

Abstract
Background:
Glucocorticoids (GCs) are key hormones used for the treatment of acute lymphoblastic leukemia (ALL) in children, but their cytotoxic effects are not well defined. The aim of this study was to evaluate the association between polymorphisms in NR3C1 encoding for protein involved in the GCs metabolism and its role in the development of ALL and the toxicity outcome, in terms of liver toxicity, glucose abnormality and infections, in ALL Saudi children.
Methods:
The following polymorphisms BCII rs41423247, ER22/23 EK rs6189 and rs6190 and N363S rs6195 in NR3C1 were analyzed in 70 children with ALL treated according to the ALL 2000 study protocol in comparison to 60 control subjects. Treatment toxicities and their association with genotypes were evaluated according to Common Toxicity Criteria (NCI-CTC).
Results:
This study demonstrated that the NR3C1 did not contribute to the development of childhood ALL. Homozygous ER22/23EK polymorphism was not found in both ALL patients and in control group whereas the heterozygous polymorphism was only observed in the control group (6.66%). The toxicology data in this study showed a significant difference between ALL patients carrying N363S polymorphism and wild type (40% and 6.51% respectively, P= 0.009) and a high-risk factor in the toxicity of glucose abnormality (OR=10.167; 1.302-79.339).BCII shows increased risk factors towards the liver toxicity (OR=2.667; 0.526-7.330) as well as the glucose abnormality (OR=7.5; 1.039-54.116).
Conclusion:
This study suggested that the polymorphisms in NR3C1 were not associated with the development of ALL in children. N363S polymorphism was sensitive to glucocorticoids and it may contribute to the glucose abnormality for these patients.
Keywords: Glucocorticoid toxicity, acute lymphoblastic leukemia, glucocorticoid receptor gene polymorphisms, SNPs
Introduction
Acute lymphoblastic leukemia (ALL) is characterized by the overproduction of immature cells of the white blood (lymphoblasts) in place were they produce, the bone marrow, and when it continuously multiplies causing death by inhibiting the production of normal cells if it is untreated (Travlos, 2006). Recovery rates for ALL patients have progressed from less than 15% to 90% (Pui et al., 2012). Although GCs are essential for ALL treatment, they can generate critical short and long term side effects and about 20% of patients are associated of treatment failure (Pui et al., 2012). However, the annual count with leukemia patients who suffer from relapse after giving an initial therapy stays a major issue (American Cancer Society, 2018).
Individual response to the treatment relies on many agents, including genetic and biologic characteristics of the leukemic blasts at the time of diagnosis and quality of the patient. Moreover, malignant cells are capable to integrate phenotypic alterations like mad that make them resistant to a group of constructional various agents of chemotherapeutic
Genetic variations, including nucleotide repeats, deletions, insertions, and single nucleotide polymorphism (SNPs) can change the amino acid sequence of the encoded proteins, RNA splicing, and transcription of gene. Such genetic polymorphisms have been actively explored in drug-metabolizing enzymes, molecular targets, and transporters regarding functional changes in phenotype (altered expression levels and/or activity of the encoded proteins) and their participation in variable drug responses (Lee et al., 2006).
Glucocorticoids (GCs) have different and essential effects on the human body and also play an important role for its survival. GCs also affect glucose and lipid metabolism, anti-inflammatory, growth, immunosuppressive affect, and central nervous system (Ciriaco et al., 2013). Cortisol is the most abundant form of GCs in human and its elevation leads to neuropsychiatric disorder and cognitive impairment. The cortisol secretion in the body is controlled by the hypothalamic -pituitary-adrenal glands(HPA). Cortisol itself is the main induction of the negative feedback mechanism for the HPA axis (Kadmiel and Cidlowski, 2013).
The actions of GCs are mediated by the nuclear receptor protein called the glucocorticoid receptors (GR). This receptor is expressed by the GR gene (NR3C1) located on chromosome 5. This gene has a three-domain structure: amino transactivation domain, DNA-binding domain, and carboxy terminal ligand binding. During unliganded form of the GR, it is located in the cytoplasm in a large complex form of protein, after binding to cortisol, it dissociates and a conformational change occurs then the GR receptor translocate to the nucleus where it acts as a transcription factor to regulate the transcription of GR gene by several mechanisms. The first mechanism is interacting with specific DNA sequence (transactivation) and play a very important role on the side effect of GC treatment by transrepression occurs (Manenschijn et al., 2009; Revollo and Cidlowski, 2009). Secondly, the transcription factor interacts with other transcription factors such as nuclear factor κ B or activator protein1 and represss their activity, leading to inhibition of the pro-inflammatory transcription factor (Oh et al., 2017). This mechanism explains the anti-inflammatory effect of GCs and extensively used GCs during inflammation and autoimmune disease. The third mechanism is nongenetic mediated and takes place through activation of nitric oxide synthase of endothelial cells which leads to a rapid effect of inflammation.
Mutation in GR receptor leading to disturbance of negative feedback mechanisms on HPA axis (reduced) and also loss its function resulting increase the secretion of cortisol and mineralocorticoid by the adrenal gland due to increasing secretion of ACTH adrenocorticotrophic hormone. On the other words, when GR receptor gene mutated, the cell will be more sensitive to the action of GC (Chen and Li, 2012). The sensitivity and resistance of the cell to GCs mechanisms are poorly understood till now. According to previous studies, some mutations in GR gene are linked with the general sensitivity syndromes (Yang et al., 2012; Vitellius et al., 2016). Beside the mutation in GR gene, there are also many polymorphisms of this gene (GR) has been discovered within this gene.
The gene polymorphisms in GR changes the sensitivity and resistance of the cell to GCs treatment. The N363S polymorphism (rs6195) is located in codon 363 of exon2 and consists of an A to G substitution (AAT→AGT) this change yields a change an amino acid in asparagine (N) to serine (S). This polymorphism shows an increase of transactivation and sensitivity to GCs in vivo (Lauten et al., 2003). Several studies on this polymorphism showed an association between the N363S polymorphism and increased body mass index and tendency toward decreased bone mineral density in tubular bone (Schoor et al., 2007). However, on the other hand, another study showed no association ..and BMI, serum lipid levels, and glucose tolerance status in male carries of the N363S (Savas and van Rossum, 2018). Since the carries of N363S polymorphism are sensitive to the GC action, it has been expected to be less susceptible to autoimmune disease. However, previous studies revealed no association between the N363S polymorphism and autoimmune disease (Herrera et al., 2018).
The BclI polymorphism (rs41423247) was described for the first as an RFLP of a fragment of 2-3kb and fragment of 4.5kb (Decorti et al., 2006). This polymorphism consists of a C to G substitution, 646 nucleotide downstream from exon 2 forming fragments of 2.2kb and 3.9kb with a sequence TGATCA to TGATGA. The cell that has this polymorphism is characterized by increased sensitivity to GC with respect to the adrenal negative feedback to the pituitary. The increased sensitivity to GC leads to high systolic blood pressure, hyperinsulinemia, increased BMI, and high abdominal fat distribution (Murray et al., 1987; van Rossum et al., 2002). The C allele is the most frequent occurring and thus can be considered the wild type allele and G allele considered the polymorphic type. The carries of G allele of the BclI polymorphism showed a greater suppression after injected by 1 mg and 0.25mg of DEX. This correlation suggests a hypersensitivity to GC in relation to the negative feedback mechanism at the pituitary level. Also, several studies reported the association between the BclI polymorphism and autoimmune disease. It was found thatpatients with BclI polymorphism had a mild ophthalmopathy compared with patients with the wild-type and it was explained by the enhance the sensitivity to endogenous GC which leads to suppressing of immune system and inflammatory reaction (Russcher et al., 2005; Boyle et al., 2008). There is also an association between BclI polymorphism and autoimmune inflammatory bowel disease and Crohn’s disease (De Ludicibus, 2007). As well as, the patients carrying the BclI polymorphism responded better to GCs treatment and were less likely to need an additional course of steroid treatment (Halsall et al., 2000). Patients with cystic fibrosis and skin cancer also had an association between them and BclI polymorphism (Corvol et al., 2007), as well as, a relationship between lung disease development and BclI polymorphism in cystic fibrosis patients and they explained that by alteration to the susceptibility to inflammation. Also, patients treated with GCs and developing a non-melanoma skin cancer (SCC) and polymorphism carrier BclI (Karagas et al., 1999; Sørensen et al., 2004; Savas and van Rossum, 2018). It was also demonstrated that patient with prolonged use of GCs had a higher risk of developing squamous cell carcinoma (SCC). They found no association between BclI polymorphism and the risk of developing SCC in the patient that are not taking GCs (Sørensen et al., 2004).
The ER22/23EK polymorphism is also located in transcription domain in exon2 of the GR gene in two codons 22 and 23. The wild type of this polymorphism is GAG in codon 22 AGG in codon 23 and the exact sequence alteration is GAG AGG to GAA AAG, which is translated to produce a glutamic acid-Arginine (ER) to glutamic acid-lysine (EK) also known as (rs6189 and rs6190) (Lauten et al., 2003). Previous studies showed a correlation betweenthe ER22/23 EK polymorphism and relative GCs-resistant. The ER-22/23EK polymorphism was associated with cortisol resistant and were found a correlation between the ER22/23EK polymorphism and favorable metabolic proliferation such as lower insulin level, increased insulin sensitivity and lower total and low density lipoprotein (LDL)- cholesterol level (Decorti et al., 2006) and this lower the risk of developing type2 diabetes mellitus or cardio vascular disease. The ER22/23EK polymorphism carrier show tendency to lower of c-reactive protein (CRP) as well as a decreased of total LDL-cholesterol level in elderly persons which is beneficial in developing inflammation and cardiovascular disorder (Koeijvoets et al., 2006). GCs are well known to repress in immune system through different many mechanisms as so that it is used in the treatment of autoimmune disease. Interestingly, heterozygous ER-22/23EK polymorphism carriers increased risk of getting infected by staphylococcus aureus (S. aureus) this is can explain by ER-22/23EK polymorphism carrier leads to increased transrepression and there by an increase suppression of the immune system (van den Akker et al., 2006).
Although the clinical importance of the polymorphism of GC receptor genes on the childhood who suffer the ALL and their response to the treatment, there are a few studies on this gene worldwide and particularly this study can be considered the first investigation among the Saudi children. There is few published articles talking about the association between NR3C1 polymorphisms and treatment response in ALL children. Therefore, we demonstrated the possible association between three GR gene polymorphism and development of childhood ALL as well as the occurrence of side effects such as liver toxicity, glucose abnormality, and infection in Saudi children with ALL.
Materials and Methods
Study population
There were 70 children, 43 male and 27 female subjects, with Pre-B ALL, standard risk, between 2009-2013 and 60 healthy control were included in this study. Fifty of the patients subject were treated at the oncology unit of the king Faisal Hospital and research center (KFHRC), and 20 patients received their treatment at the King Abdulaziz University Hospital (KAUH). Fifty one patients were less than 10 years old and 19 were older than 10 years. This study was approved by the Medical Ethics Committee of KFHRC and KAU Hospital. Informed consents were obtained from ALL patients, or their parents, before recruiting for the study. Inclusion criteria: Patients were eligible for the treatment protocol of this study if they fulfilled the following criteria: (1) aged less than 18, (2) all patients with bone marrow blast cells ≥ 20%, and (3) absence of other active malignancy. Patients were excluded if they died during induction therapy or lostfollow up routine examinations.
This study was performed using bone marrow aspirates that taken at diagnosis, on the 15th day and after completion of induction phase (treatment day 33). Clinical data were obtained retrospectively, reviewed by a pediatric oncologist, and were analyzed blindly for the genotype results. Toxicity data were obtained from the patient’s records and graded according to the NCI common toxicity criteria scales included in ALL 2000 protocols (CTC Version 2.0 1999). The common toxicities associated with GC treatment, including glucose abnormalities, liver toxicities, and infections were evaluated. Toxicities grades were used to dichotomize toxicities as “present” versus “absent”. The worst toxicity grade observed per patient during remission induction and reinduction was considered in this study.
Glucose abnormalities defined as diabetes requiring insulin treatment with significant glycosuria for at least two days or requiring medical intervention or hyperglycemia (fasting glucose >11.1 mmol (200mg/dl)). Hepatotoxicity was defined as elevated activities of alanine transferase (ALT) and aspartate transferase (AST), as well as high level of total bilirubin (TBIL). Severe toxicity was defined as AST or ALT higher than 2.5 folds and /or TBIL higher than 1.5 fold of the normal level (grade 3 toxicity) according to Common Toxicity Criteria report (NCI-CTC). Considering different side effects induced by glucocorticoids treatment, infection has the highest incidence that can be due to the use of steroids. Patients were classified according to NCI-CTC criteria for incidence of infection and evaluated with different genotype.
Genetic Analysis
This study used bone marrow embedded in paraffin wax and bone marrow smear slides for the patients and EDTA blood samples for the control. For paraffin embedded sample, the blocks of fixed bone marrow biopsy, paraffin wax embedded were analyzed. For each case, four 5 µm thick slices were cut in triplicate. The area to be sectioned was examined carefully to ensure the inclusion of tissue and that an equal amount of tissue was included in each set. Thorough cleansing was performed between cases and a new set of cutting instruments was used for each case. All polymerase chain reactions (PCR) were carried out using stringent precautions to avoid cross contamination. DNA extraction, PCR reagent preparation, amplification, and amplicon analysis were performed in separate rooms. Disposable aero-guard pipette tips were used. A negative control was included after each fifth sample, and this included the DNA extraction and PCR reagents. In addition, all positive samples were repeated in a separate PCR run and all were reproducible. The commercial QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) was used according to the manufacturer’s protocol. Bone marrow smears samples were collected at a complete remission. DNA was obtained by the standard ethanol method (DeMelo et al., 1992). In this method, DNA was isolated from unstained archived glass slide smears. Typically, one or two glass slide smears were scraped into a sterile 1.5 ml Eppendorf tube using a new razor place. The DNA was extracted from resulting powder with commercial DNA extraction kits (Wizard® Genomic DNA Purification Kit from Promega, WI, USA). The scraped material was re-suspended in 400 µL of 6 M guanidinium hydrochloride, 30 µL of 20 % of sodium sarcosyl, 30 µL of 7.5 M ammonium acetate, and 10 µL of proteinase K (10 mg/ml). The mixture was heated to 60 oC for 1 hr. If it was not completely dissolved, an additional 10 µL of proteinase K was added and the mixture was heated for another hour at 60 oC. The DNA was precipitated by addition of 1 ml of cold ethanol, gently homogenized, and the mixture left at -20o C overnight. After centrifugation for 20 minutes, the supernatant was discarded and the pellet re-suspended in 50 µL of water. The polymorphisms of the genes were determined by polymerase chain reaction and the applicant were sequenced for nucleotide by Dideoxy Sanger method. For the primers sequences and PCR conditions are in table 1. DNA fragments from PCR reaction were separated in 1% agarose gel and examined under UV light gel documentation system. The sequences of primers used for amplification of genes involved in this study are listed in the Table 1.
Table 1.
Primers Used for Amplification of PCR Fragments
| NR3C1 | N363S | F 5’-AGTACCTCTGGAGGACAGAT-3’ |
| R 5’-GTCCATTCTTAAGAAACAGG-3’ | ||
| BCLL | F 5’-TGCTGCCTTATTTGTAAATTCGT-3’ | |
| R 5’-AAGCTTAACAATTTTGGCCATC-3’ | ||
| ER22/23EK | F 5’-GATTCGGAGTTAACTAAAAG-3’ | |
| R 5’-ATCCCAGGTCATTTCCCATC-3’ |
PCR program, consisting of 95C for 7 min, 40 cycles of 94C for 1 min, 68C for 1 min, 72C for 1 min, and 1 cycle of 72C for 7 min (Bachmann et al., 2005).
The PCR products were purified and sequenced at Macrogen Inc., Seoul, Republic of Korea. The sequence results were aligned with gene bank using nucleotide BLAST (blastn) software in http://ncbi.nlm.nih.gov/blast.
Statistical Analysis
Allele frequencies of the studied genes were tested for Hardy- Weinberg equilibrium (HWE). The distribution of genotype of ALL patients and control subject were compared using Chi-square test or Fishers exact test. Any possible association between polymorphism in each gene and the glucose metabolism abnormalities, liver toxicity, and susceptibility to infection was investigated using Chi-square test. Unconditional logistic regression was used to calculate the odds ratio (OR) and 95% confidence intervals (CI) and using two-sided Fischer’s exact test, using SPSS (versions 23)). P>0.05 (two-tailed) was considered statistically significant.
Results
In this study, several polymorphisms were analyzed in genes of the possible relevance of glucocorticoids pharmacogenomics and or pharmacokinetics. For all examined polymorphism of genes, no significant differences were found ALL patients and control subjects.
Glucocorticoid Receptor Gene (NR3C1)
This study was performed in three SNPs in NR3C1 glucocorticoid receptor gene, including ER22/23 EK, BclI, and N363S. In the following, …
A) ER22/23EK SNP
The percentage of allele frequency polymorphism in ER22/23K SNPs in glucocorticoid receptor of ALL patients and control subject are represented in Figure 1. Figure 2 depicts different genotype sequences. Results on Allele and genotype frequencies of ER22/23Ek SNPs were consistent to Hardy-Weinberg equilibrium as shown in Table 2. Two different genotype patterns were detected, including wild type genotype and heterozygous genotype as reported in Table 1. The frequency of wild type waa 100% and 93.33% in ALL patients and control subjects, respectively. whereas it is estimated to 6.66% for heterozygous in control subjects only. No mutated sequence in ALL patients and control subject.
Figure 1.
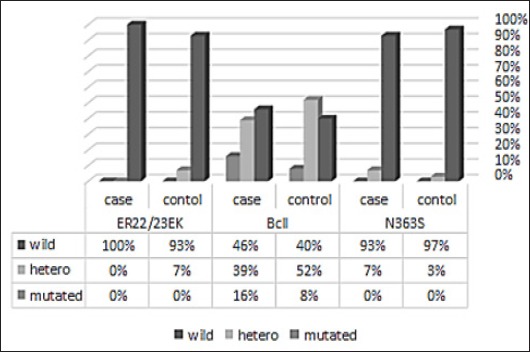
Genotype Distributions of Different SNPs in NR3C1 Gene of Control and All Patients
Figure 2.
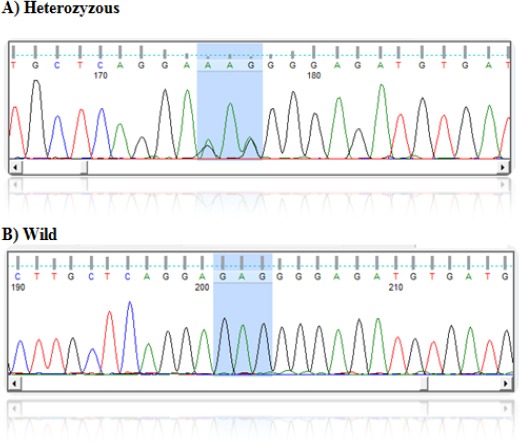
DNA Sequence of Different SNP in ER22/23EK (GR Gene) in All Patients
Table 2.
Allelic Frequencies and Genotype Distribution of the Studied SNPs of NR3C1 (GR) in ALL Children (n=70) and Control Subjects (n=60).
| SNP | NR3C1 GENE | OR (95%Cl) | P value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Allelic Frequency | OR (95%Cl) | P value | Genotype | |||||||
| WT | MUT | WT | HET | MUT | ||||||
| ER22/23EK | Case | 140 (100%) | 0 (0%) | 0.286 (0.049-1.666) | 0.16 | 70 (100%) | 0 (0%) | 0% | ||
| Control | 116 (96.66%) | 4 (3.33%) | 56 (93.33%) | 4 (6.66%) | 0% | |||||
| BclI | Case | 91 (65%) | 49 (35%) | 0.367 (0.129-1.047 | 0.064 | 32 (45.7%) | 27 (38.57% | 11 (15.71%) | 1.886 (0.694-5.122) | 0.285 |
| Control | 79 (65.83%) | 41 (34.16%) | 24 (40%) | 31 (51.66%) | 5(8.33%) | |||||
| N363S | Case | 135 (96.4%) | 1 (3.6%) | 0.429 (0.027-6.739) | 0.511 | 65 (92.8%) | 5 (7.2%) | 0 | ||
| Control | 118 (98.3%) | 2 (1.7%) | 58 (97%) | 2 (3%) | 0 | |||||
B) BclI SNP
The percentage of allelic and genotype in BclI polymorphism of GC receptor (NR3C1) in ALL patients and control subjects are represented in Figure 1. and different genotype sequence in Figure 3. The incidence of wild type (CC) pattern was 45% and 40% in ALL patients and control subjects, respectively, whereas, incidence of the heterozygous pattern was 38.57% and 51.66%, respectively. The mutated genotype (GG) was almost two times higher in ALL patients when compared with the control group (15.71% and 8.33%, respectively) (Table 2 and Figures 1 and 3).
Figure 3.
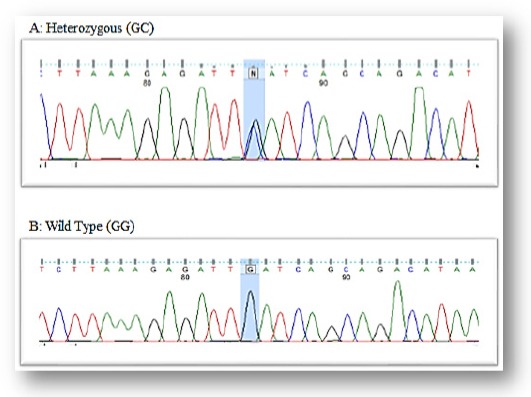
DNA Sequence of Different SNP in BclI (GR Gene) in All Patients.
C) N363S SNP
Results on Alleles and genotype frequencies for the N363S SNPs of glucocorticoid receptor were consistent to Hardy-Weinberg equilibrium as shown in Table 2. According to our results, two different alleles were exhibited in ALL patients with different alleles frequencies (Figure 1 to Figure 4). The frequency of genotype AA wild type was 92.8% in ALL patients and it was 97% in control subjects. The frequency of heterozygous genotype AG was 7.2% and 3% in ALL patients and control subjects, respectively. No mutated GG genotype was observed either the ALL group or the control group.
Figure 4.
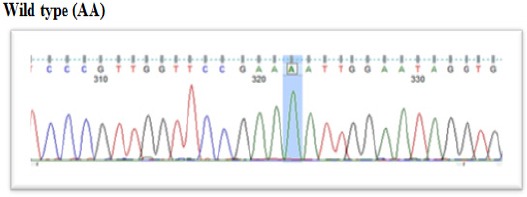
DNA Sequence of Different SNP in N363S (GR Gene) in All Patients.
Glucocorticoids-induced Side Effect (Toxicity)
The following clinical and laboratory data were collected retrospectively and scrutinized for potential adverse effects induced by glucocorticoids therapy.
1. Hepatic Toxicity
Hepatotoxicity was defined as elevated alanine transferase (ALT), aspartate transferase (AST) activities, and total bilirubin (TBIL) concentration. Severe toxicity was defined as AST or ALT higher than 2.5 folds and /or TBIL higher than 1.5 fold of the normal level (grade 3 toxicity) according to Common Toxicity Criteria report (NCI CTC).
The incidence of hepatic toxicity was insignificantly different between the studied three SNPs (p< 0.05) (Table 3).
Table 3.
Genotype (Wild vs.Hetero+Mutated) in ALL Children (n=70) with Liver Toxicity induced by Glucocorticoid Therapy
| SNP | Liver toxicity in wild type | Liver toxicity in SNP carrier | P Value (0.05) | OR* (95% CI) |
|---|---|---|---|---|
| ER22/23K | 50.70% | 0.00% | 1 | 0.971 (0.918-1.028) |
| BclI | 46.90% | 83.30% | 0.198 | 2.667 (0.526-7.330) |
| N363S | 47.70% | 40% | 0.356 | 1.097 (0.961-1.251) |
patients with liver toxicity versus total number of patients
2. Glucose Metabolism Abnormalities
Glucose metabolism abnormalities were defined as diabetes requiring insulin treatment, significant glycosuria for at least two days, or requiring medical intervention or hyperglycemia (fasting glucose >200mg/dl).
Glucose metabolism abnormalities were observed more frequently among the patients carrying the N363S polymorphism (GR) than the non-carrier (2/5 and 4/65; 40% and 6.15% respectively, P= 0.009, OR= 10.167; 1.302-79.339) (Table 4). Insignificant relationship was reported between the occurrence of glucose metabolism abnormalities and other studied gene polymorphisms and between age and gender.
Table 4.
Genotype (Wild Vs. hetero +Mutated) in ALL Children (n=70) Experienced Glucose Toxicity Induced by Glucocorticoids Therapy
| SNP | Glucose abnormality in wild type | Glucose abnormality in SNP carrier | P Value | OR* (95% CI) |
|---|---|---|---|---|
| ER22K | 13.50% | 0.00% | 0.758 | 0.984 (0.954-1.015) |
| BCII | 9.70% | 33.30% | 0.079 | 7.500 (1.039-54.116) |
| N363S | 6.15% | 40% | 0.009* | 10.167 (1.302-79.339) |
, Significant level P≤ 0.05; **, patients with glucose toxicity versus total number of patients.
3. Incidence of Infection
The incidence of infection was frequent that can be attributed to use of steroids. The incidence of infection was classified according to NCI-CTC criteria and evaluated with different genotype. This study showed non-significant differences concerning the incidence of infection between the patients who carried wild type of the genes versus heterozygous and mutated genotype (Table 5).
Table 5.
Genotype (Wild vs. Hetero+ Mutated) in ALL Children Suffered From Infection Induced by Glucocorticoid Therapy
| Gene | infection incidence in wild type | infection incidence in SNP carrier | P value (0.05) | OR* (95% CI) |
|---|---|---|---|---|
| ER22EK | 49.30% | 0.00% | 0.514 | 0.972 (0.920-1.027) |
| BCII | 4.70% | 50% | 0.635 | 1.065 (0.200-5.676) |
| N363 | 50.80% | 50% | 0.739 | 1.061 (0.064-17.657) |
Discussion
Glucocorticoids are important drugs for the treatment of ALL patients with multi drug protocols known to induce apoptosis of the blasts. In glucocorticoid sensitive cell, the apoptosis induction takes place in progressive steps. The first step is binding of glucocorticoids to glucocorticoid receptor. As the results of this binding a transactivation and/or transrepression of glucocorticoid responding gene. Leading to induce the apoptosis in glucocorticoid sensitive patients. In spite of GC benefits and efficacy, it often needs to be adjusted by intra-individual variability and can lead to treatment failure or induction of side effect. Despite the fact that glucocorticoid resistance is one of the main causes of the treatment failure in ALL patients, little information is known about the possible mechanism of resistant. Although a number of studies reflected the relation between the genetic polymorphisms and the drug outcomes in ALL patients, very few studies reflected the side effects induced by this drug.
This study investigated the presence of common SNPs in the GC receptor gene (NR3C1). Three polymorphisms in the coding region of GR gene were analyzed in 70 ALL patients and 60 control subjects. These genetic variations were identical to previously reported polymorphism ER22/23EK, BCII, and N3635. Previous studies showed that this polymorphism increased corticoid sensitivity (VanRossum and Lamberts, 2004; Rosmond et al., 2001).
ER22/23EK polymorphism is located in transactivation domains of NC3R1 in codons 22 and 23 and they are usually studied together. Lately, the action of this polymorphism has been illustrated; it changes the balance of the NC3R1 protein translation isoforms A and B. This polymorphism prefers to translate initiation from GR-A than from GR-B, and consequently, a decrease in transactivation of this gene with relative glucocorticoid resistance and higher insulin sensitivity (Tissing et al., 2003). This polymorphism is more frequent in elderly individuals and it may have a useful effect on survival. This study did not observe homozygous mutated ER22/23EK polymorphism in neither in ALL group nor in control group. Moreover, the heterozygous polymorphism was not found in ALL patients but in the control group with 6.66%. This result is in agreement with previous study that was found the minor allele of the ER22/23EK polymorphism in a lower percentage than in a healthy population of the same ethnicity (4% versus 7.4%, respectively). As well as the same finding was reported by (Tissing et al., 2005) who found the ER22/23EK polymorphism in only 4% of the ALL patients as compared with 6% and 7.4% in the normal and healthy population from the same ethnicity. In a Saudi population (in this study) 6.66% the incidence of the ER22/23EK genotype is considered higher when it is compared with the Caucasian and Brazilian subpopulation (2%) and with Africans, Chinese, and other subset of Asians population where this polymorphism dot detected in this ethnicity (0%). The concept of the ER22/23EK carrier is resistant to the effects of glucocorticoids in children (central and systemic) has been getting attention by many authors. Finkin et al., (2007) recorded that that are carrying an ER22/23EK polymorphism showed complete growth between the age of three months and one year and also children with this polymorphism is taller than noncarrying this SNP. This study also showed that fasting insulin levels were lower in the ER22/23EK carrier. Both studies support the concept of GC-resistant of the carrier of this SNP.
BCII polymorphism is located in exon 2 of the NR3C1 gene. The nucleotide alteration was identified as a C → G substitution, 646 nucleotides downstream of exon 2 (intronic polymorphism). There is an association between the BCII polymorphism and younger age onset. Girls carrying BCII SNPs suffer from juvenile arthritis had shown higher inflammation rate (Kostik et al., 2011). Also, the homozygous mutated genotype of BCII (GG) reported more severe damage in their lungs in cystic fibrosis (Corvol et al., 2007). This finding supporting the possible decrease in glucocorticoid sensitivity in the children carrying this polymorphism. However, BCII GG genotype recorded an increase to response to glucocorticoid in children with Crohn’s disease. So, the BCII GG genotype can exert different behavior depending on the disease and ethnicity of the afflicted individual. BcII polymorphism is related to increased sensitivity to glucocorticoid as measured with the response of serum cortisol level after admistration of 1 mg of dexamethasone evening before in healthy adults (Van Rossum et al., 2003; Huizenga et al., 1998). This study results showing that heterozygous BcII genotype was the most prevalence genotype (51.66%) whereas the most prevalence genotype in ALL patients was the wild type (45.7%). However, the GG genotype (mutated) in ALL patients was recorded two times more than that of the control group 15.71% and 8.33% respectively .Statistics analysis showed no significant difference between two groups P= 0.285. In other words, BcII polymorphism had not contributed to the development in the childhood ALL. The results of this study are in agreement with a previous study done by Tissing et al., (2005) and Marino et al., (2009) on a different population. The incidence of G allele in the Saudi population (8.33%) was too low when compared with Asian population (33.33%) but its incidence was similar to Caucasian population (7%), African population (6%), and Brazilian population (7.5%) (Manoel Carlos et al., 2014). This study data show, it is clear that the Saudi population, like other Caucasian /African/ Brazilian populations, is significantly different than Asian population for genotype and allele frequency of the BcII polymorphism. BcII polymorphism is the most pertinent polymorphism of the GC gene clinically as reported in the literature (Tissing et al., 2005). In the past, this polymorphism was detected by RFLP-based technique, but now, the C/G exact mutation was found as a single nucleotide polymorphism in intron 2, 646bp downstream from exon 2 by a sequencing techniques as well as a specific Real time PCR process has been developed. In the many studies, BcII polymorphism showed to increase the sensitivity of dexamethasone suppression test with the development of side effects of glucocorticoid (body mass index, abdominal obesity, and blood pressure (Rosmond et al., 2000; Ukkola et al., 2001). However, this study toxicity data in a Saudi population. The BcII polymorphism was not associated with the liver toxicity, glucose abnormality, and infection in ALL patients.
N363S (A1220G) GR gene polymorphism is located in exon 2 of GC gene codon 363and results of this polymorphism the amino acid changes from asparagine (N) to serine (S). This polymorphism increases the sensitivity of GC receptor to interact with glucocorticoid hormone and also increase the action of the hormone. The N363S was detected in low frequencies (3% to 6%) in different populations and in the heterozygous form. The results of this showed the absence of mutated homozygous (GG) genotype in both control group and ALL group. The incidence of heterozygous allele was 7.2% in ALL patients and 3% in the control group, which is in accordance with the findings of different studies in randomly selected in the different healthy population before and also in the relation of this polymorphism with the childhood ALL (Van Rossum et al., 2003). As previously mentioned, no N363 polymorphism was detected in our study population, Brazilian subjects by Asian origin and also South Asian subjects living in Europe whom showing very low frequency of N363S polymorphism (0.2%) (Syed et al., 2004) .Additionally, Japanese and Chinese population had not carried this polymorphism in the GC receptor gene (Koyano et al., 2005).
The different study linked between this polymorphism and different diseases around the world. In Australia, one study linked the N363S polymorphism with the raised body mass index and with the coronary artery diseases (Lin et al., 2003). As well as, N363Spolymorphism showed association with central obesity in males from England (Dobson et al., 2001), and overweight with Type 2 diabetes mellitus in French men (Roussel et al., 2003). However, N363S polymorphism did not show any correlation with GCs sensitivity in Danish and Swedish populations (Echwald et al., 2001). The toxicology data in this study revealeda significant difference between ALL patients carrying N363S polymorphism and wild type (P= 0.009) and the high risk factor OR (CI 95%) = 10.167 (1.302-79.339) in toxicity of glucose abnormality. Glucose abnormality during glucocorticoid treatment is steroid induce diabetes mellitus. There is a lack of the studies and literature on the role of higher sensitivity to glucocorticoids and N363S polymorphism and development of type 2 diabetes mellitus or other glucose metabolism abnormality. However, glucocorticoids might promote glucose production in the liver through gluconeogenesis and simultaneous insulin resistance leading to hyperglycemia (Tappy et al., 1998).
In conclusion, in this study, the three studied SNPs in GR gene showed no contribution and association among them and development of ALL in the children. The ER22/23EK and BCII polymorphisms did not have any correlation with liver toxicity, glucose abnormality, and infection in ALL children during treatment by GCs. Patients carrying N363S polymorphism (homozygous and heterozygous) had a tendency to develop hyperglycemia during treatment with…. Nevertheless, this finding needs to be confirmed by conducting a study on larger population and establishing a strong relationship and associations between these polymorphisms genotypes that display this side effect toxicities of glucocorticoids therapy.
Conflict of interest
No conflict of interest to declare.
Acknowledgments
This study was financially supported by the King Abdulaziz City for Science and Technology (Grant Number AT-393-30).
References
- 1.American Cancer Society. Cancer facts and figures. Atlanta: American cancer society; 2018. available at: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2018.html . [Google Scholar]
- 2.Bachmann AW, Sedgley TL, Jackson RV, et al. Glucocorticoid receptor polymorphisms and post-traumatic stress disorder. Psychoneuroendocrinology. 2005;3:297–6. doi: 10.1016/j.psyneuen.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Therapy Evaluation Program. Common toxicity criteria (CTC):CTC Version 2.0. Rockville, ML: CTEP; 1999. available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcv2nom-4-30-99-final3.pdf . [Google Scholar]
- 4.Chen H-L, Li L-R. Glucocorticoid receptor gene polymorphisms and glucocorticoid resistance in inflammatory bowel disease:a meta-analysis. Dig Dis Sci. 2012;57:3065–5. doi: 10.1007/s10620-012-2293-2. [DOI] [PubMed] [Google Scholar]
- 5.Ciriaco M, Ventrice P, Russo G, et al. Corticosteroid-related central nervous system side effects. J Pharmacol Pharmacother. 2013;4:94–8. doi: 10.4103/0976-500X.120975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corvol H, Nathan N, Charlier C, et al. Glucocorticoid receptor gene polymorphisms associated with progression of lung disease in young patients with cystic fibrosis. Respir Res. 2007;8:88. doi: 10.1186/1465-9921-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ctcv2nom-4-30-99-final3.pdf. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcv2nom-4-30-99-final3.pdf .
- 8.De Iudicibus S, Stocco G, Martelossi S, et al. Association of BclI polymorphism of the glucocorticoid receptor gene locus with response to glucocorticoids in inflammatory bowel disease. Gut. 2007;56:1319. doi: 10.1136/gut.2006.116160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Melo MB, Sales TS, Lorand-Metze I, Costa FF. Rapid method for isolation of DNA from glass slide smears for PCR. Acta Haematol. 1992;87:214–5. doi: 10.1159/000204771. [DOI] [PubMed] [Google Scholar]
- 10.Decorti G, De Iudicibus S, Stocco G, et al. Glucocorticoid receptor polymorphisms in inflammatory bowel disease. Gut. 2006;55:1053–4. [PMC free article] [PubMed] [Google Scholar]
- 11.Dobson MG, Redfern CP, Unwin N, Weaver JU. The N363S polymorphism of the glucocorticoid receptor:potential contribution to central obesity in men and lack of association with other risk factors for coronary heart disease and diabetes mellitus. J Clin Endocrinol Metab. 2001;86:2270–4. doi: 10.1210/jcem.86.5.7465. [DOI] [PubMed] [Google Scholar]
- 12.Echwald SM, Sørensen TI, Andersen T, Pedersen O. The Asn363Ser variant of the glucocorticoid receptor gene is not associated with obesity or weight gain in Danish men. Int J Obes Relat Metab Disord. 2001;25:1563–5. doi: 10.1038/sj.ijo.0801744. [DOI] [PubMed] [Google Scholar]
- 13.Encío IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;266:7182–8. [PubMed] [Google Scholar]
- 14.Ferrari P. Genetics of the mineralocorticoid system in primary hypertension. Curr Hypertens Rep. 2002;4:18–24. doi: 10.1007/s11906-002-0048-8. [DOI] [PubMed] [Google Scholar]
- 15.Halsall DJ, Jian'an Luan NH. Glucocorticoid receptor variant and body mass index. BMJ. 2018. [accessed 11 Apr 2018]. https://www.bmj.com/rapid-response/2011/10/28/glucocorticoid-receptor-variant-and-body-mass-index .
- 16.Herrera C, Marcos M, Carbonell C, et al. Association between allelic variants of the human glucocorticoid receptor gene and autoimmune diseases:A systematic review and meta-analysis. Autoimmun Rev. 2018 doi: 10.1016/j.autrev.2017.11.034. doi:10.1016/j.autrev.2017.11.034. [DOI] [PubMed] [Google Scholar]
- 17.Huizenga NA, Koper JW, De Lange P, et al. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83:144–1. doi: 10.1210/jcem.83.1.4490. [DOI] [PubMed] [Google Scholar]
- 18.Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–30. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karagas MR, Greenberg ER, Spencer SK, Stukel TA, Mott LA. Increase in incidence rates of basal cell and squamous cell skin cancer in New Hampshire, USA. New Hampshire skin cancer study group. Int J Cancer. 1999;81:555–9. doi: 10.1002/(sici)1097-0215(19990517)81:4<555::aid-ijc9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 20.Koeijvoets KCMC, van Rossum EFC, Dallinga-Thie GM, et al. A functional polymorphism in the glucocorticoid receptor gene and its relation to cardiovascular disease risk in familial hypercholesterolemia. J Clin Endocrinol Metab. 2006;91:4131–6. doi: 10.1210/jc.2006-0578. [DOI] [PubMed] [Google Scholar]
- 21.Koper JW, Stolk RP, de Lange P, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99:663–8. doi: 10.1007/s004390050425. [DOI] [PubMed] [Google Scholar]
- 22.Kostik MM, Klyushina AA, Moskalenko MV, Scheplyagina LA, Larionova VI. Glucocorticoid receptor gene polymorphism and juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2011;9:2. doi: 10.1186/1546-0096-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koyano S, Saito Y, Sai K, et al. Novel genetic polymorphisms in the NR3C1 (glucocorticoid receptor) gene in a Japanese population. Drug Metab Pharmacokinet. 2005;20:79–4. doi: 10.2133/dmpk.20.79. [DOI] [PubMed] [Google Scholar]
- 24.Lauten M, Cario G, Asgedom G, Welte K, Schrappe M. Protein expression of the glucocorticoid receptor in childhood acute lymphoblastic leukemia. Haematologica. 2003;88:1253–8. [PubMed] [Google Scholar]
- 25.Lee W, Lockhart AC, Kim RB, Rothenberg ML. Cancer Pharmacogenomics:Powerful Tools in Cancer Chemotherapy and Drug Development. Oncologist. 2005;10:104–1. doi: 10.1634/theoncologist.10-2-104. [DOI] [PubMed] [Google Scholar]
- 26.Lin RCY, Wang XL, Morris BJ. Association of coronary artery disease with glucocorticoid receptor N363S variant. Hypertension. 2003;41:404–7. doi: 10.1161/01.HYP.0000055342.40301.DC. [DOI] [PubMed] [Google Scholar]
- 27.Manenschijn L, van den Akker ELT, Lamberts SWJ, van Rossum EFC. Clinical features associated with glucocorticoid receptor polymorphisms. Ann N Y Acad Sci. 2009;1179:179–98. doi: 10.1111/j.1749-6632.2009.05013.x. [DOI] [PubMed] [Google Scholar]
- 28.Marino S, Verzegnassi F, Tamaro P, et al. Response to glucocorticoids and toxicity in childhood acute lymphoblastic leukemia:role of polymorphisms of genes involved in glucocorticoid response. Pediatr Blood Cancer. 2009;53:984–1. doi: 10.1002/pbc.22163. [DOI] [PubMed] [Google Scholar]
- 29.Murray JC, Smith RF, Ardinger HA, Weinberger C. RFLP for the glucocorticoid receptor (GRL) located at 5q11-5q13. Nucleic Acids Res. 1987;15:6765. doi: 10.1093/nar/15.16.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh KS, Patel H, Gottschalk RA, et al. Anti-inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity. 2017;47, 298-309:e5. doi: 10.1016/j.immuni.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelt AC. Glucocorticoids:effects, action mechanisms, and therapeutic uses. New York: Nova Science Publishers; 2011. available at: https://www.novapublishers.com/catalog/product_info.php?products_id=12652 . [Google Scholar]
- 32.Pui C-H, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia:where are we going and how do we get there? Blood. 2012;120:1165–4. doi: 10.1182/blood-2012-05-378943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–8. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 34.Rosmond R, Bouchard C, Björntorp P. Tsp509I polymorphism in exon 2 of the glucocorticoid receptor gene in relation to obesity and cortisol secretion:cohort study. BMJ. 2001;322:652–3. doi: 10.1136/bmj.322.7287.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosmond R, Chagnon YC, Holm G, et al. A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic-pituitary-adrenal axis. Obes Res. 2000;8:211–8. doi: 10.1038/oby.2000.24. [DOI] [PubMed] [Google Scholar]
- 36.Roussel R, Reis AF, Dubois-Laforgue D, et al. The N363S polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin Endocrinol (Oxf) 2003;59:237–1. doi: 10.1046/j.1365-2265.2003.01831.x. [DOI] [PubMed] [Google Scholar]
- 37.Russcher H, Smit P, van den Akker ELT, et al. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid-regulated gene expression. J Clin Endocrinol Metab. 2005;90:5804–50. doi: 10.1210/jc.2005-0646. [DOI] [PubMed] [Google Scholar]
- 38.Savas M, van Rossum EFC. In Module in Biomedical Sciences. Elsevier; 2018. Impact of glucocorticoid receptor polymorphisms on glucocorticoid action. doi:10.1016/B978-0-12-801238-3.65339-4. [Google Scholar]
- 39.Schoor NMV, Dennison E, Lips P, Uitterlinden AG, Cooper C. Serum fasting cortisol in relation to bone, and the role of genetic variations in the glucocorticoid receptor. Clin Endocrinol. 2007;67:871–8. doi: 10.1111/j.1365-2265.2007.02978.x. [DOI] [PubMed] [Google Scholar]
- 40.Sørensen HT, Mellemkjaer L, Nielsen GL, et al. Skin cancers and non-hodgkin lymphoma among users of systemic glucocorticoids:a population-based cohort study. J Natl Cancer Inst. 2004;96:709–1. doi: 10.1093/jnci/djh118. [DOI] [PubMed] [Google Scholar]
- 41.Souza MCLA, Martins CS, Silva-Junior IM, et al. NR3C1 polymorphisms in Brazilians of Caucasian, African, and Asian ancestry:glucocorticoid sensitivity and genotype association. Arq Bras Endocrinol Metabol. 2014;58:53–1. doi: 10.1590/0004-2730000002868. [DOI] [PubMed] [Google Scholar]
- 42.Syed AA, Irving JAE, Redfern CPF, et al. Low prevalence of the N363S polymorphism of the glucocorticoid receptor in South Asians living in the United Kingdom. J Clin Endocrinol Metab. 2004;89:232–5. doi: 10.1210/jc.2003-030995. [DOI] [PubMed] [Google Scholar]
- 43.Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab. 1994;79:1063–9. doi: 10.1210/jcem.79.4.7962275. [DOI] [PubMed] [Google Scholar]
- 44.Tissing WJE, Meijerink JPP, den Boer ML, et al. Genetic variations in the glucocorticoid receptor gene are not related to glucocorticoid resistance in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2005;11:6050–6. doi: 10.1158/1078-0432.CCR-04-2097. [DOI] [PubMed] [Google Scholar]
- 45.Tissing WJE, Meijerink JPP, den Boer ML, Pieters R. Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia. Leukemia. 2003;17:17–5. doi: 10.1038/sj.leu.2402733. [DOI] [PubMed] [Google Scholar]
- 46.Tomlinson JW, Stewart PM. Cortisol metabolism and the role of 11β-hydroxysteroid dehydrogenase. Baillieres Best Pract Res Clin Endocrinol Metab. 2001;15:61–8. doi: 10.1053/beem.2000.0119. [DOI] [PubMed] [Google Scholar]
- 47.Travlos GS. Normal structure, function, and histology of the bone marrow. Toxicol Pathol. 2006;34:548–5. doi: 10.1080/01926230600939856. [DOI] [PubMed] [Google Scholar]
- 48.Ukkola O, Rosmond R, Tremblay A, Bouchard C. Glucocorticoid receptor Bcl I variant is associated with an increased atherogenic profile in response to long-term overfeeding. Atherosclerosis. 2001;157:221–4. doi: 10.1016/s0021-9150(00)00712-7. [DOI] [PubMed] [Google Scholar]
- 49.van den Akker ELT, Nouwen JL, Melles DC, et al. Staphylococcus aureus nasal carriage is associated with glucocorticoid receptor gene polymorphisms. J Infect Dis. 2006;194:814–8. doi: 10.1086/506367. [DOI] [PubMed] [Google Scholar]
- 50.van Raalte DH, van Leeuwen N, Simonis-Bik AM, et al. Glucocorticoid receptor gene polymorphisms are associated with reduced first-phase glucose-stimulated insulin secretion and disposition index in women, but not in men. Diabet Med. 2012;29:e211–6. doi: 10.1111/j.1464-5491.2012.03690.x. [DOI] [PubMed] [Google Scholar]
- 51.van Rossum EFC, Koper JW, Huizenga NATM, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes. 2002;51:3128–4. doi: 10.2337/diabetes.51.10.3128. [DOI] [PubMed] [Google Scholar]
- 52.van Rossum EFC, Koper JW, van den Beld AW, et al. Identification of the BclI polymorphism in the glucocorticoid receptor gene:association with sensitivity to glucocorticoids in vivo and body mass index. Clin Endocrinol (Oxf) 2003;59:585–2. doi: 10.1046/j.1365-2265.2003.01888.x. [DOI] [PubMed] [Google Scholar]
- 53.van Rossum EFC, Lamberts SWJ. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59:333–7. doi: 10.1210/rp.59.1.333. [DOI] [PubMed] [Google Scholar]
- 54.Vitellius G, Fagart J, Delemer B, et al. Three novel heterozygous point mutations of NR3C1 causing glucocorticoid resistance. Hum Mutat. 2016;37:794–3. doi: 10.1002/humu.23008. [DOI] [PubMed] [Google Scholar]
- 55.Wall AM, Rubnitz JE. Pharmacogenomic effects on therapy for acute lymphoblastic leukemia in children. Pharmacogenomics J. 2003;3:128–5. doi: 10.1038/sj.tpj.6500174. [DOI] [PubMed] [Google Scholar]
- 56.Werner SC. Modification of the classification of the eye changes of Graves'disease:recommendations of the Ad Hoc Committee of the American Thyroid Association. J Clin Endocrinol Metab. 1977;44:203–4. doi: 10.1210/jcem-44-1-203. [DOI] [PubMed] [Google Scholar]
- 57.Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids. 2012;77:1041–9. doi: 10.1016/j.steroids.2012.05.007. [DOI] [PubMed] [Google Scholar]