

Abstract
Periodic episodes of excessive alcohol consumption (“binge drinking”) occur frequently among adolescents, and early binge drinking is associated with an increased risk of alcohol use disorders later in life. The PFC undergoes significant development during adolescence and hence may be especially susceptible to the effects of binge drinking. In humans and in animal models, adolescent alcohol exposure is known to alter PFC neuronal activity and produce deficits in PFC-dependent behaviors, such as decision making, response inhibition, and working memory. Using a voluntary intermittent access to alcohol (IA EtOH) procedure in male mice, we demonstrate that binge-level alcohol consumption during adolescence leads to altered drinking patterns and working memory deficits in young adulthood, two outcomes that suggest medial PFC dysfunction. We recorded from pyramidal neurons (PNs) in the prelimbic subregion of the medial PFC in slices obtained from mice that had IA EtOH and found that they display altered excitability, including a hyperpolarization of the resting membrane potential and reductions in the hyperpolarization-activated cation current (Ih) and in intrinsic persistent activity (a mode of neuronal firing that is dependent on Ih). Many of these effects on intrinsic excitability were sustained following abstinence and observed in mice that showed working memory deficits. In addition, we found that resting membrane potential and the Ih-dependent voltage “sag” in prelimbic PFC PNs are developmentally regulated during adolescence, suggesting that adolescent alcohol exposure may compromise PFC function by arresting the normal developmental trajectory of PN intrinsic excitability.
SIGNIFICANCE STATEMENT Binge alcohol drinking during adolescence has negative consequences for the function of the developing PFC. Using a mouse model of voluntary binge drinking during adolescence, we found that this behavior leads to working memory deficits and altered drinking behavior in adulthood. In addition, we found that adolescent drinking is associated with specific changes to the intrinsic excitability of pyramidal neurons in the PFC, reducing the ability of these neurons to generate intrinsic persistent activity, a phenomenon thought to be important for working memory. These findings may help explain why human adolescent binge drinkers show performance deficits on tasks mediated by the PFC.
Keywords: adolescence, alcohol, Ih, prefrontal cortex, working memory
Introduction
Adolescence is a critical developmental period during which the PFC undergoes significant integration and maturation (Casey et al., 2008). Risk-taking behaviors, such as binge alcohol drinking, are common during human adolescence (Sussman et al., 2008), and this period of enhanced risk-taking corresponds with the development of PFC-mediated control of motivated behavior (Casey, 2015). Alcohol depresses PFC activity acutely (Kähkönen et al., 2003), and a growing body of evidence supports the idea that chronic binge alcohol drinking during adolescence can significantly disrupt PFC maturation and long-term function (Squeglia et al., 2014; Müller-Oehring et al., 2018; Pfefferbaum et al., 2018). Understanding the effects of alcohol on the developing PFC is important, as excessive drinking during adolescence increases the likelihood that an individual will develop an alcohol use disorder (AUD) and/or other neuropsychiatric disorders in later life (Grant and Dawson, 1997; Kessler et al., 1997; Nixon and McClain, 2010).
The PFC is involved in coordinating a diverse range of higher-order behaviors, including decision-making, behavioral inhibition, and working memory (Miller and Cohen, 2001; Gläscher et al., 2012). Neuroadaptations in the PFC resulting from repeated episodes of binge drinking may disrupt the organization and execution of PFC-dependent behaviors. Binge alcohol drinkers generally perform more poorly on tasks of executive function (Townshend and Duka, 2005; Gil-Hernandez and Garcia-Moreno, 2016), and reduced PFC neuronal activity has been observed in adolescent binge drinkers during working memory tasks (Crego et al., 2010; Koskinen et al., 2011). Rodent models of adolescent binge drinking have demonstrated similar effects, including disruptions in attention (Gass et al., 2014), risk preference (McMurray et al., 2016), decision-making (Schindler et al., 2016), and working memory (Vargas et al., 2014). These higher-order behaviors are directed largely by the output of the principal projection neurons of the PFC, the pyramidal neurons (PNs) of layer 5, which project to other cortical areas and to subcortical structures that drive behavior, including the thalamus, amygdala, and striatum (Riga et al., 2014). Disruption of layer 5 PN output is therefore likely to contribute to the negative behavioral states observed following adolescent alcohol exposure, but the specific mechanisms involved in this process of neuroadaptation are not well understood.
Investigations into the effects of alcohol on PFC physiology have primarily focused on plasticity of excitatory synaptic transmission. Several studies have reported alterations in PFC PN synaptic currents following acute and chronic ethanol exposure (Tu et al., 2007; Weitlauf and Woodward, 2008; Abernathy et al., 2010; Kroener et al. 2012; Badanich et al., 2013; Trantham-Davidson et al., 2014; Centanni et al., 2017). The contribution of plasticity of intrinsic excitability of PFC neurons to the effects of chronic alcohol is less known. A variety of voltage-gated ion channels contribute to PN intrinsic excitability by influencing the probability, amplitude, duration, and frequency of postsynaptic responses and action potentials (Beck and Yaari, 2008). Alterations in the function of these ion channels in PNs can profoundly affect the generation of oscillations and synaptic integration within cortical neuronal ensembles (Schmidt et al., 2017). Plasticity of intrinsic excitability therefore represents a plausible neurobiological mechanism contributing to the pathophysiology of addiction (Kourrich et al., 2015; Nimitvilai et al., 2016).
The present studies were designed to test the hypothesis that binge drinking during adolescence alters PFC PN intrinsic excitability and related behaviors. Using an intermittent access to alcohol schedule (IA EtOH) (Melendez, 2011), we found that binge alcohol intake disrupts performance on a PFC-dependent working memory task and has a modest effect on drinking patterns observed during adulthood. We further report that the intrinsic excitability of layer 5 PFC PNs is altered by adolescent binge alcohol intake, as evidenced by changes in resting membrane potential (RMP), the hyperpolarization-activated cation current (Ih), and other measures of resting excitability. These changes may reflect the interruption or delay of the developmental trajectory of Ih, a critical determinant of RMP and excitability in the mature PFC, and contribute to the subsequently observed disruption of performance in a PFC-dependent working memory task.
Materials and Methods
Subjects.
Male C57BL/6J (The Jackson Laboratory) mice between postnatal day (PD) 25 and PD 95 were the subjects of this study. Experimental procedures were performed under the guidelines set forth in the National Institute of Health Guide for the care and use of laboratory animals and with the approval of the Institutional Animal Care and Use Committee of Columbia University. Group-housed mice arrived in the laboratory at PD 21–24 and were allowed to acclimate to the housing environment with minimal handling for 1 week. Mice participating in alcohol drinking experiments (including controls) were singly housed from PD 28 ± 3 onwards and were weighed and handled daily by experimenters and given nestlets for environmental enrichment. In one experiment in which the intrinsic properties of PNs were measured across adolescence, mice were either singly or group-housed (2–5 per cage) during adolescence; housing and age were then used as independent variables.
Behavioral procedures.
The two-bottle choice drinking procedure was used for these studies. Briefly, adolescent mice (PD ~30–60) were given two autoclaved Nalgene bottles (Thermo Fisher Scientific) with double ball-bearing sippers (Ancare). Mice from the control group were given two bottles of water; mice from the IA EtOH group were given one bottle of alcohol (15% v/v diluted from 95% ethanol, Decon Laboratories) with one bottle of water every other day, and two bottles of water on the intervening days. Bottles and mice were weighed daily, and bottle position (left/right) was alternated between each alcohol access session. Mice were given 15 sessions of access to alcohol in total. The daily alcohol dose consumed (g/kg), fluid intake, and alcohol preference were calculated for each mouse. On the 13th alcohol access day, blood was collected from separate mice at 1, 3, 6, and 9 h into the dark cycle to measure blood alcohol levels as previously described (Salling et al., 2016). Trunk blood was collected in a subset of mice used in the early (t = 0) electrophysiological experiments at the time of death to confirm that no alcohol was present before recordings.
Following the IA EtOH procedure, a group of experimental and control mice were tested for both anxiety-like behavior using the elevated plus maze (EPM) and working memory performance using the nonmatch to sample (NMS) task in the T-maze. Following EPM and T-maze exposure, brain tissue was collected from a subset of mice for immunohistochemistry or slice electrophysiology experiments. To determine the effect of adolescent IA EtOH on adult alcohol drinking behavior, limited access alcohol drinking was measured in separate groups of experimental and control mice during adulthood. Finally, several cohorts of mice were used for slice electrophysiology experiments but did not undergo behavioral analyses.
Recordings of active and passive membrane properties were also performed on alcohol-naive mice at different time points across adolescence. For these experiments, mice were either group-housed or singly housed and tissue was collected for electrophysiological recordings between PD 28 and PD 62, which corresponds to the period of IA EtOH in the experiments described above.
EPM.
An EPM was constructed from white Plexiglas affixed to a wooden platform raised 50 cm from the floor. Each arm was 10 × 25 cm, and two of the arms were enclosed by walls 20 cm high. The lighting was adjusted to ~100 lux measured at the center. The maze was located in a room that the mice had never been in, with minimal cues near the maze. Approximately 72 h after the last alcohol bottle was removed, mice were brought into the room and placed on the center of the maze facing an open arm, opposite from the observer. A camera above the maze was used to record activity within the maze (velocity, distance traveled, entries and time spent in each arm) for 5 min, and these behaviors were analyzed using Anymaze software (Stoelting). Before each trial, the maze was cleaned with Quatricide and dried. Time spent in the open arms was then expressed as a percentage of the total time spent on the maze.
T-maze.
The EPM was converted to a T-maze by adding Plexiglas walls and removable doors. Mice were food restricted to ~90% of their free feeding weight and were acclimated to a reward (50% sweetened condensed milk diluted in water) and to the T-maze for 2 d. On the third day, mice began the first block of the acquisition phase. The experimenter was blind to the condition of each mouse for all trials. To obscure the smell of the reward and prior mouse choice, open containers of milk were placed around the T-maze and a fresh paper sheet was placed on the maze between every mouse before each run. Each NMS trial is composed of two phases. During the sample phase, one of the T-maze arms was blocked and mice were forced to turn in a single direction (e.g., left) where they received access to the milk reward. Following consumption of the first reward, mice were placed back in the start box and the door to the opposite arm (e.g., right) was removed. In the choice phase, mice either turned toward the correct arm (right) and consumed another reward or incorrect arm (left) and were blocked in for 10 s without a reward. A choice was defined as having occurred when a mouse had four legs breaking the plane of one arm. On each day, mice underwent 10 trials that alternated pseudo-randomly, so that five forced trials began to the left and five to the right with no more than two trials in a row in the same direction.
Working memory performance was assessed using the NMS task in the T-maze; following acclimation, mice were given 10 d to acquire the task (defined a priori as 2 d in a row of 80% correct performance or better). Mice that did not acquire the task in 10 d were given up to 3 extra days to reach this criterion. A subset of these mice were then tested on a delayed NMS task (DNMS), during which they were presented with increasing latencies between forced and choice phases (0, 30, 60, 90, and 180 s) in a pseudo-random order for 10 trials. Each mouse experienced a right and left forced trial at each latency on each day to counterbalance experimental variables.
Limited access drinking sessions.
Limited access to alcohol sessions (LA EtOH) took place in a modified home cage (18 × 28 cm) with bedding and food available ad libitum. LA EtOH sessions occurred every other night, three times per week. Mice were placed in drinking cages 1–2 h before the start of the dark cycle and taken out ~16 h later. Bottles were positioned at the end of the cage attached to sipper tubes projecting through two small holes (1.5 cm diameter, 9 cm apart). The water bottle sipper was always available throughout the 16 h session. The alcohol bottle was positioned 2.5 cm inside the hole where the mouse could not reach and was attached to a servo motor arm. An Arduino Uno microcontroller (Sparkfun) was programmed to control the motor to move the ethanol bottle into the cage 3 h into the dark cycle and then pull the tube out of the cage 3 h later. A lickometer was fashioned by attaching a wire to each sipper tube and to a perforated metal platform placed under the sipper tubes, so that, when the mouse licked the sipper tube, it completed a circuit. Lick responses were recorded using commercial software (Lafayette Instruments). Cumulative responses on both bottles were collected every 5 min to assess the pattern of drinking behavior. All mice were given a total of 10 LA EtOH sessions over 22 d; the first nine sessions were used to assess drinking behavior, and the 10th session was used to determine blood ethanol levels (measured 2 h into the session).
Electrophysiology.
Mice were fully anesthetized with isoflurane and decapitated into ice-cold (4°C) ACSF containing the following (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, and 10 glucose. Brains were dissected and sectioned in cold ACSF using a VT1000S vibrating microtome (Leica Biosystems) into coronal slices (300 μm) that contained the prelimbic (PrL) subregion of the mPFC (between −2.8 to −1.78 mm from bregma) (Paxinos and Franklin, 2004). Slices were then incubated at 32°C in oxygenated (bubbled with 95% O2-5% CO2) ACSF for 30–45 min and were moved to room temperature (22°C) at least 45 min before recordings began. Brain tissue was visualized under an upright light microscope (BX51WI, Olympus) coupled to a camera (C8484, Hamamatsu), and cells were visualized using infrared and differential interference contrast microscopy. PFC cortical layers were identified under a 4× objective (layer V, between 350 and 500 μm from the pial surface) and PNs were identified under a 40× objective by their characteristic size and shape. Pipettes (open tip resistance 3–6 mΩ) were prepared from borosilicate glass (World Precision Instruments) using a pipette puller (Sutter Instruments). Data were collected using a Multiclamp 700B amplifier (Molecular Devices) and Clampex 10.2 Software (Molecular Devices) from identified PNs (Salling and Harrison, 2014) after reaching a >1 GΩ seal and following minimization of capacitive currents. Data were collected at 10 kHz and low-pass filtered at 2 kHz. For passive and active membrane properties under current-clamp and voltage-clamp conditions, whole-cell recordings were performed using K gluconate-based intracellular pipette solution (in mm as follows: 135 K gluconate, 5 KCl, 10 HEPES, 4 NaCl, 0.1 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 10 phosphocreatine). For measurement of intrinsic persistent firing, a potassium methylsulfate-based solution was used (in mm as follows: 130 KMeS03, 5 NaCl, 2 MgCL2, 10 HEPES, 0.2 EGTA, 4 Mg ATP, 0.3 Na GTP, and 10 phosphocreatine). For measurement of tonic GABA currents, a high chloride intracellular solution was used (in mm as follows: 140 CsCl, 4 NaCl, 1 MgCl2, 10 HEPES, 0.05 EGTA, 2 ATP Mg2+, and 0.4 GTP Mg2+).
RMP was measured following break-in once the recording stabilized. Following equilibration with the intracellular pipette solution (~3–5 min), passive membrane properties (input resistance, membrane capacitance, membrane time constant) were measured using 10 pA command increments (6 steps starting at −30 pA, 500 ms). This was done twice: once from the true RMP for each neuron, and again from a standard potential of −70 mV. Active membrane properties were assessed using 20 pA command increments (21 steps starting at −200 pA, 500 ms). The “sag ratio” was measured using a −200 pA hyperpolarizing current step in which the difference between the peak negative membrane potential (peak voltage) was subtracted from the steady-state potential, then measured at the end of the electrotonic potential (steady-state voltage); this value was then divided by the difference between the baseline potential (base voltage) and peak voltage and expressed as a percentage (Narayanan and Johnston, 2007). ZD7288, which is a selective inhibitor of the hyperpolarization-activated cation (HCN) channel that conducts the Ih, was used to measure Ih. In voltage clamp, Ih was measured by holding the neuron at −50 mV and then hyperpolarizing the membrane potential in −10 mV increments (10 steps, 500 ms). ZD7288 (Tocris Bioscience) was dissolved in ACSF to 50 μm and applied for at least 3 min. Membrane currents were measured before and after application of ZD7288 and then subtracted to determine ZD7288-sensitive currents. Access resistance was monitored for all cells in voltage clamp via 10 mV hyperpolarizing test pulses, and any cell with access resistance that changed >25% from the start of data collection or reached >20 mΩ was omitted from the data analysis.
Persistent firing experiments were performed under conditions similar to those previously reported (Thuault et al., 2013) at a higher bath temperature of 32°C. A mixture of blockers of synaptic transmission (APV 50 μm, NBQX 10 μm, and picrotoxin 50 μm) were included in the extracellular solution for these experiments. Following stabilization of the RMP, 10 μm carbachol was continuously perfused over the slice, and a series of current injections of increasing magnitude and duration were presented until persistent activity was observed: 200 pA 0.5 s, 400 pA 0.5 s, 400 pA 1 s, 400 pA 2 s, 400 pA 4 s, 600 pA 2 s, 600 pA 4 s, 600 pA 6 s, 800 pA 4 s, and 800 pA 6 s. The presence of persistent firing in these experiments was defined as a >5 s epoch of sustained action potentials following cessation of stimulation. In a subset of hyperpolarized PNs from the alcohol group (n = 5) that did not demonstrate persistent activity, sustained depolarizing current injection was used to restore RMP to ~−68 mV and the stimulus procedure was repeated. Recordings used to measure tonic inhibition mediated by GABA were performed as previously reported (Salling and Harrison, 2014).
Immunohistochemistry.
Following adolescent IA EtOH or water exposure and behavioral testing (EPM and T-maze) corresponding to 30 d from when alcohol bottles were removed, a subset of mice (n = 5 per group) were anesthetized with isoflurane (4%) and transcardially perfused with PBS followed by 4% PFA in PBS. Skulls were removed and fixed overnight; then brains were removed from the skull, washed, and sectioned on a vibratome (Leica) into 50 μm sections. Sections (4 hemispheres per mouse) containing the PrL PFC were processed for immunohistochemistry by blocking in 5% goat serum and incubating in primary antibodies overnight: HCN1 1:200 (monoclonal Igg1, Neuromab catalog #75–110, RRID:AB_2115181), TRIP8B 1:250 (monoclonal IgG2a, Neuromab catalog #75–244, RRID:AB_10698035), and MAP2 1:10,000 (polyclonal IgY, Abnova catalog #ab5392, RRID:AB_2138153). Sections were then incubated in the following IgG-specific secondary antibodies at 1:500 for 1 h (Invitrogen): AlexaFluor-546 goat anti-mouse IgG1 (catalog #A21123, RRID:AB_141592), AlexaFluor-647 goat anti-mouse IgG2a (catalog #A41241, RRID:AB_141698), and AlexaFluor-488 IgY goat anti-chicken (catalog #A11039, RRID:AB_2534096). Sections were washed, mounted, and imaged at 20× on a confocal microscope (A1, Nikon Instruments). Measurements of fluorescence values in the PrL PFC were performed by an experimentally blinded observer using FIJI software to select a rectangular region of interest across layers I–V that fit into the PrL region (300 × 540 μm) for each hemisphere. Surface plot expression levels across the PrL PFC layers were then compared between groups.
Experimental design and statistical analysis.
Off-line analysis of current-clamp and voltage-clamp data were performed using Clampfit 10.3.02 software. Graphing and statistical analyses were performed using GraphPad Prism 7 and R programming. Performance on the T-maze was analyzed using a two-way repeated-measures ANOVA, and criterion acquisition was compared using a survival analysis Mantel–Cox test. Number of licks and dose consumed in limited access alcohol drinking sessions were compared using a two-way ANOVA, whereas licks throughout session were collected in 5 min bins and compared using a two-way repeated-measures ANOVA. A Grubb's test was used to facilitate the detection of statistical outliers for measures of passive and active membrane properties. Differences in passive and active membrane properties between water and IA EtOH were compared using a two-way ANOVA, followed by Bonferroni's post hoc analyses or Student's t test. Input–output curves and I–V plots were analyzed using a two-way repeated-measures ANOVA. RMP and sag ratio measured across adolescent development were analyzed using an ANCOVA, in which the housing condition was the covariate. For all measures, statistical significance was assigned at p values <0.05.
Results
Binge alcohol consumption during adolescence affects working memory performance
Mice (PD ~30–60) that were given intermittent access to 15% alcohol (v/v) or water every other day during adolescence were compared with mice that drank only water using behavioral and/or electrophysiological measurements, according to the experimental designs illustrated in the figures (e.g., Fig. 1A). The cohorts of mice used in the behavioral (n = 35) and electrophysiology (n = 31) experiments showed similar levels of alcohol consumption (two-way repeated-measures ANOVA, F(1,62) = 0.061, p = 0.8) and preference (two-way repeated-measures ANOVA, F(1,62) = 0.024, p = 0.88). Fluid consumption was also not different between alcohol and water groups (two-way ANOVA, F(1,3070) = 1.05, p = 0.19 with no interaction, F(28,3070) = 0.84, p = 0.7). Alcohol consumption resembled an “inverted-U” trajectory indicating peak consumption in mid-adolescence, with initial values at ~11 g/kg on drinking session one followed by escalated intake that peaked at 17–19 g/kg during drinking session eight then stabilized at ~15 g/kg over the last five sessions (Fig. 1B). Preference for the alcohol solution started at ~50% on session one and escalated to ~80% by session six where it was maintained (Fig. 1C). Mean blood alcohol concentrations (BACs) reached 89 ± 16 mg/dl at 6 h into the dark cycle (Fig. 1D), demonstrating that under these conditions mice will consume alcohol in quantities ≥80 mg/dl (the threshold for defining binge-level drinking in humans) (Becker, 2013).
Figure 1.

IA EtOH during adolescence leads to working memory impairment in adulthood. A, Timeline illustrating the experimental design for IA EtOH exposure, EPM, and T-maze testing. B, C, Adolescent mice (30–60 d of age) escalated alcohol drinking during the IA EtOH procedure, both in terms of EtOH consumed and preference for the EtOH bottle. Mice used for behavior and electrophysiology experiments showed similar alcohol consumption and preference. D, BAC measurements demonstrate that mice reach a binge level of BAC (mean ± SEM, 89 ± 15 mg/dl) at 6 h into the alcohol access period, which is higher than the levels recorded 1 and 3 h into the drinking session (one-way ANOVA, p < 0.005; post hoc Tukey's: **p < 0.01, *p < 0.01). The binge level (dotted line) is defined as 80 mg/dl (left axis), or 17 mm (right axis). E, EPM was used to assess anxiety ~72 h following IA EtOH. F, There was no effect of IA EtOH on time spent in the open arms or open arm entries. G, Following EPM, mice were trained on the NMS task in the T-maze apparatus, in which a rewarded forced trial (black arrow) was followed by a choice trial where the opposite arm was rewarded (red arrow). H, Performance (% correct choices, mean ± SEM) of each group during training. There was a near significant difference in performance (two-way repeated-measures ANOVA, p = 0.06). I, Survival analyses for acquisition of the NMS task revealed that IA mice took longer to reach the criterion (Mantel–Cox test, *p < 0.05). J, A subset of these mice (n = 16 per group) were compared on the DNMS task, with pseudo-random delays (0, 30, 60, 90, and 180 s) between the forced and choice trials. Task performance decreased in both groups as delay was increased (main effect for latency, repeated-measures ANOVA, p < 0.0001). The IA EtOH mice showed a deficit on the DNMS task (main effect for group, *p < 0.05; two-way repeated-measures ANOVA, no effect at specific delays, post hoc Bonferroni's comparison).
A set of behavioral assays was performed following a prolonged period of withdrawal. Elevated anxiety can be a consequence of chronic alcohol consumption (Lee et al., 2014), which may influence cognitive tasks, so we first measured anxiety on the EPM at ~72 h following alcohol bottle removal (Fig. 1E). IA EtOH mice (n = 22) did not differ in terms of anxiety-like behavior compared with age-matched water-drinking controls (n = 21) in time spent in open arms (t(41) = 1.05, p = 0.30) or number of open arm entries (t(41) = 0.543, p = 0.59, unpaired t test) (Fig. 1F). Following EPM testing, the same cohort of mice was food restricted and tested for working memory performance using the NMS task in the T-maze. All mice (n = 21 water, n = 22 IA EtOH) were then tested on acquisition of the T-maze task by performing 10 trials per day for 10 d with minimal delay (<5 s) between forced and choice trials (Fig. 1G). A comparison of task performance using a two-way repeated-measures ANOVA showed that there was no main effect of alcohol treatment on performance (F(1,41) = 3.621, p = 0.064, repeated-measures ANOVA), a main effect of session (F(9,369) = 19.4, p < 0.0001) indicating a progressive increase in performance over sessions, but no interaction (F(9,369) = 0.7, p = 0.709) (Fig. 1H). The majority of mice from each group successfully acquired the task by the 10th session (3 consecutive days of ≥80% accuracy for each day); however, 8 mice needed up to 2 additional days of training to reach this criterion. A survival analysis revealed that the mice in the alcohol group (median = 10) were significantly slower in reaching a priori criterion than the water group (median = 7) (Mantel–Cox test, p = 0.0013) (Fig. 1I). Following acquisition of the task, delays were introduced in a subgroup of mice (n = 14 per group) by increasing latencies (0, 30, 60, 90, and 180 s) between forced and choice trials. Subsequent analyses with a two-way repeated-measures ANOVA revealed a main effect of condition (F(1,26) = 5.83, p = 0.023) and of latency (F(4,104) = 7.28, p < 0.0001), with no interaction (F(4,104) = 0.39, p = 0.82) (Fig. 1J), indicating that IA EtOH mice performed more poorly overall, and that longer delays lead to decreased performance in both groups. These data support the notion that binge alcohol drinking during adolescence negatively impacts working memory, particularly when working memory load is increased.
Adolescent binge drinking leads to altered pattern of alcohol consumption in adulthood
We tested the hypothesis that alcohol consumption and patterns of alcohol intake during adulthood are altered in mice that drank alcohol during adolescence. Following the IA EtOH procedure (PD 30–60), both alcohol (n = 13) and water control mice (n = 11) were given nine limited access alcohol sessions, during which mice had access to alcohol and water 3–6 h into the dark cycle, 3 nights per week (Fig. 2A). Unexpectedly, we found no significant differences between the groups in dose consumed (Fig. 2B) (two-way repeated-measures ANOVA (F(1,180) = 1.96, p = 0.164), blood alcohol levels (Fig. 2C) (unpaired t test, t(22) = 0.301, p = 0.761), or cumulative licks on the water bottle (Fig. 2D) (two-way ANOVA, F(1,194) = 3.25, p = 0.288). We did observe a significant difference in terms of cumulative licks on the ethanol bottle (two-way ANOVA, F(1,190) = 4.37, p = 0.038), but no effect was found from post hoc analyses for session (Bonferroni's comparison), including the initial session when they were experiencing the taste of alcohol for the first time (p = 0.3). There was a main effect of session for licks on the water bottle (F(8,194) = 3.25, p = 0.0017), but not the alcohol bottle, demonstrating that mice from both groups decreased their water intake only over the nine sessions (Fig. 2D). To better assess the pattern of alcohol consumption, the number of bottle licks were recorded every 5 min throughout the session. Each week was separated by a 2 d period of abstinence; and because we did not observe differences between daily sessions for each week on the alcohol bottle for IA EtOH (two-way repeated-measures ANOVA, week 1: p = 0.4; week 2: p = 0.5; week 3: p = 0.06) and alcohol-naive groups (week 1: p = 0.54; week 2: p = 0.76; week 3: p = 0.14), we averaged each week for analyses (sessions 1–3, 4–6, and 7–9). Post hoc analyses revealed that IA EtOH mice made significantly more licks on the alcohol bottle during the first 5 min of bottle presentation during each of the 3 weeks of testing (Bonferonni's comparison, week 1: p < 0.0001; week 2: p < 0.0001; week 3: p = 0.0006); the alcohol-naive mice made more licks on the alcohol bottle during the second 5 min access period during the last 2 weeks (week 2: p = 0.02; week 3: p = 0.0005). These data indicate that mice with a history of adolescent binge drinking appear more motivated to seek alcohol at the start of the session (within first 5 min), and this pattern is evident all 3 weeks. This phenomenon, known as “front-loading,” has been previously described in other drugs of abuse and during binge alcohol sessions in mice (Wilcox et al., 2014). Alcohol-naive mice begin to show this front-loading behavior during the last 2 weeks of access, but at a longer latency from the start of the session (after 5 min).
Figure 2.

Alcohol exposure in adolescence results in alcohol “front-loading” in adulthood. A, Timeline illustrating the experimental design during IA EtOH and limited access to alcohol in both water (n = 11) and IA EtOH (n = 13) groups. B, Previous experience of binge-like drinking (adolescent IA EtOH vs adolescent naive) did not affect the EtOH dose consumed during limited access sessions in adulthood (two-way ANOVA, p = 0.16). C, BAC measured in the 10th limited access session (t test, p = 0.76). D, Comparison of cumulative licks during the 3 h limited access session revealed that both groups decreased their licks on the water bottle over the 9 sessions (two-way ANOVA, main effect of session, p < 0.01). The IA EtOH mice (913 ± 28, mean ± SEM) had higher average licks on the alcohol bottle than the adolescent alcohol-naive mice (814 ± 40) for each session (two-way ANOVA, main effect of group, *p < 0.05). E, To compare drinking patterns, licks throughout the limited access session were recorded and counted in 5 min bins, averaged over weeks 1–3 (3 sessions per week). Analyses revealed no overall effect of drinking history, but a significant interaction of time and history for each week (two-way ANOVA, week 1: *p < 0.05; week 2: ***p < 0.0001; week 3: *p < 0.05). Post hoc analyses revealed that the mice in the IA EtOH group had significantly more licks on the alcohol bottle in the first 5 min of bottle presentation for weeks 1–3, and the water group had significantly more licks on the water bottle in the 5–10 min bin for weeks 2–3 (Inset). Mice that consumed binge levels of alcohol in adolescence therefore consumed more alcohol, faster upon presentation of alcohol bottle in the adult limited access paradigm.
The intrinsic excitability of layer 5 PNs of the PrL PFC is altered in adult mice following binge-level alcohol consumption in adolescence
In our behavioral analyses, we found that adolescent binge drinking led to deficits in performance on the T-maze and a modest effect on drinking pattern in adulthood. Previously, lesions of the PrL region of medial PFC led to deficits in the T-maze in mice (Kellendonk et al., 2006). Because the PrL PFC has also been implicated during reward predictive cues, showing increased activation during reward availability (Ishikawa et al., 2008; Burgos-Robles et al., 2013),we chose to perform our electrophysiological measurements on PNs in layer 5 of the PrL PFC.
Whole-cell patch-clamp recordings of PrL PFC PNs from IA EtOH (n = 77 neurons, n = 21 mice) and age-matched water control mice (n = 91 neurons, n = 23 mice) were taken at several time points following 30 d of the IA EtOH procedure (Fig. 3A). Slices from both groups containing the mPFC were prepared either 2–12 h (t = 0; water: n = 33 neurons, n = 11 mice, IA EtOH: n = 42 neurons, n = 10 mice, 10–15 d, t = 10–15; water: n = 29 neurons, n = 6 mice, IA EtOH: n = 32 neurons, n = 9 mice, or 30–35 d, t = 30–35; water: n = 15 neurons, n = 4 mice, IA EtOH: n = 17 neurons, n = 4 mice) following removal of the alcohol bottle. Current-clamp recordings of electrotonic potentials from layer 5 PNs in response to rectangular negative and positive current injections were used to measure passive and active membrane properties (Fig. 3B). Subsequent analysis of RMP of PNs at each time point revealed that RMP was significantly hyperpolarized in the mice following IA EtOH (two-way ANOVA, F(1,162) = 25.18, p < 0.0001). Bonferonni's post hoc analyses revealed significant effects at t = 0 (p = 0.0001) and t = 10–15 (p = 0.015), with near significance at t = 30–35 (p = 0.095) (Fig. 3C). In addition, measurements of passive membrane properties showed an increase in input resistance (RIN) for IA EtOH measured at −70 mV (two-way ANOVA, F(1,133) = 12.16, p = 0.0007), with significant effects at the t = 0 time point (water: 111 ± 12 mΩ, IA EtOH: 146 ± 10 mΩ, Bonferrroni's comparison, p < 0.05). We also observed an overall increase in membrane time constant in the IA EtOH group (two-way ANOVA, F(1,134) = 19.98, p < 0.0001) with significant effects across all time points (Bonferrroni's comparison, t = 0, water: 18 ± 2 ms, IA EtOH: 25 ± 2 ms, p = 0.025, t = 10–15; water: 15 ± 2 ms, IA EtOH: 25 ± 3 ms, p = 0.027, t = 30–35; water: 12 ± 2 ms, IA EtOH: 22 ± 3 ms, p = 0.039) in PrL PFC PNs from alcohol-drinking mice. These data reveal that several passive membrane properties (RMP, RIN, and time constant) are significantly affected by adolescent IA EtOH and that many of these changes persist into abstinent adulthood.
Figure 3.

Sustained effects on the intrinsic excitability of mPFC PNs of binge-like alcohol consumption during adolescence. A, Timeline illustrating the experimental design and mouse age during IA EtOH and subsequent electrophysiological recordings from water and IA EtOH groups. B, Representative recordings from layer 5 PNs in the medial PFC in slices taken from water-drinking control mice (black, left) and IA EtOH (red, right) mice (t = 0, ~60 d of age) that illustrates differences observed in RMP. C, RMP was compared between groups at 3 time points. A significant main effect of IA EtOH on RMP was observed (two-way ANOVA, p < 0.0001), and post hoc analyses revealed significant effects at t = 0 (***p = 0.0001), t = 10–15 (*p < 0.05), but not at t = 30–35 (p = 0.095). D, PN recording demonstrating the characteristic voltage “sag,” from peak deflection to steady state, in the electrotonic potential in response to a hyperpolarizing current injection. E, Comparison of voltage sag ratio between groups revealed a reduction in sag ratio in the IA EtOH group (two-way ANOVA, p < 0.0001). Post hoc analyses revealed significant effects at t = 0 (**p < 0.01), t = 30–35 (*p < 0.05), but not at t = 10–15 (p = 0.15). F, The excitability of PNs was compared by eliciting APs during 500 ms, 20 pA current steps and examining the input–output curves. When PrL PFC PNs from the t = 0 condition were tested at true RMP (typically hyperpolarized in the IA EtOH group), there was no effect. Calibration: vertical, 20 mV; horizontal, 200 ms. G, When these same neurons were tested at a common voltage (−70 mV), the PNs from the IA EtOH group were more excitable (two-way ANOVA, main effect for group, *p = 0.02; interaction between current and group, p < 0.0001). Post hoc analyses revealed significant differences at steps ≥120 pA (**p < 0.01).
In most of the layer 5 PFC PNs, we observed that hyperpolarizing current steps led to a depolarizing voltage relaxation or “sag” in the electrotonic potential, which is characteristic of the presence of the hyperpolarization-activated cation current, Ih, a prominent contributor to RMP and RIN in cortical PNs (Shah, 2014). We quantified the extent of voltage sag by measuring the “sag ratio” following a −200 pA current step (Fig. 3D). Compared with water controls, we found that PNs from the IA EtOH group had an overall reduction in the sag ratio (F(1,143) = 20.9, p < 0.0001) compared with water controls. Post hoc analyses using Bonferonni's comparison showed significant effects at t = 0 (p = 0.009) and t = 30 (p = 0.011) time points; however, no significant differences were observed at the t = 15 time point (p = 0.14). (Fig. 3E). These results suggest that the effects on intrinsic excitability observed following adolescent binge drinking may reflect a reduction in Ih. To determine whether an increase in tonic GABA current contributed to the hyperpolarization, we patched additional PrL PFC PNs from both groups at the t = 0 time point where we observed our most robust effect on RMP using an intracellular solution with high chloride (to enhance GABA currents), cesium (to block Hcn channels), and applying gabazine (20 μm) to PrL PFC PNs to detect a shift in holding current after reaching a stable baseline. We found that, although there was an observable tonic GABA current in these neurons, there was no difference between groups (water = 13 ± 2 pA, mean ± SEM, n = 5 neurons, n = 2 mice; IA EtOH = 13 ± 2 pA, n = 8 neurons, n = 2 mice; t test, t(11) = 0.112, p = 0.913); therefore, extrasynaptic GABA-A receptors are unlikely to play a role in any changes in excitability observed after IA EtOH.
To determine whether adolescent alcohol affected excitability of PrL PFC PNs, input–output curves were compared at RMP (hyperpolarized in the EtOH group) and a common voltage (−70 mV). At t = 0, we found that there were no significant differences detected between groups at RMP (Fig. 3F); however, when excitability was assessed from a standard membrane potential of −70 mV (PNs in both groups injected with current to maintain membrane potential), PNs from the IA EtOH group (n = 38 neurons, n = 11 mice) were more excitable than water controls (Fig. 3G) (n = 27 neurons, n = 10 mice) as indicated by a main effect of group (repeated-measures ANOVA, F(1,63) = 6.01, p = 0.017) and significant interaction (F(9,567) = 6.99, p < 0.0001). Post hoc analyses revealed significant effects at ≥80 pA. We also observed a similar effect at the t = 30–35 time point (main effect for group, F(1,30) = 9.94, p = 0.0045), but there was no main effect of alcohol at the t = 10–15 time point (F(1,27) = 1.12, p = 0.3). These data confirm that IA EtOH affects the active membrane properties of PFC PNs, and that some of these changes persist through abstinence in adulthood.
Ih is reduced following binge alcohol consumption during adolescence
To determine whether HCN channels underlying the macroscopic Ih current are responsible for the changes in excitability that are observed in alcohol-drinking mice, the HCN channel blocker ZD7288 was applied to PrL PFC PNs from alcohol-drinking and control mice at t = 0, at which time point we had observed large and consistent effects of IA EtOH exposure on RMP and sag ratio. In this experiment, we first made current-clamp measurements of passive and active membrane properties from RMP or −70 mV, before and after application of 50 μm ZD7288. In PrL, PFCZD7288 significantly hyperpolarized the RMP in PNs from control (n = 13 neurons, n = 8 mice; t(12) = 5.72, p < 0.0001) and IA EtOH (n = 18 neurons, n = 8 mice; t(17) = 2.86, p = 0.011) mice (Fig. 4B). The magnitude of ZD7288's effects on RMP in PNs from control mice (5.4 ± 0.9 mV) versus IA EtOH mice (2 ± 0.7 mV) was significantly more than from IA EtOH mice (t(29) = 2.95). Furthermore, ZD7288 significantly reduced the voltage sag ratio in PNs from control (t(12) = 5.49, p < 0.0001) and IA EtOH (t(17) = 4.34, p = 0.0004) mice (Fig. 4C). The magnitude of this effect (water = 14.7 ± 3%; IA EtOH = −3.5 ± 0.8%) was significantly different between groups (t(29) = 4.11, p = 0.0003). We also observed differences of the effect of ZD7288 application on RIN (water = +139 ± 18 mΩ, IA EtOH = +83 ± 19 mΩ; t(29) = 2.12, p = 0.04) as well as time constant (water = +25.6 ± 3.6 ms, IA EtOH = +10.9 ± 3 ms; t(29) = 3.14, p = 0.004). ZD7288 also increased excitability in PNs from water control mice held at −70 mV (F(1,18) = 12.6, p < 0.005) but did not alter excitability in IA EtOH mice (F(1,32) = 1.64, p = 0.21) (Fig. 4D). These results confirm that IA EtOH exposure leads to a reduction in the intrinsic properties that are mediated by Ih, and thereby partially occludes the effects of the Ih blocker ZD7288 on intrinsic excitability.
Figure 4.

A reduction in Ih is coincident and consistent with the effect of adolescent alcohol on intrinsic excitability of PrL PNs. A, A representative current-clamp recording from a layer 5 PN in mPFC of an alcohol-naive mouse. The application of the Ih blocker ZD7288 hyperpolarizes the RMP, abolishes the voltage sag, and increases both membrane time constant and input resistance (RIN). B, RMP was reduced in IA EtOH PNs (n = 18 neurons, n = 8 mice) compared with water PNs (n = 13 neurons, n = 8 mice, unpaired t test, ***p < 0.005). Comparison of effects of ZD7288 on RMP reveals that ZD7288 significantly hyperpolarizes the RMP of PNs in the water-drinking controls (paired t test, ****p < 0.0001) and to a lesser degree in PNs from IA EtOH mice (*p < 0.05). C, Voltage sag ratio is significantly decreased by ZD7288 in PNs from control mice (paired t test, ****p < 0.0001) and to a lesser degree in IA EtOH mice (***p < 0.001) mPFC PNs. D, Input–output curves show that ZD7288 increases the excitability of PNs from water-exposed mice when measured from −70 mV (two-way repeated-measures ANOVA, main effect of group, **p = 0.005; and interaction, ****p < 0.0001), with no significant effect on excitability in PNs from the IA EtOH group. E, Representative voltage-clamp recordings of currents recorded from mPFC PNs from water-drinking controls (black, left) and IA EtOH (red, right) groups in response to sequential −10 mV commands from −50 mV to −150 mV, before (top) and after (middle) ZD7288. The ZD-sensitive current (bottom) represents Ih measured at the soma. F, PrL PFC PNs from the IA EtOH group show a significant reduction in Ih compared with water-drinking controls (two-way repeated-measures ANOVA, main effect of group, ***p < 0.01; significant interaction, p < 0.0001; post hoc Bonferonni's, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001 for all steps from −100 to −150).
In addition, we measured Ih directly using voltage-clamp recordings of membrane current from the soma of layer 5 PrL PFC PNs before and after application of ZD7288. PNs were held at −50 mV and stepped down in −10 mV increments to −150 mV. Using this protocol in slices from IA EtOH and control mice at t = 0, we observed a characteristic slow inward relaxation of current to a steady state. The slow relaxation was absent in the presence of 50 μm ZD7288, consistent with the presence of Ih in PrL PFC PNs (Fig. 4E). In our analyses, we found that the ZD7288-sensitive Ih current was significantly reduced in PFC PNs from the IA EtOH mice (F(1,31) = 9.1, p = 0.005) with a significant interaction (F(10,310) = 7.2, p < 0.0001, post hoc Bonferroni's comparison revealed significant effects from −100 to −150 mV steps; Fig. 4E,F). These data confirm that Ih is greatly reduced in PrL PFC PNs in mice that consumed binge levels of alcohol during adolescence.
Decreased intrinsic persistent activity of PrL PFC neurons following adolescent alcohol consumption
Genetic ablation of Hcn1 within the mPFC has been shown to negatively affect working memory performance (Thuault et al., 2013). Electrophysiological characterization of Hcn1 knock-out mice revealed that the loss of HCN1 in layer 5 mPFC PNs resulted in a hyperpolarized RMP that prevented the induction of intrinsic persistent activity (IPA) in brain slices following muscarinic AChR activation by carbachol (Thuault et al., 2013). To examine whether the reduction of Ih observed following adolescent alcohol consumption and concomitant hyperpolarization of RMP affects IPA in the PrL PFC, we recorded in layer 5 PNs in slices obtained from water (n = 21 neurons, n = 7 mice) and IA EtOH (n = 21 neurons, n = 7 mice) at the end of the adolescent drinking period.
Following mAChR stimulation with carbachol (10 μm), depolarizing currents of increasing magnitude (200–800 pA) and duration (0.5 to 6 s) were injected into the layer 5 PNs to generate a sustained IPA that followed the current step (Fig. 5B). Under these conditions, we were able to observe IPA in control PFC PNs. In addition, we were able to replicate our previous findings that RMP is significantly hyperpolarized in IA EtOH mice using a KMeSO3-based intracellular solution (unpaired t test, t(40) = 30.1, p = 0.004, t test) (Fig. 5B). When recording from hyperpolarized PrL PFC PNs from the IA EtOH group, IPA was less frequently observed, even after longer, larger depolarizing current injections (Fig. 5C, middle). The loss of IPA following Hcn1 deletion can be rescued whether the RMP is restored to its normal value by injecting depolarizing current in layer 5 PNs (Thuault et al., 2013). In our case, a sustained current injection was used to restore RMP to control levels (−68 to −70 mv), and IPA was rescued in 50% of neurons studied (Fig. 5C, bottom). A survival analysis of charge and persistent activity revealed that water and IA EtOH PNs were significantly different (Gehan-Breslow-Wilcoxon test, p = 0.048) (Fig. 5D). Although PFC PNs from both groups displayed IPA, the fraction of neurons capable of generating sustained firing was much smaller in the IA EtOH mice compared with the water controls. When RMP was restored by depolarizing current injection in hyperpolarized PNs from the IA EtOH group, the fraction of neurons displaying IPA was similar to controls (Fig. 5E).
Figure 5.
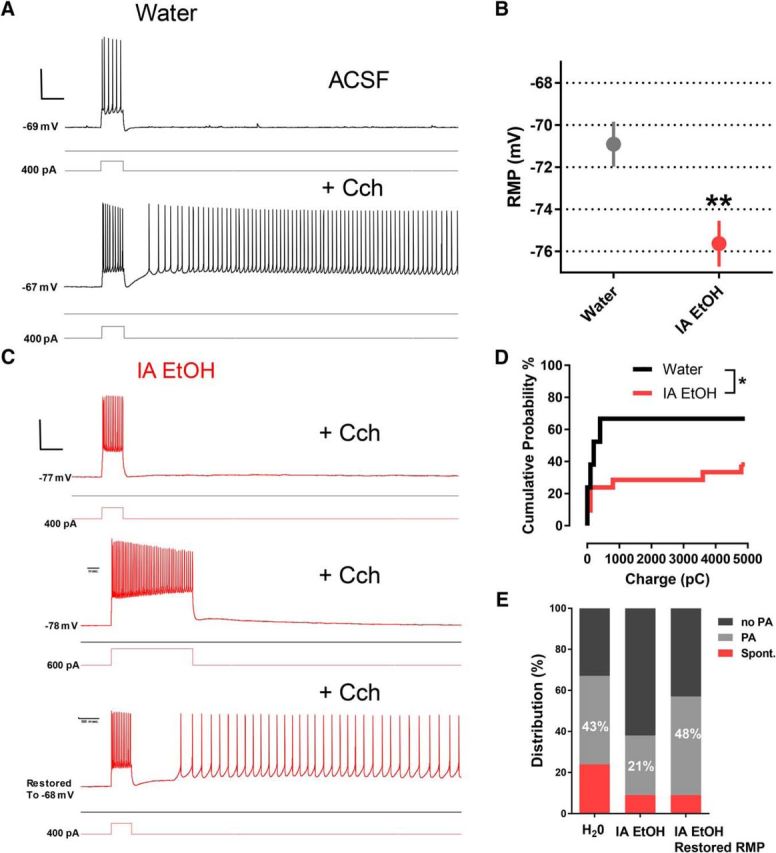
A reduction in the generation of IPA in PFC PNs after binge-like alcohol drinking during adolescence. A, Top, PN from the medial PFC following a 400 pA current injection. Bottom, When 10 μm carbachol (Cch) was applied to the same neuron in the presence of synaptic blockers (APV, NBQX, picrotoxin), the PN exhibited several seconds of IPA. Calibration: vertical, 40 mV; horizontal, 500 ms. B, RMP of PNs in the mPFC was observed to be hyperpolarized in IA EtOH (n = 21 neurons, n = 7 mice) group compared with water controls (n = 21 neurons, n = 7 mice, unpaired t test, **p < 0.01). C, Recording from a mPFC PN in the IA EtOH group, RMP = −77 mV. The addition of Cch did not result in IPA following a 500 ms, 400 pA current injection (top). The same PN did not show persistent activity even following a 2 s, 600 pA current injection (middle). When RMP of the same PN was manually restored to −68 mV using steady current injection, a single 500 ms, 400 pA stimulus elicited IPA (bottom). D, Survival analysis reveals a significant effect of IA EtOH in the presence of IPA (Gehan-Breslow-Wilcoxon test, *p < 0.05). E, The distribution of IPA observations in mPFC PNs from water-drinking control mice (H2O) and IA EtOH mice, indicating the percentage of PNs that showed spontaneous firing (Spont.), intrinsic persistent activity (PA) dependent on current injection stimulus, and those that did not exhibit IPA even at the highest current injection (6 s; 800 pA). When RMP was restored manually to ~−68 mV in IA EtOH PNs that showed a hyperpolarized RMP, the distribution of PNs exhibiting activity spontaneously or in response to a depolarizing stimulus was nearly equivalent to that in neurons from the alcohol-naive mice.
Reduction in RMP, voltage sag, Ih, and Trip8b expression in the PrL PFC in mice that consumed alcohol during adolescence and displayed working memory deficits
To examine the connection between the deficits in behavior and altered PrL PFC PN properties in more detail, a cohort of mice that underwent the IA EtOH procedure during adolescence and T-maze testing were subsequently killed at PD 90–95 d for electrophysiology or PD 90 for immunohistochemistry (Fig. 6A). The mice used in these experiments (water, n = 11; IA EtOH, n = 11) were representative of the cohort of mice used in Figure 1 as they showed a significant difference in days to criterion (Mantel–Cox, p = 0.002) and a main effect of group on the DNMS task (F(1,20) = 7.18, p = 0.014).
Figure 6.

Mice that consumed binge-like alcohol during adolescence and showed deficits on the T-maze demonstrate altered excitability, a reduction in Ih, and decreased expression of Trip8b in the PrL subregion of the medial PFC. A, Timeline of experimental design showing the order of procedures. The mice in this study were from 2 of the cohorts of mice described in Figure 1. B, mPFC PNs from IA EtOH mice (n = 10 neurons, n = 5 mice) were significantly hyperpolarized compared with PNs from water controls (n = 9 neurons, n = 5 mice, unpaired t test, **p < 0.01), and application of ZD7288 significantly affected RMP only in the water control group (paired t test, **p < 0.01). C, PNs had a reduction in sag ratio following IA EtOH procedure (unpaired t test, **p < 0.01). ZD7288 had a larger effect on mPFC PNs from the water group (paired t test, ***p < 0.001) group than PNs from the IA EtOH group (paired t test, **p < 0.01). D, In these same PNs, ZD7288-sensitive current was compared following −10 mV voltage steps from −50 mV, and a significant difference was revealed (repeated-measures ANOVA, **p < 0.01). Post hoc analyses revealed a significant effect at −150 mV (Bonferroni's comparison, *p < 0.05). E, A subset of mice was processed for immunohistochemistry (20 total sections from 5 mice from each of the groups), and sections containing the PrL PFC were probed for Hcn1 (red), Trip8b (blue), and Map2 (green) protein expression. Images collected show the typical pattern of expression across layers of the PFC, with high expression and colocalization of Hcn1 and Trip8b observed in layer 1 dendrites and in layer 5 somas as well as the expression of Map2 along the dendritic fibers across all layers. F, There was no effect of IA EtOH on the expression of Hcn1 (left) or Map2 (right); however, there was a main effect of group on the expression of Trip8b (*p = 0.016).
PrL PFC PNs from IA EtOH mice (n = 10 neurons, n = 5 mice) were significantly hyperpolarized compared with water controls (n = 9 neurons, n = 5 mice; unpaired t test, p = 0.0041), and application of ZD7288 significantly hyperpolarized RMP in the water control group only (paired t test, p = 0.002) (Fig. 6B). In addition, PrL PFC PNs showed a reduction in sag ratio following IA EtOH procedure (unpaired t test, p = 0.005). The effect of ZD7288 had a larger effect on PrL PFC PNs from the water group (paired t test, p < 0.001) than the IA EtOH group (paired t test, p < 0.01) (Fig. 6C). In these same PNs, ZD7288-sensitive current was compared following −10 mV voltage steps from a −50 mV holding potential, and a significant interaction was revealed (repeated-measures ANOVA, F(10,170) = 2.59, p = 0.006). Post hoc analyses revealed a significant difference between currents measured at −150 mV (Bonferroni's comparison, p = 0.024) (Fig. 6D). In this experiment, we were able to replicate our previous findings of a reduction in Ih in a cohort of IA EtOH mice that showed deficits in T-maze performance.
To further examine the strength of the relationship between observed changes in physiology and cognitive performance, we looked for associations between RMP, sag ratio, and maximal ZD-sensitive currents with the number of errors to criterion and errors in the DNMS phase of the T-maze task. We found that RMP was significantly correlated with acquisition errors (Pearson's correlation (n = 27 neurons, n = 12 mice, r = −0.585, p = 0.0013), but not DNMS errors (p = 0.24). We found no association of voltage sag with either acquisition errors (n = 27 neurons, n = 12 mice, r = −0.24, p = 0.22) or DNMS errors (p = 0.77). For ZD-sensitive current, we found a significant correlation with DNMS errors (n = 19 neurons, n = 11 mice, r = 0.58, p = 0.009), but not acquisition errors (p = 0.3).
Next, we considered whether the reduction in Ih is due to a reduced expression of HCN1 or an auxiliary subunit to HCN channels TRIP8B (the tetratricopeptide repeat containing Rab4 interacting protein; product of the PEX5L gene). A subset of T-maze mice was processed for immunohistochemistry (20 total sections from 5 mice from each group), and sections containing the PFC were probed for HCN1 (red), TRIP8B (blue), and MAP2 (green) protein expression (Fig. 6E). Images collected show the typical pattern of expression observed across layers of the PFC. High expression and colocalization of HCN1 and TRIP8B were observed in layer 1 (mainly dendrites) and in layer 5 (mainly somas). We included an analysis of MAP2 to rule out gross structural differences in the dendrite and soma where Hcn1 and Trip8b are coexpressed. While there was no effect on the expression of HCN1 (repeated-measures ANOVA, F(1,38) = 1.22, p = 0.58) or MAP2 (F(1,38) = 3.46, p = 0.217), there was a main effect of IA EtOH on the expression of TRIP8B (repeated-measures ANOVA, F(1,38) = 6.35, p = 0.016), indicating that TRIP8B expression was reduced across the layers of the PrL PFC (Fig. 6D) in the IA EtOH mice. TRIP8B is required for membrane insertion and establishing the HCN1 gradient in PNs (Santoro et al., 2009); therefore, a reduction in expression could be contributing to the observed reduction in Ih.
Intrinsic excitability of layer 5 PNs develops during adolescence
In the hippocampus, both Ih and the expression of HCN channel subunits are dynamically regulated during adolescence (Brewster et al., 2007). This led us to consider whether Ih is also continuing to develop in the PFC during this period. The mice in our drinking experiments were all singly housed during adolescence, and so we also wanted to determine whether the housing condition during adolescence affected PN excitability, as has been reported in the amygdala of rats (Rau et al., 2015). Brain slices containing the PrL PFC were collected for electrophysiology from alcohol-naive mice that were either singly or group-housed from the onset of adolescence (PD 28) to early adulthood (PD 62). We measured RMP and sag ratio of layer 5 PNs of the PrL PFC across adolescent development (PD 28–62). RMP comparisons using an ANCOVA with housing condition as the covariate revealed no significant effect of housing (F(1,140) = 0.344, p = 0.56) or housing × age interaction (F(1,140) = 3.69, p = 0.057), but we did detect a significant main effect of age (F(1,132) = 20.2, p < 0.0001). For sag ratio, the same analyses indicated no effect of housing (F(1,140) = 0.18, p = 0.67) or housing × age interaction (F(1,132) = 0.31, p = 0.58), but a significant effect of age (F(1,140) = 6.4, p = 0.013). These data demonstrate that RMP and sag ratio are age-dependent but not affected by housing condition in the mouse, with RMP of PrL PFC PNs becoming more depolarized with age (Fig. 7A) and sag ratio increasing with age during adolescence (Fig. 7B), consistent with an emergence of increased Ih.
Figure 7.

The development of intrinsic properties of mPFC PNs during adolescence is influenced by drinking history but not by housing condition. A, Current-clamp recordings from mPFC PNs were made throughout adolescence (ages 28–62) in both single and group-housed mice. Analyses using ANCOVA with housing as the covariate revealed that RMP is significantly affected by adolescent age (****p < 0.0001), becoming more depolarized during adolescence, but not by housing. B, Voltage sag was also significantly influenced by age (*p < 0.05) but not by housing condition. The sag ratio (SR) from mPFC PNs increased with age. C, Left, The distribution of RMP values of PNs during early adolescence, ages 28–45 (left) and late adolescence, ages 46–62. Right, The distribution of RMP values from water-drinking controls and IA EtOH mice (from the experiment in Fig. 3C). These data demonstrate not only the rightward (depolarizing) shift of RMP from early to late adolescence but also a relative leftward (hyperpolarizing) shift in adulthood in the IA mice (red). D, Distribution of voltage SRs from early and late adolescence and following binge-like alcohol drinking. As mice age during adolescence, the SR distribution becomes more bimodal, with two peaks apparent in the SR distribution: at 0%–8% (low) and 16%–28% (high). Following adolescent binge-like alcohol drinking (red), SR in adults is reduced, with a large decrease in the population of mPFC PNs within the 16%–28% (high) SR range and an increase in the fraction of PNS in the low SR range.
When comparing the distributions of RMP and sag ratio at early (PD 28–45) and late (PD 46–62) adolescence to our alcohol drinking mice, we observed some intriguing similarities. The distribution of RMP values shifts to the right between early and late adolescence (Fig. 7C), regardless of housing condition. The distribution of data collected following adolescent drinking experiments (all time periods from Fig. 3C,E) shifts to the left for the group of mice that drank alcohol during adolescence. For the sag ratio, the distribution becomes more bimodal from early to late adolescence where a population of high sag ratio PNs emerge along with a smaller group of PNs with low values. In contrast, alcohol drinking during adolescence appears to prevent the emergence of this high sag ratio population of PNs, creating a distribution that is skewed to the left and coinciding with the low sag ratio population among water drinkers (Fig. 7D). These data are consistent with previous hypotheses concerning a mechanism by which adolescent drinking might delay or arrest brain maturation (Crews et al., 2016).
Discussion
Adolescent alcohol is associated with working memory deficits in mice
In this study, we investigated a possible link between adolescent binge drinking in mice and changes in working memory and intrinsic properties of PrL PFC PNs. Our behavioral experiments indicate that adolescent binge-like alcohol drinking in mice is associated with a reduction in working memory performance on the T-maze, findings that parallel observations made in human adolescents. The magnitude of the working memory deficit observed here approaches what has previously been observed in rodents following lesion of the medial PFC (Kellendonk et al., 2006) or optogenetic inhibition of the inputs from medial PFC to mediodorsal thalamus (Bolkan et al., 2017). Furthermore, our findings are in agreement with several rodent studies in which performance on PFC-dependent tasks was negatively influenced by chronic alcohol exposure (Gass et al., 2014; Vargas et al., 2014; McMurray et al., 2016; Schindler et al., 2016). This is the first study demonstrating a working memory deficit following a voluntary alcohol consumption procedure in mice. In addition to working memory deficits, voluntary binge-like alcohol consumption during adolescence led to modest effects on drinking patterns in adulthood, characterized by robust licking on the alcohol bottle in first 5 min of a limited access session. This “front-loading” behavior has also been observed in humans with an increased vulnerability to developing an AUD (Gowin et al., 2017), further supporting the face validity of the mouse IA EtOH procedure. Working memory and alcohol consumption have previously been negatively correlated and are likely to involve similar modalities (Squeglia et al., 2012; Houck and Feldstein Ewing, 2018; Moser et al., 2017).
Adolescent alcohol is associated with changes in excitability in PrL PFC PNs
The PrL PFC of rodents undergoes significant development during adolescence, is a known target of both acute and chronic alcohol, and mediates both working memory and reward-seeking behavior (Dalley et al., 2004; Abernathy et al., 2010; Koss et al., 2014). It is therefore a likely neural substrate for the behavioral changes observed following IA EtOH during adolescence. In parallel experiments, we found a specific pattern of changes in intrinsic excitability in the PrL PFC following IA EtOH cessation, and this persisted into the period of abstinence during which the behavioral analyses were performed. A reduction in Ih was implicated in these effects of IA EtOH, as blocking Ih with ZD7288 causes a similar change in RMP and increased excitability in PrL PFC PNs. These effects on excitability are similar to those observed in the hippocampus, cerebellum, and mPFC in Hcn1 KO mice (Nolan et al., 2003; Thuault et al., 2013), which suggests that the Hcn1 subunit might be perturbed by the IA EtOH procedure.
When the Hcn1 subunit is genetically deleted in medial PFC PNs (Thuault et al., 2013), the majority of the hyperpolarization-activated inward current is lost and RMP becomes hyperpolarized from −68 mV to −73 mV. Importantly, this hyperpolarization prevents mPFC PNs from generating IPA following muscarinic stimulation with carbachol. IPA has been demonstrated in both the entorhinal cortex and PFC and is implicated in both spatial and working memory (Egorov et al., 2002). mPFC Hcn1 KO mice show working memory deficits, a hyperpolarized RMP, and loss of IPA ex vivo (Thuault et al., 2013). IA EtOH mice also showed a reduced probability of generating IPA, and that this was due to membrane hyperpolarization as restoring RMP to normal levels rescued IPA in hyperpolarized IA EtOH PNs. This finding brings up the possibility that restoring RMP by targeting HCN channels in vivo may rescue IPA and the working memory deficits seen in IA EtOH mice. Although recent research has questioned the role of persistent active states of PFC PNs in working memory (Lundqvist et al., 2016), there are well-established lines of research that support a role for persistent firing (Baeg et al., 2003; Riley and Constantinidis, 2015), cholinergic modulation (Ragozzino and Kesner, 1998; Croxson et al., 2011), and synaptic integration (Wang, 2001) within the mPFC in working memory tasks, and all are significantly affected by reductions in Ih (Thuault et al., 2013; Schmidt et al., 2017).
Potential mechanisms for changes in Ih and excitability in PrL PFC PNs
There are a number of candidate mechanisms that might explain the decrease in Ih, including reduced HCN expression, reduced membrane insertion, or changes in HCN channel function, either directly or via effector molecules, such as cyclic nucleotides. Using immunohistochemistry, we found that, in IA EtOH mice that displayed deficits in the T-maze, there was no significant difference in change in HCN1 or MAP2 expression, but rather a reduction in TRIP8B expression, an accessory subunit of HCN channels that is required for surface membrane insertion and for establishing the characteristic distribution of HCN channels along the dendrites and soma (Santoro et al., 2009). In Trip8b KO mice, HCN channels are mislocalized; the magnitude of Ih is greatly reduced in these mice, and the resulting phenotype is similar to that of Hcn1 KO mice (Han et al., 2017). TRIP8B is a regulatory subunit of HCN channels and interacts with a tripeptide sequence on the C-terminal domain of the HCN subunit, which negatively influences cAMP from binding to the channel (Han et al., 2011). A reduction in Trip8b expression would be expected to prevent HCN channels from being properly inserted into the membrane and might alter HCN function by increasing the dependence on intracellular cAMP for activation of the channel.
Significance of changes in Ih for alcohol effects in the PrL PFC
Our findings complement previous work examining how alcohol exposure effects neurotransmission in the rodent mPFC. Intermittent binge-level alcohol vapor inhalation during early adolescence in rats leads to a reduction in tonic GABA currents that persists into adulthood (Centanni et al., 2017). Intermittent alcohol exposure during early adulthood in mice has been shown to increase synaptic NMDA receptor-mediated currents (Kroener et al., 2012) and glutamate levels in the mPFC of mice (Hwa et al., 2015). These studies indicate that intermittent alcohol exposure shifts the balance of excitatory and inhibitory neurotransmission toward excitation. We propose that the reduction in Ih observed here might be a homeostatic response to increased excitatory activity (Shah et al., 2004) during cyclic alcohol exposure or perhaps to the direct actions of alcohol on HCN channels (Chen et al., 2012) or its effectors (Yoshimura and Tabakoff, 1995). In animal models of pathophysiology (e.g., hyperalgesia and epilepsy), experimental interventions that increase cortical activity during adolescence have also been shown to affect PN properties via a persistent decrease in Ih (Shah et al., 2004; Cordeiro Matos et al., 2015; Santello and Nevian, 2015).
An emerging hypothesis, supported by both human and animal studies, is that alcohol exposure during adolescence delays or arrests aspects of brain development, resulting in the persistence of an adolescent phenotype into adulthood (Crews et al., 2016). We observed that RMP and voltage sag continue to develop during postnatal age 28–62 d in the mouse, indicating developmental regulation of Ih during adolescence. These results complement observations in the hippocampus, where Hcn1 expression increases whereas Hcn2 and Hcn4 expression decreases from ages PD 21 to PD 56, indicating a shift from more cAMP-sensitive to cAMP-insensitive HCN channel expression (Bender et al., 2005; Seo et al., 2015). When comparing the distributions of PrL PFC PN measurements, we found evidence that a population of high-voltage sag neurons emerges during late adolescence. This finding is not surprising as subpopulations of high Ih PNs have been observed in other areas of the cortex as well as the adult mPFC (Dembrow et al., 2010), but the age dependence of this effect has not previously been reported in the mouse. The high-voltage sag population of mPFC PNs project preferentially to subcortical targets, such as the mediodorsal thalamus (Gee et al., 2012), and have an important role in maintaining and amplifying activity during working memory performance through bidirectional connectivity with the mPFC (Bolkan et al., 2017; Schmidt et al., 2017). PNs with high Ih measurements in the mPFC exhibit high levels of expression of dopamine D2 receptors and inputs from parvalbumin-positive interneurons (Lee et al., 2014). Both D2Rs and parvalbumin interneurons within the mPFC have well-established roles in working memory tasks and are influenced by alcohol exposure (Robbins and Arnsten, 2009; Trantham-Davidson et al., 2014; Murray et al., 2015). Ih would appear to represent another target of alcohol in the mPFC that could influence working memory, although it should be noted that there is disagreement regarding the role of Ih in working memory among animal models (Wang et al., 2007).
In conclusion, because the majority of adult individuals with AUDs initiate their excessive drinking during adolescence (Grant and Dawson, 1997), the identification of the neurobiological alterations arising from adolescent binge drinking is critical for understanding the etiology of AUDs. We conclude that voluntary alcohol consumption in mice alters the excitability of PrL PFC PNs, in part through a reduction in Ih, which could prevent them from entering the sustained neuronal activity necessary to perform working memory tasks. Adolescence is a vulnerable period during which excessive alcohol exposure may delay or arrest the normal developmental trajectory of intrinsic excitability in subpopulations of PNs of the PFC with higher levels of Ih. These findings suggest that cortical HCN channels may represent novel targets for treating the neurocognitive symptoms associated with adolescent binge drinking.
Footnotes
References
- Abernathy K, Chandler LJ, Woodward JJ (2010) Alcohol and the prefrontal cortex. Int Rev Neurobiol 91:289–320. 10.1016/S0074-7742(10)91009-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badanich KA, Mulholland PJ, Beckley JT, Trantham-Davidson H, Woodward JJ (2013) Ethanol reduces neuronal excitability of lateral orbitofrontal cortex neurons via a glycine receptor dependent mechanism. Neuropsychopharmacology 38:1176–1188. 10.1038/npp.2013.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeg EH, Kim YB, Huh K, Mook-Jung I, Kim HT, Jung MW (2003) Dynamics of population code for working memory in the prefrontal cortex. Neuron 40:177–188. 10.1016/S0896-6273(03)00597-X [DOI] [PubMed] [Google Scholar]
- Beck H, Yaari Y (2008) Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci 9:357–369. 10.1038/nrn2371 [DOI] [PubMed] [Google Scholar]
- Becker HC. (2013) Animal models of excessive alcohol consumption in rodents. Curr Top Behav Neurosci 13:355–377. 10.1007/7854_2012_203 [DOI] [PubMed] [Google Scholar]
- Bender RA, Galindo R, Mameli M, Gonzalez-Vega R, Valenzuela CF, Baram TZ (2005) Synchronized network activity in developing rat hippocampus involves regional hyperpolarization-activated cyclic nucleotide-gated (HCN) channel function. Eur J Neurosci 22:2669–2674. 10.1111/j.1460-9568.2005.04407.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, Harris AZ, Gordon JA, Kellendonk C (2017) Thalamic projections sustain prefrontal activity during working memory maintenance. Nat Neurosci 20:987–996. 10.1038/nn.4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AL, Chen Y, Bender RA, Yeh A, Shigemoto R, Baram TZ (2007) Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex 17:702–712. 10.1093/cercor/bhk021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Bravo-Rivera H, Quirk GJ (2013) Prelimbic and infralimbic neurons signal distinct aspects of appetitive instrumental behavior. PloS one 8:e57575. 10.1371/journal.pone.0057575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BJ. (2015) Beyond simple models of self-control to circuit-based accounts of adolescent behavior. Annu Rev Psychol 66:295–319. 10.1146/annurev-psych-010814-015156 [DOI] [PubMed] [Google Scholar]
- Casey BJ, Jones RM, Hare TA (2008) The adolescent brain. Ann N Y Acad Sci 1124:111–126. 10.1196/annals.1440.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centanni SW, Burnett EJ, Trantham-Davidson H, Chandler LJ (2017) Loss of δ-GABA receptor-mediated tonic currents in the adult prelimbic cortex following adolescent alcohol exposure. Addict Biol 22:616–628. 10.1111/adb.12353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu P, Fan X, Chen H, Yang J, Song T, Huang C (2012) Ethanol enhances human hyperpolarization-activated cyclic nucleotide-gated currents. Alcohol Clin Exp Res 36:2036–2046. 10.1111/j.1530-0277.2012.01826.x [DOI] [PubMed] [Google Scholar]
- Cordeiro Matos S, Zhang Z, Séguéla P (2015) Peripheral neuropathy induces HCN channel dysfunction in pyramidal neurons of the medial prefrontal cortex. J Neurosci 35:13244–13256. 10.1523/JNEUROSCI.0799-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crego A, Rodriguez-Holguín S, Parada M, Mota N, Corral M, Cadaveira F (2010) Reduced anterior prefrontal cortex activation in young binge drinkers during a visual working memory task. Drug Alcohol Depend 109:45–56. 10.1016/j.drugalcdep.2009.11.020 [DOI] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP, Broadwater MA, Robinson DL (2016) Adolescent alcohol exposure persistently impacts adult neurobiology and behavior. Pharmacol Rev 68:1074–1109. 10.1124/pr.115.012138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxson PL, Kyriazis DA, Baxter MG (2011) Cholinergic modulation of a specific memory function of prefrontal cortex. Nat Neurosci 14:1510–1512. 10.1038/nn.2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW (2004) Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev 28:771–784. 10.1016/j.neubiorev.2004.09.006 [DOI] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D (2010) Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci 30:16922–16937. 10.1523/JNEUROSCI.3644-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA (2002) Graded persistent activity in entorhinal cortex neurons. Nature 420:173–178. 10.1038/nature01171 [DOI] [PubMed] [Google Scholar]
- Gass JT, Glen WB Jr, McGonigal JT, Trantham-Davidson H, Lopez MF, Randall PK, Yaxley R, Floresco SB, Chandler LJ (2014) Adolescent alcohol exposure reduces behavioral flexibility, promotes disinhibition, and increases resistance to extinction of ethanol self-administration in adulthood. Neuropsychopharmacology 39:2570–2583. 10.1038/npp.2014.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS (2012) Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci 32:4959–4971. 10.1523/JNEUROSCI.5835-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Hernandez S, Garcia-Moreno LM (2016) Executive performance and dysexecutive symptoms in binge drinking adolescents. Alcohol 51:79–87. 10.1016/j.alcohol.2016.01.003 [DOI] [PubMed] [Google Scholar]
- Gläscher J, Adolphs R, Damasio H, Bechara A, Rudrauf D, Calamia M, Paul LK, Tranel D (2012) Lesion mapping of cognitive control and value-based decision making in the prefrontal cortex. Proc Natl Acad Sci U S A 109:14681–14686. 10.1073/pnas.1206608109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowin JL, Sloan ME, Stangl BL, Vatsalya V, Ramchandani VA (2017) Vulnerability for alcohol use disorder and rate of alcohol consumption. Am J Psychiatry 174:1094–1101. 10.1176/appi.ajp.2017.16101180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA (1997) Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the national longitudinal alcohol epidemiologic survey. J Subst Abuse 9:103–110. 10.1016/S0899-3289(97)90009-2 [DOI] [PubMed] [Google Scholar]
- Han Y, Noam Y, Lewis AS, Gallagher JJ, Wadman WJ, Baram TZ, Chetkovich DM (2011) Trafficking and gating of hyperpolarization-activated cyclic nucleotide-gated channels are regulated by interaction with tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) and cyclic AMP at distinct sites. J Biol Chem 286:20823–20834. 10.1074/jbc.M111.236125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Heuermann RJ, Lyman KA, Fisher D, Ismail QA, Chetkovich DM (2017) HCN-channel dendritic targeting requires bipartite interaction with TRIP8b and regulates antidepressant-like behavioral effects. Mol Psychiatry 22:458–465. 10.1038/mp.2016.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck JM, Feldstein Ewing SW (2018) Working memory capacity and addiction treatment outcomes in adolescents. Am J Drug Alcohol Abuse 44:185–192. 10.1080/00952990.2017.1344680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Nathanson AJ, Shimamoto A, Tayeh JK, Wilens AR, Holly EN, Newman EL, DeBold JF, Miczek KA (2015) Aggression and increased glutamate in the mPFC during withdrawal from intermittent alcohol in outbred mice. Psychopharmacology (Berl) 232:2889–2902. 10.1007/s00213-015-3925-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL (2008) Dorsomedial prefrontal cortex contribution to behavioral and nucleus accumbens neuronal responses to incentive cues. The J Neurosci 28:5088–5098. 10.1523/JNEUROSCI.0253-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kähkönen S, Wilenius J, Nikulin VV, Ollikainen M, Ilmoniemi RJ (2003) Alcohol reduces prefrontal cortical excitability in humans: a combined TMS and EEG study. Neuropsychopharmacology 28:747–754. 10.1038/sj.npp.1300099 [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER (2006) Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 49:603–615. 10.1016/j.neuron.2006.01.023 [DOI] [PubMed] [Google Scholar]
- Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC (1997) Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the national comorbidity survey. Arch Gen Psychiatry 54:313–321. 10.1001/archpsyc.1997.01830160031005 [DOI] [PubMed] [Google Scholar]
- Koskinen SM, Ahveninen J, Kujala T, Kaprio J, O'Donnell BF, Osipova D, Viken RJ, Näätänen R, Rose RJ (2011) A longitudinal twin study of effects of adolescent alcohol abuse on the neurophysiology of attention and orienting. Alcohol Clin Exp Res 35:1339–1350. 10.1111/j.1530-0277.2011.01470.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss WA, Belden CE, Hristov AD, Juraska JM (2014) Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse 68:61–72. 10.1002/syn.21716 [DOI] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ, Bonci A (2015) Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci 16:173–184. 10.1038/nrn3877 [DOI] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ (2012) Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One 7:e37541. 10.1371/journal.pone.0037541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, Sohal VS (2014) Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron 81:61–68. 10.1016/j.neuron.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundqvist M, Rose J, Herman P, Brincat SL, Buschman TJ, Miller EK (2016) Gamma and beta bursts underlie working memory. Neuron 90:152–164. 10.1016/j.neuron.2016.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MS, Amodeo LR, Roitman JD (2016) Consequences of adolescent ethanol consumption on risk preference and orbitofrontal cortex encoding of reward. Neuropsychopharmacology 41:1366–1375. 10.1038/npp.2015.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI. (2011) Intermittent (every-other-day) drinking induces rapid escalation of ethanol intake and preference in adolescent and adult C57BL/6J mice. Alcohol Clin Exp Res 35:652–658. 10.1111/j.1530-0277.2010.01383.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001) An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24:167–202. 10.1146/annurev.neuro.24.1.167 [DOI] [PubMed] [Google Scholar]
- Moser DA, Doucet GE, Ing A, Dima D, Schumann G, Bilder RM, Frangou S (2017) An integrated brain-behavior model for working memory. Mol Psychiatry. Advance online publication. Retrieved December 11, 2017. doi: 10.1038/mp.2017.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Oehring EM, Kwon D, Nagel BJ, Sullivan EV, Chu W, Rohlfing T, Prouty D, Nichols BN, Poline JB, Tapert SF, Brown SA, Cummins K, Brumback T, Colrain IM, Baker FC, De Bellis MD, Voyvodic JT, Clark DB, Pfefferbaum A, Pohl KM (2018) Influences of age, sex, and moderate alcohol drinking on the intrinsic functional architecture of adolescent brains. Cereb Cortex 28:1049–1063. 10.1093/cercor/bhx014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Woloszynowska-Fraser MU, Ansel-Bollepalli L, Cole KL, Foggetti A, Crouch B, Riedel G, Wulff P (2015) Parvalbumin-positive interneurons of the prefrontal cortex support working memory and cognitive flexibility. Sci Rep 5:16778. 10.1038/srep16778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D (2007) Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56:1061–1075. 10.1016/j.neuron.2007.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, Lopez MF, Mulholland PJ, Woodward JJ (2016) Chronic intermittent ethanol exposure enhances the excitability and synaptic plasticity of lateral orbitofrontal cortex neurons and induces a tolerance to the acute inhibitory actions of ethanol. Neuropsychopharmacology 41:1112–1127. 10.1038/npp.2015.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon K, McClain JA (2010) Adolescence as a critical window for developing an alcohol use disorder: current findings in neuroscience. Curr Opin Psychiatry 23:227–232. 10.1097/YCO.0b013e32833864fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A (2003) The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115:551–564. 10.1016/S0092-8674(03)00884-5 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2004) The mouse brain in stereotaxic coordinates, Ed 2 Amsterdam, the Netherlands: Elsevier Academic. [Google Scholar]
- Pfefferbaum A, Kwon D, Brumback T, Thompson WK, Cummins K, Tapert SF, Brown SA, Colrain IM, Baker FC, Prouty D, De Bellis MD, Clark DB, Nagel BJ, Chu W, Park SH, Pohl KM, Sullivan EV (2018) Altered brain developmental trajectories in adolescents after initiating drinking. Am J Psychiatry 175:370–380. 10.1176/appi.ajp.2017.17040469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME, Kesner RP (1998) The effects of muscarinic cholinergic receptor blockade in the rat anterior cingulate and prelimbic/infralimbic cortices on spatial working memory. Neurobiol Learn Mem 69:241–257. 10.1006/nlme.1998.3823 [DOI] [PubMed] [Google Scholar]
- Rau AR, Chappell AM, Butler TR, Ariwodola OJ, Weiner JL (2015) Increased basolateral amygdala pyramidal cell excitability may contribute to the anxiogenic phenotype induced by chronic early-life stress. J Neurosci 35:9730–9740. 10.1523/JNEUROSCI.0384-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riga D, Matos MR, Glas A, Smit AB, Spijker S, Van den Oever MC (2014) Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci 8:230. 10.3389/fnsys.2014.00230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley MR, Constantinidis C (2015) Role of prefrontal persistent activity in working memory. Front Syst Neurosci 9:181. 10.3389/fnsys.2015.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Arnsten AF (2009) The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu Rev Neurosci 32:267–287. 10.1146/annurev.neuro.051508.135535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, Harrison NL (2014) Strychnine-sensitive glycine receptors on pyramidal neurons in layers II/III of the mouse prefrontal cortex are tonically activated. J Neurophysiol 112:1169–1178. 10.1152/jn.00714.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, Faccidomo SP, Li C, Psilos K, Galunas C, Spanos M, Agoglia AE, Kash TL, Hodge CW (2016) Moderate alcohol drinking and the amygdala proteome: identification and validation of calcium/calmodulin dependent kinase II and AMPA receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol Psychiatry 79:430–442. 10.1016/j.biopsych.2014.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Nevian T (2015) Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron 86:233–246. 10.1016/j.neuron.2015.03.003 [DOI] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA (2009) TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron 62:802–813. 10.1016/j.neuron.2009.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler AG, Soden ME, Zweifel LS, Clark JJ (2016) Reversal of alcohol-induced dysregulation in dopamine network dynamics may rescue maladaptive decision-making. J Neurosci 36:3698–3708. 10.1523/JNEUROSCI.4394-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SL, Dorsett CR, Iyengar AK, Fröhlich F (2017) Interaction of intrinsic and synaptic currents mediate network resonance driven by layer V pyramidal cells. Cereb Cortex 27:4396–4410. 10.1093/cercor/bhw242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, Seol MJ, Lee K (2015) Differential expression of hyperpolarization-activated cyclic nucleotide-gated channel subunits during hippocampal development in the mouse. Mol Brain 8:13. 10.1186/s13041-015-0103-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM. (2014) Cortical HCN channels: function, trafficking and plasticity. J Physiol 592:2711–2719. 10.1113/jphysiol.2013.270058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D (2004) Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44:495–508. 10.1016/j.neuron.2004.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squeglia LM, Pulido C, Wetherill RR, Jacobus J, Brown GG, Tapert SF (2012) Brain response to working memory over three years of adolescence: influence of initiating heavy drinking. J Stud Alcohol Drugs 73:749–760. 10.15288/jsad.2012.73.749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squeglia LM, Jacobus J, Tapert SF (2014) The effect of alcohol use on human adolescent brain structures and systems. Handb Clin Neurol 125:501–510. 10.1016/B978-0-444-62619-6.00028-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman S, Skara S, Ames SL (2008) Substance abuse among adolescents. Subst Use Misuse 43:1802–1828. 10.1080/10826080802297302 [DOI] [PubMed] [Google Scholar]
- Thuault SJ, Malleret G, Constantinople CM, Nicholls R, Chen I, Zhu J, Panteleyev A, Vronskaya S, Nolan MF, Bruno R, Siegelbaum SA, Kandel ER (2013) Prefrontal cortex HCN1 channels enable intrinsic persistent neural firing and executive memory function. J Neurosci 33:13583–13599. 10.1523/JNEUROSCI.2427-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townshend JM, Duka T (2005) Binge drinking, cognitive performance and mood in a population of young social drinkers. Alcohol Clin Exp Res 29:317–325. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, Burnett EJ, Gass JT, Lopez MF, Mulholland PJ, Centanni SW, Floresco SB, Chandler LJ (2014) Chronic alcohol disrupts dopamine receptor activity and the cognitive function of the medial prefrontal cortex. J Neurosci 34:3706–3718. 10.1523/JNEUROSCI.0623-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Kroener S, Abernathy K, Lapish C, Seamans J, Chandler LJ, Woodward JJ (2007) Ethanol inhibits persistent activity in prefrontal cortical neurons. J Neurosci 27:4765–4775. 10.1523/JNEUROSCI.5378-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas WM, Bengston L, Gilpin NW, Whitcomb BW, Richardson HN (2014) Alcohol binge drinking during adolescence or dependence during adulthood reduces prefrontal myelin in male rats. J Neurosci 34:14777–14782. 10.1523/JNEUROSCI.3189-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF (2007) Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 129:397–410. 10.1016/j.cell.2007.03.015 [DOI] [PubMed] [Google Scholar]
- Wang XJ. (2001) Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci 24:455–463. 10.1016/S0166-2236(00)01868-3 [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Woodward JJ (2008) Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol Clin Exp Res 32:690–698. 10.1111/j.1530-0277.2008.00625.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, Lovinger DM, Alvarez VA (2014) Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology 39:579–594. 10.1038/npp.2013.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Tabakoff B (1995) Selective effects of ethanol on the generation of cAMP by particular members of the adenylyl cyclase family. Alcohol Clin Exp Res 19:1435–1440. 10.1111/j.1530-0277.1995.tb01004.x [DOI] [PubMed] [Google Scholar]