamada N, Ikeda DD, Yanase S, Suzuki M, Ishii N, Kobayashi Y (2008) Modulatory effect of ionizing radiation on food-NaCl associative learning: the role of gamma subunit of G protein in Caenorhabditis elegans. FASEB J 22:713–720. 10.1096/fj.07-9259com [DOI] [PubMed] [Google Scholar]
- Satterlee JS, Ryu WS, Sengupta P (2004) The CMK-1 CaMKI and the TAX-4 cyclic nucleotide-gated channel regulate thermosensory neuron gene expression and function in C. elegans. Curr Biol 14:62–68. 10.1016/j.cub.2003.12.030 [DOI] [PubMed] [Google Scholar]
- Schild LC, Zbinden L, Bell HW, Yu YV, Sengupta P, Goodman MB, Glauser DA (2014) The balance between cytoplasmic and nuclear CaM kinase-1 signaling controls the operating range of noxious heat avoidance. Neuron 84:983–996. 10.1016/j.neuron.2014.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME (1991) CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252:1427–1430. 10.1126/science.1646483 [DOI] [PubMed] [Google Scholar]
- Stetak A, Hörndli F, Maricq AV, van den Heuvel S, Hajnal A (2009) Neuron-specific regulation of associative learning and memory by MAGI-1 in C. elegans. PLoS One 4:e6019. 10.1371/journal.pone.0006019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Thiele TR, Faumont S, Ezcurra M, Lockery SR, Schafer WR (2008) Functional asymmetry in Caenorhabditis elegans taste neurons and its computational role in chemotaxis. Nature 454:114–117. 10.1038/nature06927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TR, Faumont S, Lockery SR (2009) The neural network for chemotaxis to tastants in Caenorhabditis elegans is specialized for temporal differentiation. J Neurosci 29:11904–11911. 10.1523/JNEUROSCI.0594-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomioka M, Adachi T, Suzuki H, Kunitomo H, Schafer WR, Iino Y (2006) The insulin/PI 3-kinase pathway regulates salt chemotaxis learning in Caenorhabditis elegans. Neuron 51:613–625. 10.1016/j.neuron.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Vellai T, McCulloch D, Gems D, Kovács AL (2006) Effects of sex and insulin/insulin-like growth factor-1 signaling on performance in an associative learning paradigm in Caenorhabditis elegans. Genetics 174:309–316. 10.1534/genetics.106.061499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward S. (1973) Chemotaxis by the nematode Caenorhabditis elegans: identification of attractants and analysis of the response by use of mutants. Proc Natl Acad Sci U S A 70:817–821. 10.1073/pnas.70.3.817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR (2008) Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59:914–931. 10.1016/j.neuron.2008.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Avery L, Baude E, Garbers DL (1997) Guanylyl cyclase expression in specific sensory neurons: a new family of chemosensory receptors. Proc Natl Acad Sci U S A 94:3384–3387. 10.1073/pnas.94.7.3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YV, Bell HW, Glauser D, Van Hooser SD, Goodman MB, Sengupta P (2014) CaMKI-dependent regulation of sensory gene expression mediates experience-dependent plasticity in the operating range of a thermosensory neuron. Neuron 84:919–926. 10.1016/j.neuron.2014.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Behavior of cmk-1(ok287) mutants expressing wild-type CMK-1 under ASEL- and ASER-specific promoters (biological replicate of assays shown in Figure 3C). Each circle represents the results of a single assay and bars indicate mean ± SD; open and closed circles indicate conditioning in the absence and presence of 20mM NaCl, respectively. Data were pooled from 3 independent experiments with the following strains: N2, DAG142, DAG713, DAG717, DAG720, where each experiment consisted of 6 plates per condition. A two-way ANOVA revealed a significant interaction between genotype and conditioning [F(4, 170) = 29.94, p < 0.0001] and a Sidak’s posthoc test revealed a significant effect of conditioning for all genotypes. Download Figure 3-1, EPS file (708.9KB, eps)
The effect of salt conditioning upon calcium signals evoked by a transient decrease in external salt in ASER and ASEL cell bodies. A. Average calcium signals recorded in ASER cell bodies exposed to a decrease in salt concentration from 20 mM to 0 mM. B. Average calcium signals recorded in ASER cell bodies exposed to a decrease in salt concentration from 20 mM to 0 mM. Solid and dashed lines show the average (±s.e.m.) recorded from animals conditioned in 0 mM and 20 mM NaCl. Download Figure 6-1, EPS file (1.4MB, eps)
Individual traces shown as averages in Figure 7. A. Raster plots of chemosensory calcium signals in the ASEL and ASER dendrites in wild type worms conditioned in 0 mM or 20 mM NaCl and no food and exposed to a 40 second pulse of 20mM NaCl. Each horizontal line corresponds to a recording from a single dendrite tip; the number of recordings is shown in parentheses. Color indicates the change in fluorescence relative to pre-stimulus levels: red indicates an increase and blue indicates a decrease. B. Raster plots of chemosensory calcium signals in the ASEL and ASER dendrites in cmk-1(ok287) mutants conditioned in 0 mM or 20 mM NaCl and no food and exposed to a 40 second pulse of 20 mM NaCl. Each horizontal line corresponds to a recording from a single dendrite tip; the number of recordings is shown in parentheses. Color indicates the change in fluorescence relative to pre-stimulus levels: red indicates an increase and blue indicates a decrease. The recordings shown are the source data for average traces shown in Figure 7B. Download Figure 7-1, EPS file (1.5MB, eps)