

Abstract
Nerve injury-induced hyperactivity of primary sensory neurons in the dorsal root ganglion (DRG) contributes to chronic pain development, but the underlying epigenetic mechanisms remain poorly understood. Here we determined genome-wide changes in DNA methylation in the nervous system in neuropathic pain. Spinal nerve ligation (SNL), but not paclitaxel treatment, in male Sprague Dawley rats induced a consistent low-level hypomethylation in the CpG sites in the DRG during the acute and chronic phases of neuropathic pain. DNA methylation remodeling in the DRG occurred early after SNL and persisted for at least 3 weeks. SNL caused DNA methylation changes at 8% of CpG sites with prevailing hypomethylation outside of CpG islands, in introns, intergenic regions, and repetitive sequences. In contrast, SNL caused more gains of methylation in the spinal cord and prefrontal cortex. The DNA methylation changes in the injured DRGs recapitulated developmental reprogramming at the neonatal stage. Methylation reprogramming was correlated with increased gene expression variability. A diet deficient in methyl donors induced hypomethylation and pain hypersensitivity. Intrathecal administration of the DNA methyltransferase inhibitor RG108 caused long-lasting pain hypersensitivity. DNA methylation reprogramming in the DRG thus contributes to nerve injury-induced chronic pain. Restoring DNA methylation may represent a new therapeutic approach to treat neuropathic pain.
SIGNIFICANCE STATEMENT Epigenetic mechanisms are critically involved in the transition from acute to chronic pain after nerve injury. However, genome-wide changes in DNA methylation in the nervous system and their roles in neuropathic pain development remain unclear. Here we used digital restriction enzyme analysis of methylation to quantitatively determine genome-wide DNA methylation changes caused by nerve injury. We showed that nerve injury caused DNA methylation changes at 8% of CpG sites with prevailing hypomethylation outside of CpG islands in the dorsal root ganglion. Reducing DNA methylation induced pain hypersensitivity, whereas increasing DNA methylation attenuated neuropathic pain. These findings extend our understanding of the epigenetic mechanism of chronic neuropathic pain and suggest new strategies to treat nerve injury-induced chronic pain.
Keywords: chemotherapy, DNA methylation, dorsal root ganglion, neuropathic pain, neuroepigenetics
Introduction
Chronic pain costs ~$635 billion each year in medical treatment and lost productivity in the United States alone (Institute of Medicine of the National Academies, 2011). Neuropathic pain is a complex, chronic pain state that is usually caused by damage to the nervous system. Current treatments for neuropathic pain are poorly effective because of the limited understanding of the molecular mechanism involved. A well defined characteristic of neuropathic pain is the prolonged hyperactivity of damaged primary afferent nerves and sensory neurons located in the dorsal root ganglion (DRG; Pan et al., 1999; Liu et al., 2000). The establishment of this long-term hyperactivity is upheld by sustained changes in the expression levels of many genes, which suggests the involvement of global epigenetic changes (Xiao et al., 2002; Hammer et al., 2010). We and others have demonstrated that epigenetic mechanisms are critically involved in chronic pain development after nerve injury (Matsushita et al., 2013; Laumet et al., 2015). However, most studies focus on post-translational histone modifications, whereas the changes in DNA methylation in injured DRGs during chronic neuropathic pain are not well defined.
Control of chromatin accessibility for gene expression by DNA methylation is vital during the development and cell type specificity of different tissues. DNA methylation is crucial during the differentiation of the nervous system and also has a relevant role in the function of adult postmitotic neurons (Guo et al., 2011; Sharma et al., 2016). Aberrant cytosine methylation at CpG sites may result in the dysregulation of expression of affected genes. Altered DNA methylation has been linked to memory formation, drug addiction, and several neurodegenerative diseases (Morris et al., 2014; Massart et al., 2015; Sanchez-Mut et al., 2016). Nerve injury induces DNA methylation changes in the DRG (Gölzenleuchter et al., 2015). However, the analysis was performed only 24 h after nerve injury, which predominantly reflects an acute pain state. Little is known about long-lasting changes in DNA methylation in the DRG during acute-to-chronic pain transition.
In the present study, we used a highly quantitative digital restriction enzyme analysis of methylation (DREAM; Jelinek et al., 2012; Jelinek and Madzo, 2016) to determine genome-wide changes in DNA methylation in the nervous system caused by peripheral nerve injury. We showed that nerve injury, but not chemotherapy, primarily caused consistent low-level hypomethylation in the CpG sites in the DRG during the acute and chronic phases of neuropathic pain. The CpG sites affected by these changes were located mainly outside of CpG islands, in introns, intergenic regions, and repetitive sequences. Also, reducing DNA methylation levels using a DNA methyltransferase inhibitor or methyl group donor-deficient diets caused pain hypersensitivity. This new information extends our understanding of epigenetic mechanisms underlying neuropathic pain development and suggests new strategy to treat nerve injury-induced chronic pain.
Materials and Methods
Animal models of neuropathic pain and intrathecal cannulation.
All experimental protocols and procedures used in this study were approved by the Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Spinal nerve ligation (SNL) in male Sprague Dawley rats (8–9 weeks old, ~220 g; Harlan Sprague Dawley) was used for this study as the neuropathic pain model. Animals were anesthetized with 2–3% isoflurane, and the left L5 and L6 spinal nerves were isolated and ligated tightly with 4.0 silk sutures under a surgical microscope, as described previously (Kim and Chung, 1992; Chen et al., 2000). Sham surgery (without nerve ligation) was performed on the contralateral side as the control.
Also, chemotherapy-induced peripheral neuropathy was used as another neuropathic pain model. Four intraperitoneal injections of the chemotherapeutic agent paclitaxel (2 mg/kg, every 2 d) were performed (Polomano et al., 2001; Chen et al., 2014b), and rats treated with vehicle (cremophor ethanol 1:1) were used as the control group. Both neuropathic pain models consistently induce pain hypersensitivity lasting for at least 4 weeks.
DNA methyltransferase (DNMT) inhibitor N-phthaloyl-l-tryptophan (RG108, Tocris Bioscience) was administered intrathecally. RG108 is cell permeable (Brueckner et al., 2005) and has been shown to inhibit DNMTs in neurons in vivo (Guo et al., 2011). Under anesthesia with 2% isoflurane, an intrathecal catheter (PE-10 tubing) was surgically inserted through an incision made in the cisternal membrane. The catheter was directed to the lumbar region of the spinal cord, and the external opening was sealed (Yaksh and Rudy, 1976). Rats displaying signs of the motor or neurological dysfunction were excluded and terminated immediately with an overdose of phenobarbital (200 mg/kg, i.p.). The animals were allowed to recover for 5 d before intrathecal injections. Ten micrograms of RG108 or vehicle (10% DMSO; saline) were injected intrathecally in a 5 μl volume followed by a 10 μl saline flush, as previously described (Laumet et al., 2015). The dose was selected based on data in a previous publication (Chestnut et al., 2011). Animals received intrathecal injections for 4 consecutive days.
Behavioral assessment of tactile allodynia and hyperalgesia in rats.
To quantify tactile allodynia, rats were placed in individual plastic boxes on a mesh floor and allowed to acclimate for 30–45 min. A series of calibrated von Frey filaments was applied perpendicularly to the plantar surface of the hindpaw with sufficient force to bend the filaments for 6 s, and brisk paw withdrawals or flinching was considered as a positive response. In the absence of a response, the filament of next greater force was applied. If a response occurred, the filament of next lower force was applied. The tactile stimulus producing a 50% likelihood of withdrawal was determined using the “up-down” calculating method (Chaplan et al., 1994).
We measured mechanical hyperalgesia in rats by testing the withdrawal threshold in response to a noxious pressure stimulus (Randall-Selitto test) using the paw-pressure Analgesy-Meter (Ugo Basile). When the animal displayed pain by either withdrawing the paw or vocalizing, the pedal was immediately released, and the nociceptive withdrawal threshold was read on the scale (Laumet et al., 2015; Xie et al., 2016). The investigator (G.L.) performing behavioral experiments was blinded to the treatments.
Modified-diet treatment.
To manipulate DNA methylation levels, we altered the intake of methyl group donors (folate and vitamin B12) by feeding the rats with modified diets. The original diet AIN-93M (normal folate dose, 2.1 ppm; vitamin B12 dose, 28 μg/kg) was modified by Purina Animal Nutrition LLC Technical Services to make a folate-deficient and vitamin B12-deficient diet (0.1 ppm and 3 μg/kg, respectively) and a diet five times enriched in folate and vitamin B12 (10.5 ppm and 140 μg/kg, respectively). Five-week-old rats (weight, ~120 g) were randomly placed in new cages with ad libitum regular, deficient, and enriched diets, respectively. The animals were kept on the diets for 4 weeks before SNL surgery, followed by 3 weeks after SNL. Behavioral tests were performed every 3–4 d during the diet feeding period.
Neuronal nuclear enrichment.
To prepare a neuronal nuclear enrichment, fresh whole DRG samples were cut and mashed onto an ice-cold Petri dish. Mashed tissue from 3 DRGs was pooled in one 1.5 ml tube. The tissue was resuspended in buffer NAME (20 mm Tris, pH 8; 20 mm KCl; and 1.5 mm MgCl2) with a cocktail of protease and phosphatase inhibitors (Thermo Fisher Scientific), 0.5 mm DTT, and 1 mm PMSF (Sigma-Aldrich) to obtain a single cell suspension. Cells were incubated on ice for 5 min before being centrifuged at 3000 rpm at 4°C for 2 min. The pellet was resuspended in the buffer NAME containing 0.15% NP-40. Cells were incubated on a rotator overnight to isolate nuclei. Neuronal nuclei were immunoprecipitated using anti-Fox3 (neuronal nuclear marker; catalog #104225, Abcam) in PBS, and genomic DNA extraction was performed. Neuronal nuclei were lysed with lysis buffer (0.1 m Tris, pH 8, 5 mm EDTA, 0.2% SDS, 0.2 m NaCl, and proteinase K) overnight and precipitated with isopropanol. Genomic DNA pellet was washed with 75% ethanol, dried, and resuspended in TE buffer (Tris 10 mm, pH 8, and 1 mm EDTA). The isolation purity was validated by Western blot analysis. Nuclear proteins were extracted from one batch. Cytoplasm/nucleus separation was validated by Western blot using anti-histone H3 (1/4000; catalog #9715, Cell Signaling Technology), and neuronal enrichment was validated by Western blot using the neuronal marker anti-Fox3 (catalog #104225, Abcam) and the satellite/glia markers anti-glutamine synthetase (1/200; catalog #MAB302, Millipore) and anti-GFAP (1/4000; catalog #7260, Abcam).
DNA methylation analysis.
Rats were deeply anesthetized with 5% isoflurane, and then the brain, spinal cord, and ligated or Sham-operated DRG tissues at the L5 and L6 levels were rapidly harvested. We used the DREAM method to quantify methylation status at tens of thousands CpG sites across the genome (Jelinek et al., 2012; Jelinek and Madzo, 2016). In brief, genomic DNA extracted from DRG samples was spiked with methylation standards and sequentially digested with two restriction endonucleases recognizing CCCGGG sites in DNA. SmaI cleaved only unmethylated sites, resulting in blunt-ended fragments starting with 5′-GGG. Next, XmaI cleaved the remaining methylated sites creating 5′-CmeCGG overhang sequences. Exonuclease-deficient Klenow fragment DNA polymerase was then used to fill in the 3′ recesses left by XmaI and add 3′-dA overhangs to the ends of all fragments. Thus, specific signatures 5′-GGG and 5′-CCGGG were created at unmethylated and methylated CpG sites, respectively. Illumina sequencing adapters were then ligated to the enzyme-treated DNA; sequencing libraries were generated according to Illumina protocols and run on Illumina HiSeq 2000 and 2500 instruments at the Fox Chase Cancer Center Genomics facility. Sequencing data were aligned to the rat genome (rn4), and the reads with unmethylated and methylated signatures were counted at individual CCCGGG sites. Methylation levels were calculated as the ratio of reads with the methylated signature to all reads mapping to each respective site. DNA methylation values at individual CpG sites were adjusted based on spike-in standards and filtered for the minimum sequencing depth of 100 reads. We deposited DNA methylation DREAM data into the Gene Expression Omnibus database under accession number GSE100641.
Reduced representation bisulfite sequencing.
Genomic DNA extracted from triplicate samples of DRGs 3 weeks after SNL or Sham operation was analyzed for DNA methylation changes by reduced representation bisulfite sequencing (RRBS; Gu et al., 2011) to validate DREAM results by an orthogonal method. We followed the NEB protocol for methylated adaptors. Briefly, 1 μg of genomic DNA was spiked 100 pg of lambda phage DNA as the unmethylated standard and digested with MspI. Ends of restriction fragments were filled in, 3′-dA was tailed, and methylated adaptors (catalog #E7535, NEB) were ligated to the ends of restriction fragments. Bisulfite treatment using the Epitect kit (Qiagen) followed. Bisulfite-converted libraries were amplified using EpiMark TaqDNA polymerase (NEB) and primers with barcode indices. The libraries were pooled and sequenced at Fox Chase Cancer Center Genomics Facility on an Illumina HiSeq2500 instrument using single end reads of 50 bases. We used Bismark version 0.18.1 (Krueger and Andrews, 2011) to align the sequences to the Rattus norvegicus rn4 genome assembly. We used methylKit version 1.3.3 (Akalin et al., 2012) to analyze differential methylation. We deposited RRBS data to the Gene Expression Omnibus database under accession number GSE114865.
RNA sequencing.
We harvested DRGs from deeply anesthetized rats and isolated total RNA using RNeasy (Qiagen). We made RNA libraries from 1 μg of total RNA using stranded total RNA plus Ribo-zero kits (Illumina). The sequencing was performed at the Fox Chase Cancer Center Genomics Core Facility. The detailed method has been described previously (Laumet et al., 2015). RNA sequence data are deposited in the Gene Expression Omnibus database under accession number GSE59043.
Statistical analysis.
We used one-way or two-way repeated ANOVA to analyze behavioral data for statistical differences (Prism, GraphPad Software). Statistical calculations for DNA methylation and RNA-sequencing (RNA-seq) data were performed using the R suite (R Development Core Team, 2017). Unsupervised hierarchical clustering was performed using the pvclust package (Suzuki and Shimodaira, 2006). Differences in methylation means and t test p values were used for Volcano plots. We used Fisher's exact test to calculate the odds ratios. The statistical significance of multiset intersections was computed using the SuperExactTest package (Wang et al., 2015). To compare the variation in expression and methylation level, we used two tests: a Levene's test, which was used to assess the equality of variances between the two groups with stable or variable methylation. We performed overrepresentation analysis (ORA) for genes associated with methylation or expression changes. We used the ConsensusPathDB on-line tool (Kamburov et al., 2009; Herwig et al., 2016) and gene set enrichment analysis (GSEA) software version 3.0, and Molecular Signature Database (MSigDB) version 6.0 (Subramanian et al., 2005; http://software.broadinstitute.org/gsea/index.jsp) to identify enriched pathways and gene ontology (GO) sets.
Results
Peripheral nerve injury induces DNA methylation reprogramming in DRGs
Pain hypersensitivity induced by SNL in rats occurs immediately after nerve injury and persists as a chronic pain state for at least 6 weeks. We first examined DNA methylation in DRGs at 3 d after SNL to detect early epigenetic changes. We used the highly quantitative DREAM method to determine genome-wide DNA methylation patterns. We observed changes in DNA methylation at >1000 CpG sites. Overall, 4.6% of analyzed CpG sites showed gains of methylation while 3% of sites showed losses of methylation, predominantly those sites with high levels of baseline methylation (Fig. 1A).
Figure 1.

DNA methylation changes in DRGs after SNL. A, B, Analysis of 6 SNL or Sham-operated pairs of DRGs 3 d (A) and 3 weeks (B) after surgery. Volcano plots show CpG sites differentially methylated after SNL, defined by a difference from Sham of >2% and a p value of <0.05. Dark blue dots show hypomethylated sites. Orange dots show hypermethylated sites. Percentage of differentially methylated sites is shown in parentheses. C, A scatterplot shows a high correlation of methylation differences detected at 3 d and at 3 weeks after SNL (Pearson's correlation r = 0.62; 95% CI, 0.61–0.63; t = 95.2; df = 14,566; p < 2 × 10−16). The red line shows a linear regression. D, Venn diagrams show significant overlaps of CpG sites hypomethylated (blue, hypergeometric test p = 2 × 10−102) or hypermethylated (orange, hypergeometric test p = 4 × 10−108) at 3 d (d3) and 3 weeks (w3), respectively. E, CpG sites with methylation changes 3 weeks after SNL were rarely located in CpG islands, shores, promoters, and exons. There were more likely to be located in introns, intergenic regions, and repeats. Horizontal lines show 95% CIs of odds ratios of variable/stable methylation (Fisher's exact test). Validation of the methylation changes after SNL is shown in Figure 1-1.
Prevailing DNA hypomethylation in DRGs 3 weeks after spinal nerve ligation (SNL). Related to Fig. 1. DNA methylation changes in DRGs 3 weeks after SNL in three experiments, each comparing 3 SNL/Sham pairs. (A, B) DNA methylation was analyzed by the DREAM assay. Dark blue dots show hypomethylated sites. Orange dots show hypermethylated sites. Percentage of differentially methylated sites is shown in parentheses. (C) Correlation of methylation differences in two representative experiments at 17869 CpG sites. Red solid line shows linear regression (p-value < 2e-16). (D) DNA methylation analyzed by the RRBS assay. Aquamarine blue dots show hypomethylated sites. Magenta dots show hypermethylated sites. (E) Hypomethylated CpG sites are distributed on all chromosomes. (F) RRBS and DREAM assays detected distinct sets of CpG sites with an overlap of 0.3%. Methylation values at CpG sites detected by both assays were highly concordant (linear regression R^2=0.98). Download Figure 1-1, TIF file (2.1MB, tif)
We next focused on methylation reprogramming at the chronic pain stage 3 weeks after SNL. In the first experiment, we obtained DRGs from SNL and Sham surgery sides in six individual rats and observed significant DNA methylation changes in 4.5% of sites. In contrast to the early stage, loss of methylation (i.e., hypomethylation) prevailed with 3.6% of CpG sites, and only 0.9% sites were gaining methylation (Fig. 1B). We repeated this experiment two additional times and obtained similar results (Fig. 1-1A,B). To address whether the global changes observed consistently occurred at the same CpG sites in the different experiments, we compared the two experiments with the highest number of paired samples and found that the methylation differences were highly correlated (linear regression R2 = 0.47, p < 2 × 10−16; Fig. 1-1C). To increase the statistical power, we pooled the three experiments and analyzed a total of 15 animals with high-quality data (Fig. 2A). We confirmed prevailing hypomethylation that affected 6.5% of examined CpG sites. Simultaneously, we noted gains of methylation in 1.4% of analyzed CpG sites.
Figure 2.

A–C, DNA methylation changes in DRGs (A), spinal cord (B), and prefrontal cortex (C) 3 weeks after SNL. Volcano plots show hypomethylated CpG sites in blue and hypermethylated sites in orange. CpG sites differentially methylated after SNL were defined by a difference from Sham of >2% and a t-test p value of <0.05. D, A bar chart illustrating all possible intersections among CpG sites differentially methylated in prefrontal cortex (PFC), spinal cord (SC), and DRGs. The matrix of solid and empty circles at the bottom illustrates the “presence” (solid green) or “absence” (empty) of the datasets in each intersection. The numbers to the right of the matrix are set sizes. The colored bars represent the intersection sizes with the color intensity showing the p value significance. DNA methylation changes after paclitaxel treatment are shown in Figure 2-1.
DNA methylation changes after paclitaxel treatment are minimal and distinct from those after SNL. Related to Fig. 2. (A) DNA methylation changes at 15807 CpG sites in DRGs from three paclitaxel-treated and three control rats. (B) DNA methylation changes at 15559 CpG sites in dorsal horns of the spinal cord from two paclitaxel-treated and three control rats. Dark blue dots show hypomethylated sites. Orange dots show hypermethylated sites. Percentage of differentially methylated sites is shown in parentheses. Inverse correlation of DNA methylation changes after paclitaxel treatment and SNL in (C) DRGs (Pearson's correlation r = -0.35, t = -39.347, df = 10977, p-value < 2.2e-16) and (D) the spinal cord (Pearson's correlation r = -0.17, t = -20.497, df = 13257, p-value < 2.2e-16). Download Figure 2-1, TIF file (1.1MB, tif)
Finally, we validated the observation of prevailing hypomethylation in DRGs after SNL by an orthogonal method, RRBS. Using the same criteria as for the restriction enzyme-based DREAM method, we quantified methylation at 32,688 CpG sites. We observed hypomethylation at 3659 (11.2%) sites and hypermethylation at 963 (2.9%) sites (Fig. 1-1D). We found hypomethylation changes after SNL to be distributed globally across all chromosomes (Fig. 1-1E). Since there was only a small overlap detected with the ≥100 read coverages read by both methods, the RRBS results represented an additional dataset of independent CpG sites validating the tendency to hypomethylation after SNL observed by the DREAM data. The overlapping sites showed an excellent correlation of methylation values measured by both methods (R2 = 0.98; Fig. 1-1F). Thus, chronic pain is associated with a complex methylation reprogramming that affects up to 14% of the genome and is characterized predominantly by the loss of methylation at DRGs.
We note that, while highly statistically significant, the methylation changes are relatively small (average loss of 1.7% for hypomethylated sites). There was a significant correlation between the degree of change at 3 d and at 3 weeks after SNL [Pearson's correlation r = 0.62; 95% confidence interval (CI), 0.61–0.63; t = 95.2; df = 14,566; p < 2 × 10−16; Fig. 1C], and a substantial overlap between the sites changed at 3 d and those changed at 3 weeks (hypergeometric test, p < 10−100; Fig. 1D). When comparing week 3 to day 3, the dominant pattern seems to be fewer sites with gains of methylation and many more sites with losses of methylation. Thus, the reshaping of the methylome starts early after SNL with both gains and losses of methylation, but the chronic pain phase is characterized by expansion of the hypomethylation defect.
The effects of DNA methylation on gene expression vary depending on the location of the CpG sites affected (Jones, 2012). Therefore, we mapped the CpG sites that changed 3 weeks after SNL and compared their locations to CpG sites with stable methylation (Fig. 1E). The variable CpG sites were significantly less likely to be in CpG islands (odds ratio, 0.03; 95% CI, 0.02–0.05), promoters (odds ratio, 0.07; 95% CI, 0.05–0.1), CpG island shores (odds ratio, 0.43; 95% CI, 0.36–0.51), and exons (odds ratio, 0.56; 95% CI, 0.47–0.67). The variable CpG sites were more often in introns (odds ratio, 1.2; 95% CI, 1.1–1.3), intergenic regions (odds ratio, 7.4; 95% CI, 6.5–8.3), and repeats (odds ratio, 1.5; 95% CI, 1.3–1.8).
Peripheral nerve injury induces DNA methylation reprogramming in the spinal cord and cortex
To determine whether SNL-induced DNA methylation changes are limited to DRGs, we performed DREAM analysis using spinal cord and prefrontal cortex tissues 3 weeks after SNL. As shown in Figure 2, we observed DNA methylation changes in all tissues with qualitative and quantitative differences compared with DRGs. As opposed to the substantial bias toward hypomethylation in DRGs, we observed more gains of methylation in the spinal cord and prefrontal cortex. While progressively fewer loci were affected, there was a highly significant overlap of differentially methylated CpG sites in all three tissues (super exact test, p = 10−21; Fig. 2D).
Chemotherapy-induced painful neuropathy causes minimal changes in DNA methylation
We next asked whether DNA methylation changes caused by SNL would also occur in a different neuropathic pain model. We chose the rat model of chemotherapy-induced painful neuropathy caused by paclitaxel treatment, which induces prolonged pain hypersensitivity (Polomano et al., 2001). In contrast to the traumatic nerve injury model, paclitaxel treatment caused minimal changes in DNA methylation (<2% of CpG sites) in the DRG and spinal cord (Fig. 2-1A,B). DNA methylation changes in DRGs after SNL and after paclitaxel treatment were inversely correlated (Pearson r = −0.35; p < 2e-16; Fig. 2-1C). We also saw a similar inverse correlation between methylation changes after paclitaxel treatment and SNL in the spinal cord (Pearson r = −0.17; p < 2e-16; Fig. 2-1D). There was no statistically significant overlap of genes associated with either hypomethylated or hypermethylated CpG sites between SNL and paclitaxel treatment.
Nerve injury-induced DNA methylation changes in DRGs recapitulate developmental reprogramming
DNA methylation shows significant tissue and cellular specificity, and it is possible that the changes we observed in DRGs were caused by a shift in the cellular composition (e.g., increased satellite cells and/or infiltration of inflammatory cells). Therefore, we sought to determine whether the methylation changes observed in DRGs corresponded to methylation changes occurring in neurons by analyzing DNA obtained from neuronal nuclei captured using the anti-Fox3 antibody 3 weeks after nerve ligation (Fig. 3-1). Difficulties in purifying enough nuclei for DREAM analysis limited the number of samples we could study. Nevertheless, an analysis of five samples (two “input” mixed cellular populations and three neuron-enriched pools) showed that samples from the SNL clustered together and away from samples from the Sham group (Fig. 3A). In this analysis, the neuron-enriched sample clustered very close to the unpurified DRGs, suggesting that injured DRG neurons likely undergo the same DNA methylation changes as detected in the whole DRGs.
Figure 3.

DNA methylation changes in DRGs after SNL recapitulate normal development. A, Unsupervised hierarchical clustering of DNA methylation values shows that total DRGs and purified DRG neurons cluster together, while Sham and SNL cluster separately. B, Developmental DNA methylation differences between neonatal and adult DRGs. C, Overlap of DNA methylation changes in development and after SNL. D, Top gene ontology terms enriched for methylation changes at genes common to SNL and development (blue, hypomethylated genes; orange, hypermethylated genes). Neuronal isolation Western blots are shown in Figure 3-1.
Validation of DRG neuronal isolation by western blots. Related to Fig. 3. (A) Nuclei were isolated from a pool of DRGs as evidenced by immunolabeling of histone 3 by western blots. (B) Neuronal nuclei were separated by immunoprecipitation. The separation of neuronal and non-neuronal nuclei were confirmed immunolabeling of neuronal marker Neuronal Nuclei (NeuN) and glial markers Glial fibrillary acidic protein (GFAP) and glutamine synthetase (GS). Western blots were imaged and quantified by infrared imaging system. Download Figure 3-1, TIF file (444.1KB, tif)
DNA methylation reprogramming is critically involved in neuronal development. Having shown that the methylation changes occur in injured DRG neurons, we asked whether this epigenetic change reflected reprogramming at an early developmental stage. We analyzed DNA methylation in DRGs obtained from neonatal and adult animals. Striking differences in DNA methylation were observed (Fig. 3B), affecting 17% of the methylome. Both increases and decreases in methylation occurred during development, and there was a significant overlap (hypergeometric test, p = 4 × 10−108) between the development-associated changes and those that occurred 3 weeks after SNL (Fig. 3C). Thus, DNA methylation remodeling associated with the chronic pain reflects partial reprogramming of the DRG neurons along pathways that recapitulate normal development.
CpG sites hypomethylated after SNL were associated with 788 genes, and sites hypomethylated in development were associated with 776 genes. The overlap was 262 genes (p = 10−46). ORA for the overlapping genes revealed “nervous system development” as the top GO category. CpG sites hypermethylated after SNL were associated with 167 genes, and sites hypermethylated in development were associated with 599 genes. The overlap was 62 genes (p = 1.4 × 10−9). ORA for the overlapping genes revealed “muscle organ development” as the top GO category (Fig. 3D).
Nerve injury-induced methylation reprogramming correlates with increased gene expression variability in DRGs
Gene expression in DRGs after SNL analyzed by RNA-seq showed 1684 genes upregulated (10%) and 1039 genes downregulated (6%; Fig. 4A). Downregulated genes were enriched in the neuronal system, potassium channels, neuroactive ligand–receptor interactions, and G-protein-coupled receptor downstream signaling pathways. Upregulated genes were enriched in pathways including extracellular matrix (ECM) organization, cytokine–cytokine receptor interaction, spinal cell injury, ECM–receptor interaction, and hematopoietic cell lineage (Fig. 4B).
Figure 4.
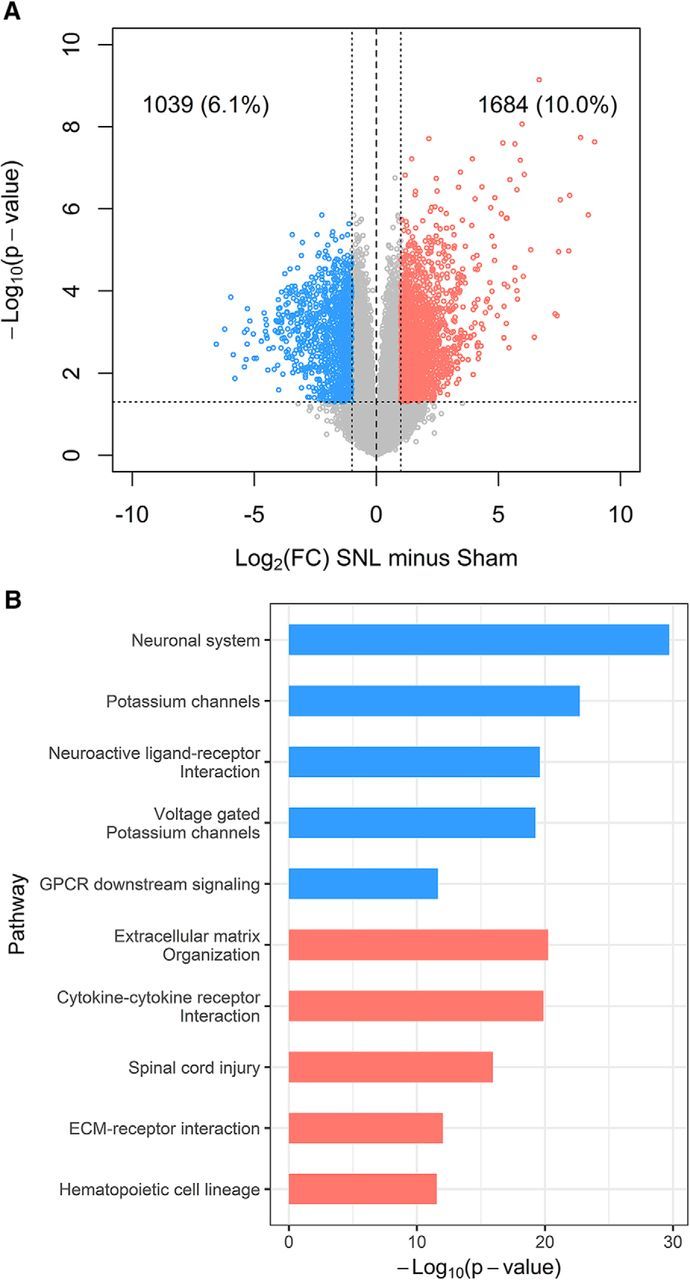
Gene expression changes in DRGs 3 weeks after SNL. A, Volcano plot shows 1684 (10%) upregulated and 1039 (6%) downregulated genes. B, Top pathways with enriched downregulated (blue) and upregulated (red) genes. Functional annotation of genes with simultaneous dysregulation of DNA methylation and gene expression is shown in Figure 4-1 and Figure 4-2.
Over-representation analysis of genes with dysregulated methylation and expression. Related to Fig. 4. Genes with expression and DNA methylation changes were enriched in extracellular matrix organization and cytokine-cytokine receptor pathways. Gene ontology functional annotation analysis showed enrichment for cell adhesion, immune response, response to wounding, channel activity, signaling receptor activity, plasma membrane, extracellular matrix, basement membrane and neuron projection. Download Figure 4-1, TIF file (1.2MB, tif)
Gene set enrichment analysis of genes associated with DNA methylation changes in DRGs after SNL. Related to Fig. 4. (A, B) Top gene ontology (GO) categories enriched after SNL. (C) Frequency of gene representation in GO subsets significantly enriched after SNL (FDR < 0.05). (D, E) Top GO categories repressed after SNL. (F) Frequency of gene representation in GO subsets significantly repressed after SNL (FDR < 0.05). Download Figure 4-2, TIF file (391.4KB, tif)
We next determined whether methylation changes detected in the DRG were correlated with the altered gene expression we reported previously using RNA-seq analysis in the SNL model (Laumet et al., 2015). DNA methylation outside of gene promoter regions can be associated with either increased or decreased expression (Jones, 2012). We analyzed the relationship between the variability of DNA methylation at CpG sites and expression of the closest gene.
We obtained RNA-seq data for 16809 genes and DNA methylation data for CpG sites associated with 7625 genes. Both datasets overlapped at 7112 genes. We observed significantly greater variability of expression of 2892 genes near the CpG sites that changed methylation compared with the CpG sites with stable methylation (Levene's test for homogeneity of variance, F = 9.38, p = 0.002). We noted concordant changes in gene expression (more than twofold, p < 0.05) and in DNA methylation (>2%, p < 0.05) at 664 genes. The odds ratio of simultaneous dysregulation of DNA methylation and gene expression in DRGs after SNL was 1.23 (95% CI, 1.09–1.38; p = 0.0005). Thus, the methylation reprogramming was associated with significant gene expression changes in the injured DRG. Genes affected by expression and DNA methylation changes were enriched for ECM organization and cytokine–cytokine receptor pathways. Gene ontology functional annotation analysis showed enrichment for cell adhesion, immune response, response to wounding, channel activity, signaling receptor activity, plasma membrane, ECM, basement membrane, and neuron projection (Fig. 4-1). GSEA of genes with variable methylation showed increased expression of genes involved in morphogenesis and development, and decreased expression of voltage-gated and ligand-gated ion channels and axon-associated genes in DRGs after SNL (Fig. 4-2).
Reducing DNA methylation causes pain hypersensitivity
Our findings showed that nerve injury predominantly causes genome-wide losses of DNA methylation in the DRG. To determine whether DNA hypomethylation is sufficient to modulate pain sensitivity, we first used a DNA methyltransferase inhibitor, RG108 (Brueckner et al., 2005). Drugs can get direct access to DRG neurons via intrathecal injections (Laumet et al., 2015; Xie et al., 2016). Intrathecal injection of RG108 (10 μg/d for 4 consecutive days) caused a large and long-lasting reduction in the mechanical withdrawal thresholds, measured with von Frey filaments and a noxious pressure stimulus (Randall-Selitto test) in naive rats (Fig. 5). Intrathecal vehicle injection produced no effect on the withdrawal thresholds.
Figure 5.
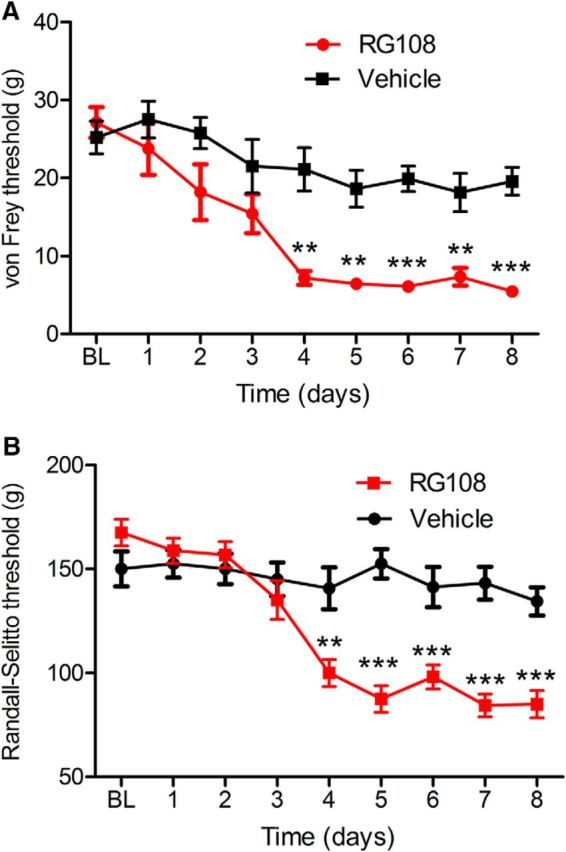
Intrathecal injection of RG108 induced pain hypersensitivity in naive rats. Pain hypersensitivity was measured 24 h after intrathecal injection of RG108 10 μg or vehicle (8 rats/group). A, B, RG108 induced significant reduction of von Frey thresholds (A) and Randall-Selitto thresholds (B) analyzed by repeated-measures two-way ANOVA (interaction drug × time: F(8,112) = 11.4, p < 0.0001 and F(8,104) = 5.16, p < 0.0001, respectively). ***p < 0.001, **p < 0.01, compared with baseline (BL). Data are presented as the mean ± SEM.
Folate and vitamin B12 are the main sources of the methyl group donors in the diet. We tested the effects of diets with a modified content of the key methyl group donors, folate and vitamin B12, on the paw withdrawal thresholds. The tactile threshold (von Frey) increased until the rats reached their adult body size, as previously described (McKelvey et al., 2015). Feeding the rats with the diet deficient in folate and vitamin B12 for 2–3 weeks significantly reduced the tactile (von Frey) withdrawal threshold before SNL surgery and in the contralateral (Sham control) hindpaw (Fig. 6). Although the withdrawal thresholds were not statistically different between the regular and enriched diet groups, the withdrawal thresholds were significantly lower in the deficient diet group than in the enriched diet group (Fig. 6). To confirm that the modified diets affect DNA methylation in the DRG, we compared global DNA methylation changes occurring 3 weeks after surgery for SNL in rats exposed for 7 weeks to the deficient, regular, and enriched diets. Interestingly, DNA hypomethylation in the DRG induced by SNL was not further enhanced in the rats being fed the folate-deficient and vitamin B12-deficient diet (Fig. 7A,C). In contrast, folate and vitamin B12 deficiencies resulted in DNA hypomethylation in DRGs obtained from the Sham-operated contralateral side (Fig. 7B,D). Together, these data suggest that nerve injury-induced DNA hypomethylation in the DRG is causally related to the development of chronic pain.
Figure 6.

Effect of folate- and vitamin B12-enriched, regular and deficient diets on pain hypersensitivity before and after nerve injury. Randall-Selitto and von Frey thresholds were measured every 3 d for 7 weeks. SNL was performed on day 26. The data were analyzed using two-way repeated-measures ANOVA followed by Bonferroni's correction for multiple tests. A, B, von Frey thresholds were measured on the ipsilateral rear paw (main diet effect: F(2,21) = 5.31, p = 0.014; significant time × diet interaction: F(26,273) = 3.96, p < 0.0001; A) and contralateral rear paw (main diet effect: F(2,12) = 10.1, p = 0.003; significant time × diet interaction: F(24,144) = 3.04, p < 0.0001; B). C, D, Randall-Selitto thresholds were measured on the ipsilateral rear paw (main diet effect: F(2,20) = 5.89, p = 0.01; C) and the contralateral rear paw (main diet effect: F(2,12) = 4.45, p = 0.036; D). Eight mice/diet group. *p < 0.05, **p < 0.01, ***p < 0.001. Significant difference between deficient diet and regular diet; #p < 0.05, ##p < 0.01, ###p < 0.001, significant difference between deficient diet and enriched diet. Data are presented as the mean ± SEM.
Figure 7.

Effect of folate- and vitamin B12-enriched, regular, and folate- and vitamin B12-deficient diets and nerve injury on DNA methylation in DRGs. Hypomethylation at the injured DRGs was not further enhanced by the diet deficient in folate and vitamin B12. On the other hand, uninjured DRGs in rats fed with a diet deficient in methyl donors showed hypomethylation changes similar to those occurring after SNL. CpG sites hypomethylated after deficient diet are shown in blue, and CpG sites with increased methylation are shown in orange. The percentage of total analyzed CpG sites is shown in parentheses. A, DNA methylation changes at the injured side, deficient vs regular diet. B, Prevailing hypomethylation at the control side, deficient vs regular diet. C, DNA methylation changes at the injured side, deficient vs enriched diet. D, Prevailing hypomethylation at the control side, deficient vs enriched diet.
Discussion
In this study, we systemically examined changes in DNA methylation in the DRG during neuropathic pain development after nerve injury. We found that hypermethylation prevailed in injured DRGs (60% of differentially methylated sites) 3 d after SNL. Similarly, an analysis using reduced representation bisulfite sequencing in rat DRGs 24 h after SNL showed that DNA methylation changes occurred at 1% of 1.4 million CpG sites (Gölzenleuchter et al., 2015). Specifically, hypermethylation and hypomethylation were detected at 79% and 21%, respectively, differentially methylated CpG sites. To determine DNA methylation changes during the transition from acute to chronic pain after SNL, the majority of our analyses were performed in the DRGs 3 weeks after SNL. At this time, hypomethylation was pronounced in injured DRGs, affecting 83% of differentially methylated CpG sites. Thus, although the early phase of epigenetic reprogramming after nerve injury mainly involves DNA hypermethylation in the DRG, the chronic phase of nerve injury-induced neuropathic pain is associated predominantly with DNA hypomethylation.
Traumatic nerve injury resulted in significant changes in the gene expression of 2723 genes. Membrane transporters and genes involved in synaptic transmission were enriched among downregulated genes. We showed that DNA methylation can be targeted, and the combined analysis of DNA methylation and gene expression revealed several pathways of potential interest for therapeutic interventions.
The persistent, abnormal activity of DRG neurons can also cause secondary structural and functional changes in the spinal cord and brain regions. Interestingly, we found less differentially methylated sites and a tendency toward increased methylation in both the spinal cord and the prefrontal cortex as opposed to prevailing hypomethylation in DRGs during the chronic phase of neuropathic pain induced by SNL. In contrast, it has been reported that sciatic nerve injury induces global hypomethylation in the mouse prefrontal cortex (Tajerian et al., 2013). This discrepancy may be explained by the difference in time after nerve injury: 3 weeks in our study versus 6 months in the study by Tajerian et al. 2013.
The large overlap between DNA methylation changes in the DRGs from neonates and the adult SNL DRGs suggests that the epigenetic status of DRG neurons returned to an early developmental state after injury. The precise mechanisms leading to nerve injury-induced DNA hypomethylation in the DRG remain unclear. The regeneration of peripheral nerves induces a hypersensitivity state of primary sensory neurons (Truini et al., 2013). A recent study (Weng et al., 2017) indicates that Tet3 demethylase plays a major role in active DNA demethylation for axon regeneration and regional hypomethylation of genes linked to nerve regeneration after peripheral nerve injury. Also, we have shown that Gadd45 in the DRGs is profoundly upregulated after SNL (Laumet et al., 2015). Because Gadd45 is actively involved in DNA demethylation (Niehrs and Schäfer, 2012; Schäfer, 2013), it may also contribute to DNA hypomethylation in the DRG induced by nerve injury.
In contrast to SNL-induced DNA hypomethylation in the DRGs, we observed minimal changes in DNA methylation in paclitaxel-treated rats. Our data are consistent with the concept that traumatic nerve injury-induced active DNA hypomethylation is a key epigenetic mechanism involved in axon regeneration (Shin and Cho, 2017; Weng et al., 2017). It is likely that traumatic nerve injury, not chemotherapy, causes neuropathic pain through DNA hypomethylation involved in hyperexcitable DRG neurons associated with nerve regeneration. Our findings suggest that different molecular mechanisms are involved in the development of neuropathic pain caused by traumatic nerve injury and chemotherapy. In support of this notion, SNL increases postsynaptic NMDA receptor activity through CK2, whereas paclitaxel treatment primarily augments presynaptic NMDA receptor activity via PKC (Chen et al., 2014a; Xie et al., 2016). We also investigated a correlation between the changes in DNA methylation with changes in gene expression occurring in the DRGs 3 weeks after SNL. We observed increased variability in the expression of genes associated with CpG sites differentially methylated after SNL. At the same time, variable methylation at CpG sites at transcription start sites was associated with decreased gene expression. Thus, DNA methylation reprogramming may contribute to altered gene expression in the DRG after nerve injury.
Our study provides new evidence that DNA hypomethylation can directly affect the pain sensitivity. We showed that intrathecal injection of the methyltransferase inhibitor RG108 or eating a folate-deficient and vitamin B12-deficient diet produced pain hypersensitivity in naïve rats. Similar to our findings, it has been reported that S-adenosylmethionine supplementation, a methyl donor, reverses nerve injury-induced neuropathic pain in mice (Grégoire et al., 2017). The methyl donor not only increases DNA methylation but also promotes histone methylation (Mentch et al., 2015; Tang et al., 2016), which may account for the small effect of the enriched diet on nociception. Because of the complex actions of the methyl diets, our conclusion about the role of DNA methylation in neuropathic pain is primarily based on the use of the DNA methylation inhibitor. Our findings of DNA methylation reprogramming in SNL-induced neuropathic pain are also in line with several clinical observations. For example, patients carrying mutations in the DNMT1 gene developed sensory neuropathy (Klein et al., 2011). Moreover, two-thirds of subjects with MECP2 mutation-Rett syndrome, a DNA methylation reader dysfunction, also show abnormal nociception (Downs et al., 2009). In addition, vitamin B12 and folate deficiencies increase the risk of the development of peripheral neuropathy in patients (Shorvon et al., 1980; Reynolds, 2006). Interestingly, we were able to reproduce these observations in a rat model. Folate or vitamin B12 deficiency might also alter the function of immune cells and glia and thus contribute to chronic pain. Therefore, folate and vitamin B12 levels and intake should be monitored in patients at risk for the development of painful neuropathy.
In summary, our study reveals that traumatic nerve injury-induced DNA hypomethylation in the DRG during chronic pain is a genome-wide event. Because reducing DNA methylation caused pain hypersensitivity, it suggests a causal role of DNA hypomethylation in neuropathic pain development. Nerve injury-induced persistent DNA hypomethylation could contribute to epigenetic reprogramming to maintain hyperactivity of sensory neurons chronic pain. This epigenetic reprogramming could underlie a persistent transcriptional change necessary to sustain the chronic pain state.
Footnotes
This work was supported the National Institutes of Health (Grants R01-DE-022015 and R01-NS-101880). We thank Dr. Carrie Schultz from Purina Animal Nutrition LLC Technical Services and Dr. Jason Raynor from TestDiet for helping with the design of the diets with modified amounts of folate and vitamin B12.
The authors declare that they have no competing financial interests.
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE (2012) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13:R87. 10.1186/gb-2012-13-10-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F (2005) Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res 65:6305–6311. 10.1158/0008-5472.CAN-04-2957 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen SR, Eisenach JC, McCaslin PP, Pan HL (2000) Synergistic effect between intrathecal non-NMDA antagonist and gabapentin on allodynia induced by spinal nerve ligation in rats. Anesthesiology 92:500–506. 10.1097/00000542-200002000-00033 [DOI] [PubMed] [Google Scholar]
- Chen SR, Zhou HY, Byun HS, Chen H, Pan HL (2014a) Casein kinase II regulates N-methyl-D-aspartate receptor activity in spinal cords and pain hypersensitivity induced by nerve injury. J Pharmacol Exp Ther 350:301–312. 10.1124/jpet.114.215855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Zhu L, Chen H, Wen L, Laumet G, Pan HL (2014b) Increased spinal cord Na(+)-K(+)-2Cl(-) cotransporter-1 (NKCC1) activity contributes to impairment of synaptic inhibition in paclitaxel-induced neuropathic pain. J Biol Chem 289:31111–31120. 10.1074/jbc.M114.600320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ (2011) Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci 31:16619–16636. 10.1523/JNEUROSCI.1639-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J, Bergman A, Carter P, Anderson A, Palmer GM, Roye D, van Bosse H, Bebbington A, Larsson EL, Smith BG, Baikie G, Fyfe S, Leonard H (2009) Guidelines for management of scoliosis in rett syndrome patients based on expert consensus and clinical evidence. Spine (Phila Pa 1976) 34:E607–E617. 10.1097/BRS.0b013e3181a95ca4 [DOI] [PubMed] [Google Scholar]
- Gölzenleuchter M, Kanwar R, Zaibak M, Al Saiegh F, Hartung T, Klukas J, Smalley RL, Cunningham JM, Figueroa ME, Schroth GP, Therneau TM, Banck MS, Beutler AS (2015) Plasticity of DNA methylation in a nerve injury model of pain. Epigenetics 10:200–212. 10.1080/15592294.2015.1006493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire S, Millecamps M, Naso L, Do Carmo S, Cuello AC, Szyf M, Stone LS (2017) Therapeutic benefits of the methyl donor S-adenosylmethionine on nerve injury-induced mechanical hypersensitivity and cognitive impairment in mice. Pain 158:802–810. 10.1097/j.pain.0000000000000811 [DOI] [PubMed] [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A (2011) Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 6:468–481. 10.1038/nprot.2010.190 [DOI] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H (2011) Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14:1345–1351. 10.1038/nn.2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer P, Banck MS, Amberg R, Wang C, Petznick G, Luo S, Khrebtukova I, Schroth GP, Beyerlein P, Beutler AS (2010) mRNA-seq with agnostic splice site discovery for nervous system transcriptomics tested in chronic pain. Genome Res 20:847–860. 10.1101/gr.101204.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwig R, Hardt C, Lienhard M, Kamburov A (2016) Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11:1889–1907. 10.1038/nprot.2016.117 [DOI] [PubMed] [Google Scholar]
- Institute of Medicine of the National Academies (2011) Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington, DC: National Academies Press. [PubMed] [Google Scholar]
- Jelinek J, Madzo J (2016) DREAM: a simple method for DNA methylation profiling by high-throughput sequencing. Methods Mol Biol 1465:111–127. 10.1007/978-1-4939-4011-0_10 [DOI] [PubMed] [Google Scholar]
- Jelinek J, Liang S, Lu Y, He R, Ramagli LS, Shpall EJ, Estecio MR, Issa JP (2012) Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 7:1368–1378. 10.4161/epi.22552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Kamburov A, Wierling C, Lehrach H, Herwig R (2009) ConsensusPathDB—a database for integrating human functional interaction networks. Nucleic Acids Res 37:D623–628. 10.1093/nar/gkn698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363. 10.1016/0304-3959(92)90041-9 [DOI] [PubMed] [Google Scholar]
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, Simon M, Lander C, Boardman LA, Cunningham JM, Smith GE, Litchy WJ, Boes B, Atkinson EJ, Middha S, B Dyck PJ, et al. (2011) Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 43:595–600. 10.1038/ng.830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27:1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, Dong Y, Jelinek J, Cesaroni M, Issa JP, Pan HL (2015) G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci 18:1746–1755. 10.1038/nn.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CN, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M (2000) Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85:503–521. 10.1016/S0304-3959(00)00251-7 [DOI] [PubMed] [Google Scholar]
- Massart R, Barnea R, Dikshtein Y, Suderman M, Meir O, Hallett M, Kennedy P, Nestler EJ, Szyf M, Yadid G (2015) Role of DNA methylation in the nucleus accumbens in incubation of cocaine craving. J Neurosci 35:8042–8058. 10.1523/JNEUROSCI.3053-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita Y, Araki K, Omotuyi Oi, Mukae T, Ueda H (2013) HDAC inhibitors restore C-fibre sensitivity in experimental neuropathic pain model. Br J Pharmacol 170:991–998. 10.1111/bph.12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKelvey R, Berta T, Old E, Ji RR, Fitzgerald M (2015) Neuropathic pain is constitutively suppressed in early life by anti-inflammatory neuroimmune regulation. J Neurosci 35:457–466. 10.1523/JNEUROSCI.2315-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW (2015) Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab 22:861–873. 10.1016/j.cmet.2015.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, Adachi M, Na ES, Monteggia LM (2014) Selective role for DNMT3a in learning and memory. Neurobiol Learn Mem 115:30–37. 10.1016/j.nlm.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, Schäfer A (2012) Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol 22:220–227. 10.1016/j.tcb.2012.01.002 [DOI] [PubMed] [Google Scholar]
- Pan HL, Eisenach JC, Chen SR (1999) Gabapentin suppresses ectopic nerve discharges and reverses allodynia in neuropathic rats. J Pharmacol Exp Ther 288:1026–1030. [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ (2001) A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 94:293–304. 10.1016/S0304-3959(01)00363-3 [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2017) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Reynolds E. (2006) Vitamin B12, folic acid, and the nervous system. Lancet Neurol 5:949–960. 10.1016/S1474-4422(06)70598-1 [DOI] [PubMed] [Google Scholar]
- Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, Schultz MD, Ansoleaga B, Garcia-Esparcia P, Pons-Espinal M, de Lagran MM, Dopazo J, Rabano A, Avila J, Dierssen M, Lott I, Ferrer I, Ecker JR, Esteller M (2016) Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry 6:e718. 10.1038/tp.2015.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A. (2013) Gadd45 proteins: key players of repair-mediated DNA demethylation. Adv Exp Med Biol 793:35–50. 10.1007/978-1-4614-8289-5_3 [DOI] [PubMed] [Google Scholar]
- Sharma A, Klein SS, Barboza L, Lohdi N, Toth M (2016) Principles governing DNA methylation during neuronal lineage and subtype specification. J Neurosci 36:1711–1722. 10.1523/JNEUROSCI.4037-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y (2017) Epigenetic regulation of axon regeneration after neural injury. Mol Cells 40:10–16. 10.14348/molcells.2017.2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon SD, Carney MW, Chanarin I, Reynolds EH (1980) The neuropsychiatry of megaloblastic anaemia. Br Med J 281:1036–1038. 10.1136/bmj.281.6247.1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542. 10.1093/bioinformatics/btl117 [DOI] [PubMed] [Google Scholar]
- Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS (2013) Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One 8:e55259. 10.1371/journal.pone.0055259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Tian B, Brasier AR, Sowers LC, Zhang K (2016) Measurement of histone methylation dynamics by one-carbon metabolic isotope labeling and high-energy collisional dissociation methylation signature ion detection. Sci Rep 6:31537. 10.1038/srep31537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truini A, Garcia-Larrea L, Cruccu G (2013) Reappraising neuropathic pain in humans—how symptoms help disclose mechanisms. Nat Rev Neurol 9:572–582. 10.1038/nrneurol.2013.180 [DOI] [PubMed] [Google Scholar]
- Wang M, Zhao Y, Zhang B (2015) Efficient test and visualization of multi-set intersections. Sci Rep 5:16923. 10.1038/srep16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng YL, An R, Cassin J, Joseph J, Mi R, Wang C, Zhong C, Jin SG, Pfeifer GP, Bellacosa A, Dong X, Hoke A, He Z, Song H, Ming GL (2017) An intrinsic epigenetic barrier for functional axon regeneration. Neuron 94:337–346.e6. 10.1016/j.neuron.2017.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG, Zhang X (2002) Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci U S A 99:8360–8365. 10.1073/pnas.122231899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Zeng WA, Pan HL (2016) Presynaptic N-methyl-d-aspartate (NMDA) receptor activity is increased through protein kinase C in paclitaxel-induced neuropathic pain. J Biol Chem 291:19364–19373. 10.1074/jbc.M116.732347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17:1031–1036. 10.1016/0031-9384(76)90029-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Prevailing DNA hypomethylation in DRGs 3 weeks after spinal nerve ligation (SNL). Related to Fig. 1. DNA methylation changes in DRGs 3 weeks after SNL in three experiments, each comparing 3 SNL/Sham pairs. (A, B) DNA methylation was analyzed by the DREAM assay. Dark blue dots show hypomethylated sites. Orange dots show hypermethylated sites. Percentage of differentially methylated sites is shown in parentheses. (C) Correlation of methylation differences in two representative experiments at 17869 CpG sites. Red solid line shows linear regression (p-value < 2e-16). (D) DNA methylation analyzed by the RRBS assay. Aquamarine blue dots show hypomethylated sites. Magenta dots show hypermethylated sites. (E) Hypomethylated CpG sites are distributed on all chromosomes. (F) RRBS and DREAM assays detected distinct sets of CpG sites with an overlap of 0.3%. Methylation values at CpG sites detected by both assays were highly concordant (linear regression R^2=0.98). Download Figure 1-1, TIF file (2.1MB, tif)
DNA methylation changes after paclitaxel treatment are minimal and distinct from those after SNL. Related to Fig. 2. (A) DNA methylation changes at 15807 CpG sites in DRGs from three paclitaxel-treated and three control rats. (B) DNA methylation changes at 15559 CpG sites in dorsal horns of the spinal cord from two paclitaxel-treated and three control rats. Dark blue dots show hypomethylated sites. Orange dots show hypermethylated sites. Percentage of differentially methylated sites is shown in parentheses. Inverse correlation of DNA methylation changes after paclitaxel treatment and SNL in (C) DRGs (Pearson's correlation r = -0.35, t = -39.347, df = 10977, p-value < 2.2e-16) and (D) the spinal cord (Pearson's correlation r = -0.17, t = -20.497, df = 13257, p-value < 2.2e-16). Download Figure 2-1, TIF file (1.1MB, tif)
Validation of DRG neuronal isolation by western blots. Related to Fig. 3. (A) Nuclei were isolated from a pool of DRGs as evidenced by immunolabeling of histone 3 by western blots. (B) Neuronal nuclei were separated by immunoprecipitation. The separation of neuronal and non-neuronal nuclei were confirmed immunolabeling of neuronal marker Neuronal Nuclei (NeuN) and glial markers Glial fibrillary acidic protein (GFAP) and glutamine synthetase (GS). Western blots were imaged and quantified by infrared imaging system. Download Figure 3-1, TIF file (444.1KB, tif)
Over-representation analysis of genes with dysregulated methylation and expression. Related to Fig. 4. Genes with expression and DNA methylation changes were enriched in extracellular matrix organization and cytokine-cytokine receptor pathways. Gene ontology functional annotation analysis showed enrichment for cell adhesion, immune response, response to wounding, channel activity, signaling receptor activity, plasma membrane, extracellular matrix, basement membrane and neuron projection. Download Figure 4-1, TIF file (1.2MB, tif)
Gene set enrichment analysis of genes associated with DNA methylation changes in DRGs after SNL. Related to Fig. 4. (A, B) Top gene ontology (GO) categories enriched after SNL. (C) Frequency of gene representation in GO subsets significantly enriched after SNL (FDR < 0.05). (D, E) Top GO categories repressed after SNL. (F) Frequency of gene representation in GO subsets significantly repressed after SNL (FDR < 0.05). Download Figure 4-2, TIF file (391.4KB, tif)