

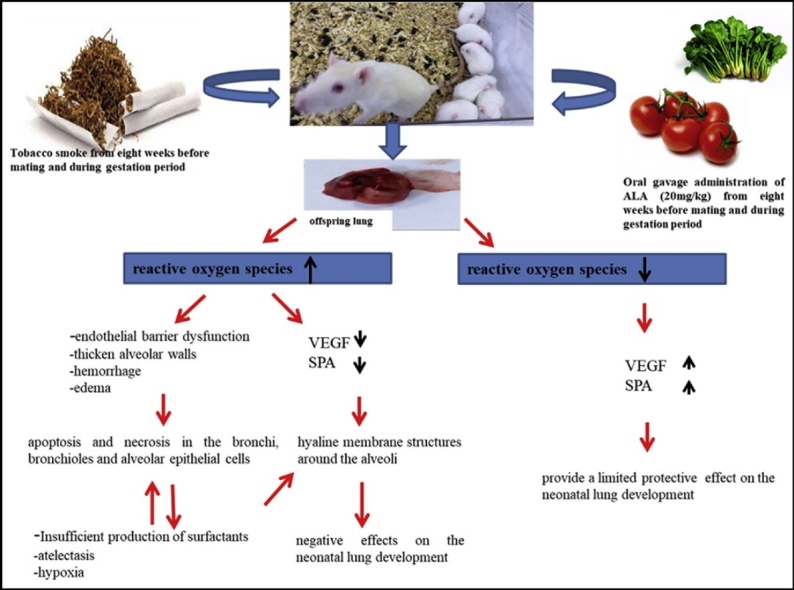
Graphical abstract
Keywords: Alpha lipoic acid, Lung, Neonate, Tobacco smoke, Pregnancy
Highlights
-
•
Exposure to tobacco smoke during the prenatal period affects many multiple organ system developments in neonatals.
-
•
Maternal tobacco smoke exposure could effect on neonatal lung development by increasing oxidative stress in the lungs.
-
•
Vascular endothelial growth factor and surfactant protein-A play a key role in lung development and damage.
-
•
Alpha lipoic acid can limited protective effect on the neonatal lung development against oxidative stress originating from tobacco smoke.
Abstract
This study was carried out to determine the changes in the lungs of the rat pups exposed to tobacco smoke during pregnancy period and to investigate the protective effects of alpha lipoic acid, which is administered during pregnancy, on these changes.
Spraque-Dawley female rats were divided into four groups: control, tobacco smoke (TS), tobacco smoke + alpha lipoic acid (TS + ALA) and alpha lipoic acid (ALA). The rats in control group were untreated. Rats were exposed to TS twice a day for one hour starting from eight weeks before mating and during pregnancy. 20 mg / kg of ALA was administered to rats. On 7th and 21st days 7 of the pups from each group were decapitated. Histological, morphometric, biochemical and quantitative real-time RT-PCR analyzes were performed.
Histopathological and biochemical changes were observed in TS group. While a significant decrease was observed both in SP-A and VEGF immunoreactivities and mRNA levels, caspase-3 immunoreactivity and TUNEL positive cells were increased in TS group.
It is suggested that prenatal TS exposure leads to morphological and histopathological changes on lung development by causing oxidative damage in lungs of neonatal rats and the maternal use of ALA can provide a limited protective effect on the neonatal lung development against this oxidative stress originating from TS. Although pregnant women are increasingly aware on health risks of smoking, environmental tobacco smoke exposure is still a widespread problem. For this reason, it is thought that this damage can be partially reduced by some antioxidant supplements in pregnancy.
1. Introduction
Exposure to tobacco smoke during the prenatal period affects multiple organ system developments in neonatals such as nervous, respiratory and cardiovascular systems [1]. The harmful chemicals in all forms of tobacco smoke, such as nicotine and carbon monoxide (CO), rapidly pass the placental barrier and accumulate overdose in the fetal compartment [2]. It is known that these chemicals cause oxidation by increasing the production of superoxide anion from free radicals, and these oxidants cause damage to alveolar-capillary membranes. Therefore, it is thought that the underlying mechanism of damage caused by tobacco smoke on the lungs is due to oxidants [3,4]. Surfactant protein-A (SP-A) is a surface tension reducing apoprotein which is very important in regulation of the surfactant concentration [5,6]. Alterations in surfactant insufficiency or composition are one of the findings associated with inadequate lung development in the prenatal and neonatal period [7]. Vascular endothelial growth factor (VEGF) plays a key role in lung development and damage, regulating endothelial cell proliferation, mitogenesis, migration, differentiation, vascular permeability, mobilization of endothelial cell precursors [8].
Programmed cell deaths, or apoptosis, of lung cells play an essential role in normal development of the respiratory system [9,10]. Caspase-3 is involved in characteristic morphological changes in apoptotic cells, such as chromatin condensation, DNA fragmentation, and membrane budding [11]. Thus, Caspase-3 is an important predictor of the change in apoptosis levels in the lungs.
Alpha lipoic acid (ALA) is a non-enzymatic, natural, metabolic antioxidant [12]. Potential antioxidant function is achieved by passing two sulfur atoms in the structure through oxidation-reduction reactions [13].
This study was fulfill to detect the histological, immunohistochemical and biochemical changes in the lungs of the rat pups exposed to tobacco smoke during gestation period, and to research the protective effects of ALA, which is administered during pregnancy, on these changes.
2. Materials-methods
2.1. Ethical approval
All experimental procedures including animals were confirmed by the local ethics committee of Firat University (07.10.2015, 2015/17-159).
2.2. Experimental design
This study was conducted at the Experimental Research Unit of Firat University (FUDAM). Rats were fed with standard rat chow and tap water freely available. 12-h light-dark cycles, 21 ± 1 °C standard temperature and humidity conditions were provided. In the study, six-weeks old 28 Spraque-Dawley female rats weighing 160 ± 10 g were used. Animals were group housed from the start of the experiment until one week before the birth and then singly housed until the end of the experiment. The minimum number of animals required for the statistical evaluation of the difference between the groups in the obtained data was preferred. Rats were randomly divided into four groups: control, tobacco smoke (TS), tobacco smoke + alpha lipoic acid (TS + ALA) and alpha lipoic acid (ALA) (n = 7). The rats in the control group were untreated. Rats in the TS and TS + ALA groups were exposed to tobacco smoke twice a day for one hour starting from eight weeks before mating and during gestation period. 20 mg / kg of ALA (cat: 29,862 Lot: 002241-20161019, DL-α-Lipoic acid, Chem-Impex Int’l Inc, USA) was administered via oral gavage to the ALA and TS + ALA groups [14]. A glass cage was planned specifically for exposing the rats to tobacco smoke. The smoke of 10 g tobacco was put in the glass cage via air pump (AP-001 Aquarium Air Pump Xilong, China). Once after the birth, all administrations were stopped. On the 7th and 21 st days, 7 of the pups from each group were decapitated under xylazine (5 mg/kg)–ketamine (50 mg/kg) anesthesia. The lungs were removed rapidly and utilized for morphometric, histological, biochemical and quantitative real-time polymerase chain reaction (qRT-PCR) analyses as described below.
2.3. Histological evaluation
Lung tissue samples were fixed in 10% buffered formaldehyde, and then embedded in paraffin. Samples were cut at 5 μm thicknesses and stained with hematoxylin and eosin (H&E), Periodic Acid Schiff (PAS) and Masson's Trichrome staining and examined under light microscope (NovelN-800 M, Ningbo, China). Severity of lung injury was semi-quantitatively assesed with the following changes: increased inflammatory cell, hemorrhagic areas, edema, interalveolar septal thickening, decrease in alveolar numbers, degeneration of some bronchi and bronchial epithelium, epithelial cells that were fallen into the lumen and hyaline membrane formation. Histoscores were given as 0= absent, 1= weak, 2= moderate and 3= strong for each parameter.
2.4. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
ApopTagPlus Peroxidase in situ Apoptosis Detection Kit (Chemicon, cat no: S7101, USA) was used for determination of apoptotic cells. In the evaluation of TUNEL staining, cells with blue nuclei were normal, while brown nuclei belonged to apoptotic cells. At least 1000 cells were counted on each field. Apoptotic index was calculated as a ratio of the TUNEL positive cell number to the total cell number (normal + apoptotic cells).
2.5. Immunohistochemical evaluation
Avidin-biotin-peroxidase complex method was used to define SP-A, VEGF, Caspase-3 immunreactivities (RB-9031-P lot No: 9031P1505 A, Polyclonal Anti-VEGF Antibody, Thermo Scientific, SC-13977 lot No: G2314, Polyclonal Anti-SP-A Antibody, Santa Cruz Biotechonology, PA5-16335 lot No: QD2017719, Polyclonal Anti-Caspase-3 Antibody, Thermo Scientific) in lung tissue. The immunohistochemical histoscore was calculated with immunoreactivity prevalence (0.1: < 25%, 0.4: 26–50%, 0.6: 51–75%, 0.9: 76–100 %) and severity (0: no, +0.5: very little, +1: little, +2: medium, +3: severe). (Histoscore = prevalence × severity).
2.6. Biochemical evaluation of lung tissue
Malondialdehyde (MDA) concentrations were assessed according to a modified method of Placer et al. [15]. Glutathione (GSH) levels and superoxide dismutase (SOD) activities were measured respectively using the methods of Beutler and Sun et al. [16,17].
2.7. Calculation of relative lung weight
After decapitation, the lung tissues of pups were removed from peripheral fat tissues and then calculated the relative lung weights (Relative lung weight = (absolute lung weight (g) / body weight) x100).
2.7. Morphometric analysis
The radial alveolar count (RAC) method was performed strictly according to the method of Cooney and Thurlbeck [18]. According to this method, a linear line was drawn from respiratory bronchiole to the closest connective tissue septum. The number of alveoli cut by this line was then counted. Ten such counts were done from each pup lung.
2.8. qRT-PCR analysis
For Total RNA isolation from lung tissues, TRIzol reagent was used (Invitrogen, Carlsbad, CA). Random primed cDNAs were produced by reverse-transcription of total RNA samples by using High Capacity RNA to cDNA Synthesis kit (Invitrogen, Carlsbad, CA). A real-time PCR analysis was applied with the ABI Prism 7500 Fast Real Time PCR Instrument (Applied Biosystems, CA) using Tag Man Master Mix (Applied Biosystems, CA). Each value was standardized to the levels of GAPDH. The samples were quantified for SP-A (Rn-04338808_g1, Thermo Scientific) and VEGF (Rn01511610_m1, Thermo Scientific), utilizing the comparative Ct (ΔΔCt) method [19].
2.9. Statistical analysis
Statistical analyses were implemented by SPSS 22.0 (Statistical Package for Social Sciences) software. The values were presented as the means ± standard deviation. The probability values (p) less than 0.05 were accepted as statistically significant. Statistical differences of the multiple groups with normal distribution were measured by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test.
3. Results
3.1. Body weight and relative lung weight values
It was shown that tobacco smoke exposure and ALA administration led to body weight changes. Significant decrease was observed in the weights of 7 days old pups in the TS group compared to the control group (p < 0.05). No significant difference was found between the other groups (p > 0.05). Also body weight and relative lung weight values are shown in Table 1.
Table 1.
A: Values of body weight, relative lung weight, radial alveolar count 7 and 21 days old pups. B: Biochemical results in 7 and 21 days old pups lung tissues.
| Control |
TS |
TS + ALA |
ALA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (days) | 7 | 21 | 7 | 21 | 7 | 21 | 7 | 21 | |
| Body weight(g) | female | 9,86 ± 0.40 | 29,00 ± 1,45 | 8,85 ± 0.30a | 27,30 ± 1,56 | 9,84 ± 0.70b | 30,6 ± 2,02 | 9,70 ± 0.57b | 32,56 ± 2,83 b |
| male | 9,80 ± 0.57 | 30,2 ± 0,27 | 8,84 ± 0.45a | 29,5 ± 0,93 | 9,37 ± 0.36b | 31,8 ± 1,15b | 9,82 ± 0.74b | 31,5 ± 1,73 | |
| Relative lung weight (g) | female | 2,36 ± 0,09 | 1,28 ± 0,10 | 3,19 ± 0,16a | 1,23 ± 0,55 | 2,86 ± 0,25a | 1,28 ± 0,06 | 2,44 ± 0,24b | 1,19 ± 0,08 |
| male | 2,41 ± 0,12 | 1,18 ± 0,01 | 3,24 ± 0,16a | 1,41 ± 0,12 | 2,88 ± 0,37a | 1,21 ± 0,13 | 2,36 ± 0,22b | 1,17 ± 0,10 | |
| Radial alveolar count (mean) | 5,33 ± 0.33 | 7,42 ± 0.20 | 3,00 ± 0.25a | 4,00 ± 0.53a | 4,5 ± 0.42 b | 6,28 ± 0.28b | 5,16 ± 0.30b | 7,28 ± 0.18b | |
3.2. Morphometric measurements
In morphometric measurements performed with the RAC method, the alveolar numbers in the lung sections from 7- and 21-day old pups of the control and ALA groups were observed to be close to each other (p > 0.05). A statistically significant decrease was found in the alveolar numbers of the TS group when compared to the control group (p < 0.05). There was a significant increase in the alveolar numbers in the TS + ALA group compared to the TS group (p < 0.05) (Table 1). In addition, an average 40% increase in the number of alveoli on days 7th and 21 st was calculated in the control, ALA and TS + ALA groups. But, this difference was 33% in the TS group.
3.3. Histological evaluation
Lung tissue slides of 7 and 21 days old pups were examined under the light microscope. In the histological evaluations; normal morphology was observed in the control group (Fig. 1a, Fig. 2a, b). Increased inflammatory cell, hemorrhage, edema, interalveolar septal thickening, degeneration of some bronchi and bronchial epithelium, epithelial cells debris in lumen and hyaline membrane formation were examined in the TS group compared to the control group (Fig. 1b–d, Fig. 2c, d). These histopathological findings were observed to decrease in the TS + ALA group (p < 0.05). Also, hyaline membrane formation was not defined in this group (Fig. 1e, Fig. 2e). On the other hand, in the ALA group similar findings were detected with the control group (p > 0.05) (Fig. 1f, Fig. 2f). The results of the histoscores are shown in Table 2.
Fig. 1.

A: Photomicrographs of lung slides of 7 days old pups examined by light microscope showing: (a) control group; normal morphology (HEx10); (b) TS group; inflammatory cell increase (star), edema (white arrow), hyaline membrane formation (plus) (HEx10); (c) TS group; degeneration of bronchi epithelium (trigon), epithelial cells debris in lumen (long arrow), hemorrhagic areas (thick arrow) (HEx20); (d) TS group; PAS (+) hyaline membrane formation (plus) (PASx40); (e) TS + ALA group; interalveolar septal thickening (heptagon), hemorrhagic areas (thin arrow), edema (white arrow), inflammatory cell increase (star) (HEx10); (f) ALA group; normal morphology (HEx10).
Fig. 2.

Photomicrographs of lung slides of 21 days old pups examined by light microscope showing: (a) control group; normal morphology (HEx10); (b) control group; perivascular connective tissue fibers (arrow) (Masson's Trichrome x20); (c) TS group; hemorrhagic areas (thin arrow), inflammatory cell increase (star), epithelial cells debris in lumen (long arrow), hyaline membrane formation (plus) (HEx10); (d) TS group; increase connective tissue (arrow) (Masson's Trichrome x40); (e) TS + ALA group; inflammatory cell increase (star), interalveolar septal thickening (heptagon), epithelial cells debris in lumen (long arrow) (HEx10); (f) ALA group; normal morphology (HEx10).
Table 2.
The histoscores in 7 and 21 days old pups lung tissues.
| Control |
TS |
TS+ALA |
ALA |
|||||
|---|---|---|---|---|---|---|---|---|
| Age (days) | 7 | 21 | 7 | 21 | 7 | 21 | 7 | 21 |
| inflammatory cell increase | 0,00±0,00 | 0,14±0,37 | 2,00±1,00a | 2,29±0,75a | 1,00±0,81ab | 1,00±0,81ab | 0,29±0,48b | 0,29±0,48b |
| hemorrhagic areas | 0,14±0,37 | 0,14±0,37 | 2,71±0,48a | 2,57±0,53a | 1,29±0,48ab | 1,29±0,75ab | 0,71±0,48b | 0,71±0,48b |
| Edema | 0,00±0,00 | 0,14±0,37 | 2,57±0,78a | 2,57±0,78a | 1,43±0,78ab | 1,14±0,69ab | 0,86±0,69b | 0,86±0,69b |
| interalveolar septal thickening | 0,29±0,48 | 0,29±0,48 | 2,29±0,75a | 2,00±1,00a | 1,00±0,57ab | 1,00±0,48ab | 0,14±0,37b | 0,43±0,53b |
| degeneration of epithelium | 0,00±0,00 | 0,00±0,00 | 2,14±0,69a | 2,29±0,75a | 0,57±0,53b | 0,57±0,53b | 0,29±0,48b | 0,29±0,48b |
| cell debris in the lumen | 0,00±0,00 | 0,00±0,00 | 2,00±1,00a | 2,00±1,00a | 0,86±0,37b | 0,86±0,69b | 0,43±0,78b | 0,43±0,78b |
| connective tissue increase | 0,00±0,00 | 0,14±0,37 | 1,28±0,48a | 2,43±0,53a | 0,71±0,48ab | 1,00±0,57ab | 0,43±0,53b | 0,57±0,53b |
3.4. Evaluation of apoptosis in lung tissues
In TUNEL positive cell numbers in the TS groups compared to the control group, a significant increase was detected (Fig. 3(A)b, (B)b). These findings decreased in the TS + ALA group (p < 0.05) (Fig. 3(A)c, (B)c). The number of TUNEL positive cells was evaluated in the ALA group similar to the control group (p > 0.05) (Fig. 3(A)d, (B)d). The results of the apoptotic index are shown in Table 3.
Fig. 3.

Photomicrographs of lung slides of 7 (A) and 21 (B) days old pups. TUNEL findings of the groups; (a) control group, a TUNEL positive cell (arrow), (b) TS group, numerous TUNEL positive cells (arrows), (c) TS + ALA group, numerous TUNEL positive cells (arrows), (d) ALA group, a few TUNEL positive cells (arrows). (x40).
Table 3.
The apoptotic index and Caspase-3, SP-A, VEGF immunoreactivities histoscores.
| Control |
TS |
TS+ALA |
ALA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (days) | 7 | 21 | 7 | 21 | 7 | 21 | 7 | 21 | |
| Apoptotic index (%) | 2,92±1.82 | 3,01±1.78 | 35,50±6.11a | 30,07±6.84a | 14,05±1.54ab | 12,54±2.43ab | 4,05±0.89b | 4,25±1.48b | |
| Caspase-3 immunreactivity | 0,35±0.15 | 0,2± 0.13 | 2,30±0.48a | 2,1±0.6a | 1,04± 0.39 ab | 0,81± 0.2 ab | 0,52±0.23b | 0,4± 0.15 b | |
| SP-A | immunreactivity | 1,43±0.17 | 2,00± 0.24 | 0,22±0.08a | 0,43±0.08a | 1,03± 0.18 b | 1,30± 0.10ab | 1,13±0.16b | 1,85±0.19b |
| mRNA level (fold change) | 1 | 1 | 0,114±0.09a | 0,594±0.03a | 2,495±0.02ab | 2,603±0.23ab | 3,173±0.01ab | 3,427±0.14ab | |
| VEGF | immunreactivity | 1,23±0.13 | 2,00±0.24 | 0,43±0.05a | 0,51±0.11a | 0,91± 0.21 b | 1,45± 0.16 b | 1,16±0.14b | 1,75±0.32b |
| mRNA level (fold change) | 1 | 1 | 0,131±0.03a | 0,327±0.12a | 2,173±0.11ab | 2,561±0.01ab | 5,189±0.32ab | 5,971±0.26ab | |
3.5. Immunohistochemical findings
SP-A immunoreactivity was observed in bronchiol epithelium and Type 2 alveolar cells, while VEGF immunoreactivity was observed in alveolar epithelial cells, vascular and bronchial smooth muscle cells (Fig. 4(A)a, (B)a; Fig. 6(A)a, (B)a). Both SP-A and VEGF immunoreactivities in the lung tissues from 7 and 21-day old pups, a significant decrease was observed in the TS group compared to the control group (p < 0.05) (Fig. 4(A)b, (B)b); Fig. 6(A)b, (B)b). On the other hand, a significant increase was observed in the TS + ALA group compared to the TS group (p < 0.05) (Fig. 4(A)c, (B)c; Fig. 6(A)c, (B)c). Caspase-3 immunoreactivity was observed in bronchi and bronchial epithelium in the lung, alveolar cells and some cells in the interalveolar septum. Caspase-3 immunoreactivity a statistically significant increase in the TS group compared to the control group (p < 0.05), but a significant decrease in the TS + ALA group compared to the TS group (p < 0.05) (Fig. 5(A)b, (B)b; (A)c, (B)c). All three immunoreactivity scores of the ALA group were similar to the control group (p > 0.05) (Fig. 4(A)d, (B)d; Fig. 5(A)d, (B)d; Fig. 6(A)d, (B)d). Immunoreactivity histoscores are shown in Table 3.
Fig. 4.

Photomicrographs of lung slides of 7 (A) and 21 (B) days old pups. SP-A immunoreactivities of the groups; (a) control, (b) TS, (c) TS + ALA, (d) ALA. (x40).
Fig. 6.

Photomicrographs of lung slides of 7 (A) and 21 (B) days old pups. VEGF immunoreactivities of the groups; (a) control, (b) TS, (c) TS + ALA, (d) ALA. (x40).
Fig. 5.

Photomicrographs of lung slides of 7 (A) and 21 (B) days old pups. Caspase-3 immunoreactivities of the groups; (a) control, (b) TS, (c) TS + ALA, (d) ALA. (x40).
3.6. qRT-PCR analysis
Following maternal tobacco smoke exposure, a significant decrease in SP-A and VEGF mRNA levels in lung of the TS group was obtained (p < 0.05). However, the ALA administration increased both SP-A and VEGF mRNA levels in the TS + ALA group (p < 0.05) (Table 3).
4. Biochemical results
MDA levels and GSH, SOD activities were shown in Table 4. In the TS group compared with the control group a significant increase was found in the levels of MDA (p < 0.05). In the TS + ALA group compared with the TS group a significant decrease was found in the levels of MDA (p < 0.05). SOD and GSH enzyme activities in the TS group significantly decreased compared to the control group (p < 0.05). Also, a significant increase in SOD and GSH enzyme activities was found in the TS + ALA group compared with the TS group.
Table 4.
Biochemical results in 7 and 21 days old pups lung tissues.
| Control |
TS |
TS + ALA |
ALA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (days) | 7 | 21 | 7 | 21 | 7 | 21 | 7 | 21 | |
| Apoptotic index (%) | 2,92 ± 1.82 | 3,01 ± 1.78 | 35,50 ± 6.11a | 30,07 ± 6.84a | 14,05 ± 1.54ab | 12,54 ± 2.43ab | 4,05 ± 0.89b | 4,25 ± 1.48b | |
| Caspase-3 immunreactivity | 0,35 ± 0.15 | 0,2 ± 0.13 | 2,30 ± 0.48a | 2,1 ± 0.6a | 1,04 ± 0.39 ab | 0,81 ± 0.2 ab | 0,52 ± 0.23b | 0,4 ± 0.15 b | |
| SP-A | immunreactivity | 1,43 ± 0.17 | 2,00 ± 0.24 | 0,22 ± 0.08a | 0,43 ± 0.08a | 1,03 ± 0.18 b | 1,30 ± 0.10ab | 1,13 ± 0.16b | 1,85 ± 0.19b |
| mRNA level (fold change) | 1 | 1 | 0,114 ± 0.09a | 0,594 ± 0.03a | 2,495 ± 0.02ab | 2,603 ± 0.23ab | 3,173 ± 0.01ab | 3,427 ± 0.14ab | |
| VEGF | immunreactivity | 1,23 ± 0.13 | 2,00 ± 0.24 | 0,43 ± 0.05a | 0,51 ± 0.11a | 0,91 ± 0.21 b | 1,45 ± 0.16 b | 1,16 ± 0.14b | 1,75 ± 0.32b |
| mRNA level (fold change) | 1 | 1 | 0,131 ± 0.03a | 0,327 ± 0.12a | 2,173 ± 0.11ab | 2,561 ± 0.01ab | 5,189 ± 0.32ab | 5,971 ± 0.26ab | |
5. Discussion
This study is the first study conducted to assess the effects of ALA, an effective antioxidant, against the possible harm that maternal tobacco smoke exposure may cause on newborn lungs. Although it is stated in the literature that ALA creates positive effect in various experimental damage models, studies about its gestational use and intrauterine development are very limited.
Oxidative stress due to increased reactive oxygen species inside the tissue constitutes the basis of the harm on newborn lungs that maternal tobacco smoke exposure caused. Studies revealed that maternal tobacco use or exposure causes failure of newborn lung functions, and has several negative effects on lung structure and metabolism [20,21]. In the study, the increase of relative lung weight of postnatal 7 days old rats exposed to tobacco smoke is thought to be edema related. It is suggested that the rapid increase in body weights during lactation period is responsible for the insignificant difference between relative lung weights of postnatal 21 days old rats.
RAC (radial alveolar count) is a morphometric method giving information about intrauterine, early-late postnatal and childhood lung developments by revealing alveoli counts in lungs [18,22]. İn rats, it is presented that cigarette exposure during intrauterine period causes reduction in alveoli counts and disturbs alveolarization with negative effects on lung development [23,24]. In the study, decrease in alveoli counts measured with the RAC method in lung tissues of postnatal 7 and 21 days old tobacco smoke group was observed. It is suggested that this decrease is an indicator of lung development retardation due to tobacco smoke exposure, and ALA administration can help alveolarization by increasing alveoli counts.
There are many studies in rodents about tobacco smoke causing histopathological changes in lungs [25,26]. Airway epithelial and endothelial cells are known to be direct targets of damage induced by tobacco smoke [27]. Nicotinic acethylcoline receptors (nAChRs) that are receptors of nicotine in tobacco smoke is detected in lung epithelial and endothelial cells and it is stated that the histopathological changes may take place in lungs by nAChR pathways [28]. Reactive oxygen species (ROS) cause endothelial barrier dysfunction, thicken alveolar walls due to fibril accumulation, and cause hemorrhage because of endothelial damage and edema by increasing inflammatory cells generating an increased macromolecule and fluid permeability [29,30]. Activated neutrophils deposited in small vessel of lungs release many toxic mediators such as protease, ROS, proinflammatory cytokine by degranulation and after these events along with the increase in vascular permeability, also loss of endothelial barrier function gradually increases. In a study with humans, environmental tobacco smoke has been shown to increase circulating inflammatory cytokine levels [31,32]. Accordingly, cell deaths occur through apoptosis and necrosis in the bronchi, bronchioles and alveolar epithelial cells [31]. Insufficient production of surfactants due to increase in type 2 alveolar epithelial cell deaths leads to atelectasis, reduced gas exchange and severe hypoxia [33]. And this hypoxic situation causes an increase in endothelial, alveolar and bronchiolar cell necrosis by creating a further damage in pulmonary cells. As a result of the vascular damages that occurred, along with the leaking plasma into alveolar space, necrotic type 2 alveolar cells come together with fibrin and form hyaline membrane structures around the alveoli [34]. Also while the alveolar epithelium normally absorbs the developing edema, in this case due to the damage to the epithelium edema absorption cannot occur properly [35]. In the study, it is suggested that a large part of these histopathological findings of lung tissue is connected to the increased ROS. In addition, the result of the evaluation conducted with TUNEL staining and caspase-3 immunoreactivity that are apoptotic cell markers, supports the increase of apoptotic cells due to ROS caused by tobacco smoke exposure. Besides, the increase of the ROS causes membrane peroxidation in cells, MDA formation and leads to a decrease in endogenous antioxidant levels [36]. In the study, it is found that increased tobacco smoke-induced ROS increases the level of MDA in the lungs, while dropping the levels of glutathione (GSH), which is one of the most important antioxidants participating in the endogenous defense system against free radicals, together with SOD activity. This find supports the view of damage caused by oxidative stress, as well.
Clear information on how tobacco smoke exposure disrupts alveolarization is not revealed but some clues give rise to the idea that it may be associated with varying angiogenesis in lungs. It is indicated that development of airways and alveoli is closely associated with vasculature development of these structures in coordination [37]. This synchronized development is controlled by growth factors such as VEGF [38]. In the study, it is suggested that decreased VEGF immunoreactivity and mRNA levels because of tobacco smoke exposure are indicators of lung damage and that the reduction in vascularization might be related with reduced alveoli counts. In addition, VEGF deficiency is being correlated with surfactant production failure in type 2 alveolar cells and hyaline membrane disease seen in newborn mice [39]. It is suggested that decreasing VEGF immunoreactivity in tobacco smoke exposure group postnatal 7 and 21 days old pulmonary sections is associated with the declining of SP-A immunoreactivity, also in connection with this, hyaline membrane structures evolved.
Tobacco smoke is known to increase the production of peroxynitrite, one of reactive oxygen species [40]. It is reported that reactive oxygen species such as nitric oxide and superoxide anion increase the levels of peroxynitrite in tissues, increasing peroxynitrites reduces the levels of VEGF produced from epithelial cells by causing oxidative damage, reduced VEGF levels increase apoptosis in endothelial cells [41]. In the light of this information, it is suggested that the reduced VEGF immunoreactivity depending on tobacco smoke also supports the finding of increased apoptosis.
Failure in the metabolism of surfactant homeostasis is thought to be caused by lipid peroxidation as a result of oxidant/antioxidant imbalances due to oxidative stress. It is reported that the damages in pulmonary surfactant are related with many pulmonary diseases such as hyaline membrane disease, asthma and COPD and that the SP-A levels may be used as an indicator for the diagnosis [42,43]. The final effect of intrauterine exposure to tobacco smoke on pulmonary surfactant is not clear. However, it is stated that oxidative stress increased with cigarette smoke exposure decreases the thyroid transcription factor-1, which is the critical transcription regulator for SP-A, and in this way can cause reduced SP-A production [44,45]. With this study, it can be said that increased oxidative stress depending on tobacco smoke reduces SP-A immunoreactivity and mRNA levels by both interrupting surfactant metabolism and causing VEGF deficiency.
Glucose is reported to be a key substance used in energy production and biosynthesis of pulmonary surfactant by alveolar type II cells [46]. Irreversible suppression of glycolytic pathway and glycogenolysis in newborn lungs due to maternal nicotine was identified by Kordom et al. [47]. In earlier studies done with rats, it is reported that cigarette smoke reduces the energy dependent surfactant produced from lamellar granules and lipid secretions by disrupting the function of pulmonary mitochondria [48,49]. In the study, it is suggested that the increased free radicals due to tobacco smoke can lead to a lack of energy required for protein phospholipid synthesis by suppressing the glycolysis, also ALA, acting as a cofactor in α-ketoglutarate dehydrogenase and pyruvate dehydrogenase enzymes in mitochondria, can contribute to energy production for surfactant synthesis by providing glucose uptake into cells and increasing glucose oxidation.
A decrease in GSH, SOD, CAT and GPx antioxidant levels of smoking individuals has been reported in humans [50]. Antioxidant deficiency is thought to be one of the essential reasons for chronic and degenerative pathological situations [51]. Thioctic acid, also known as ALA, is potential antioxidant showing biological activity in cells, involved in metal ion chelation, exogenous and endogenous antioxidant regeneration and repair of oxidized proteins. In a study with rats, it is stated that dietary intake of ALA can ameliorate the histopathological changes in lungs caused by oxidative damage due to maternal tobacco smoke exposure with a protective effect [52]. In addition, decreasing pulmonary MDA levels with increasing GSH level and SOD enzyme activities together with ALA administration is supportive of the information about ALA clearing many oxygen species such as hydroxyl, superoxide and peroxyl radicals and regenerating antioxidants such GSH, tocopherols and ascorbic acid.
6. Conclusion
In conclusion, when the results of our study are evaluated together, it is suggested that the exposure of tobacco smoke during gestation period gives rise to morphological, histopathological and functional changes on lung development by causing oxidative damage in lung tissues of rat pups and the maternal use of ALA, which is known to have strong antioxidant properties, can ensure a limited protective effect on the neonatal lung development against this oxidative stress originating from tobacco smoke. Although smoking threatens human health in all circumstances, it has a serious negative impact on the development of the baby, as well as the damage it inflicts on the mother. But although pregnant women are increasingly aware of this issue, environmental tobacco smoke exposure is still a widespread problem. For this reason, when the findings of this study are evaluated together, this damage can be partially reduced by conscious and controlled ALA supplements in pregnancy.
Conflicts of interest
The authors declare they have no conflicts of interest.
Transparency document
Acknowledgements
The study was supported by the Fırat University Scientific Research Projects Unit, Elazig, TURKEY, Project number 16.06.
Contributor Information
Elif Erdem Guzel, Email: elfer192@hotmail.com.
Nalan Kaya, Email: nalankaya@firat.edu.tr.
Gonca Ozan, Email: gozan@firat.edu.tr.
Ahmet Tektemur, Email: atektemur@firat.edu.tr.
Durrin Ozlem Dabak, Email: dozlemdabak@firat.edu.tr.
Ibrahim Enver Ozan, Email: eozan@firat.edu.tr.
References
- 1.Abbott L.C., Winzer-Serhan U.H. Smoking during pregnancy: lessons learned from epidemiological studies and experimental studies using animal models. Crit. Rev. Toxicol. 2012;42(4):279–303. doi: 10.3109/10408444.2012.658506. [DOI] [PubMed] [Google Scholar]
- 2.Rogers J.M. Tobacco and pregnancy. Reprod. Toxicol. 2009;28:152–160. doi: 10.1016/j.reprotox.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Gillespie M.N., Owasoyo J.O., Kojima S., Jay M. Enhanced chemotaxis and superoxide anion production by polymorphonuclear leucocytes from nicotine-treated and smoke-exposed rats. Toxicology. 1987;45:45–52. doi: 10.1016/0300-483x(87)90113-2. [DOI] [PubMed] [Google Scholar]
- 4.Kistler G.S., Caldwell P.R.B., Weibel E.R. Development of fine damage to alveolar and capillary lining cells in oxygen poisoned rat lungs. J. Cell. Biol. 1967;33:605–628. doi: 10.1083/jcb.32.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haagsman H.P. Surfactant proteins A and D. Biochem. Soc. Trans. 1993;22:100–106. doi: 10.1042/bst0220100. [DOI] [PubMed] [Google Scholar]
- 6.Pison U., Max M., Neuendan A., Weissbach S., Pietschmann S. Host defence capacities of pulmonary surfactant: evidence for ‘non-surfactant’ functions of the surfactant system. Eur. J. Clin. Invest. 1994;24:586–599. doi: 10.1111/j.1365-2362.1994.tb01110.x. [DOI] [PubMed] [Google Scholar]
- 7.Benowitz N.L., Brunetta P.G. Smoking hazards and cessation. In: Mason R.J., Broaddus C., Murray J.F., Nadel J.A., editors. 4th ed. Vol. 24. Elsevier Saunders; Philadelphia: 2005. pp. 53–68. (Murray and Nadels Textbook of Respiratory Medicine). [Google Scholar]
- 8.Maniscalco W.M., Bhandari V. Disruption of lung microvascular development. In: Abman S.H., editor. Bronchopulmonary Dysplasia. Informa Healthcare; New York: 2010. pp. 146–166. [Google Scholar]
- 9.Bem R.A., Bos A.P., Matute-Bello G., Van T.M., van Woensel J.B. Lung epithelial cell apoptosis during acute lung injury in infancy. Pediatr. Crit. Care Med. 2007;8:132–137. doi: 10.1097/01.PCC.0000257207.02408.67. [DOI] [PubMed] [Google Scholar]
- 10.Del Riccio V., Van Tuyl M., Post M. Apoptosis in lung development and neonatal lung injury. Pediatr. Res. 2004;55:183–189. doi: 10.1203/01.PDR.0000103930.93849.B2. [DOI] [PubMed] [Google Scholar]
- 11.Thornberry N.A., Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 12.Goraca A., Huk-Kolega H., Piechota A. Lipoic acid-biological activity and therapeutic potential. Pharmacol. Rep. 2011;63:849–858. doi: 10.1016/s1734-1140(11)70600-4. [DOI] [PubMed] [Google Scholar]
- 13.Kramer K., Hoppe P.P., Packer L. R-alpha-lipoic acid in health and disease prevention. Nutraceuticals. 2001:129–164. [Google Scholar]
- 14.Al Ghafli M.H.M., Padmanabhan R., Kataya H.H., Berg B. Effects of α-lipoic acid supplementation on maternal diabetes-induced growth retardation and congenital anomalies in rat foetuses. Mol. Cell. Biochem. 2004:1–13. doi: 10.1023/b:mcbi.0000028747.92084.42. [DOI] [PubMed] [Google Scholar]
- 15.Placer Z.A., Cushmann L.L., Johnson B.C. Estimation of products of lipid peroxidation (as malondialdehyde) in biochemical systems. Anal. Biochem. 1966;16:359–364. doi: 10.1016/0003-2697(66)90167-9. [DOI] [PubMed] [Google Scholar]
- 16.Beutler E., Duron O., Kelly B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963;61:882–888. [PubMed] [Google Scholar]
- 17.Sun Y., Oberley L.W., Li Y. A simple method for clinical assay of superoxide dismutase. Clin. Chem. 1988;34(3):497–500. [PubMed] [Google Scholar]
- 18.Cooney T.P., Thurlbeck W.M. The radial alveolar count method of Emery and mithal: a reappraisal 1-postnatal lung growth. Thorax. 1982;37:572–579. doi: 10.1136/thx.37.8.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etem E.O., Bal R., Akagac A.E., Kuloglu T., Tuzcu M., An-. drievsky G.V., Buran I., Nedzvetsky V.S., Baydas G. The effects of hydrated c(60) fullerene on gene expression profile of TRPM2 and TRPM7 in hyperhomocysteinemic mice. J. Recept Signal. Trans. Res. 2014;34:317–324. doi: 10.3109/10799893.2014.896381. [DOI] [PubMed] [Google Scholar]
- 20.Maritz G.S., Harding R. Life-long programming implications of exposure to tobacco smoking and nicotine before and soon after birth: evidence for altered lung development. Int. J. Environ. Res. Public. Health. 2011;8:875–898. doi: 10.3390/ijerph8030875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stick S.M., Burton P.R., Gurrin L., Sly P.D., LeSouëf P.N. Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet. 1996;348:1060–1064. doi: 10.1016/s0140-6736(96)04446-7. [DOI] [PubMed] [Google Scholar]
- 22.Emery J.L., Mithal A. The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch. Dis. Child. 1960;35:544–547. doi: 10.1136/adc.35.184.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maritz G.S. Maternal nicotine exposure during gestation and lactation of rats induce microscopic emphysema in the offspring. Exp. Lung Res. 2002;28:391–403. doi: 10.1080/01902140290092010. [DOI] [PubMed] [Google Scholar]
- 24.Petre M.A., Labiris R.N., Inman M.D., Holloway A.C. Annual Meeting of the American Thoracic Society Toronto. 2008. Fetal and neonatal exposure to nicotine disrupts postnatal lung development in rats: role of VEGF and its receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shraideh Z., Wajdy A.A., Darwish B. Effects of cigarette smoking on histology of trachea and lungs of albino rat. Res. Opin. Anim. Vet. Sci. 2013;3(10):356–362. [Google Scholar]
- 26.Dogan O.T., Elagoz I.S., Ozsahin S.L., Epozturk K., Tuncer E., Akkurt I. Pulmonary toxicity of chronic exposure to tobacco and biomass smoke in rats. Clinics. 2011;66(6):1081–1087. doi: 10.1590/S1807-59322011000600027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z., Wang D., Liu X., Pei W., Li J., Cao Y., Zhang J., An Nie Y., Tong J. Oxidative DNA damage is involved in cigarette smoke-induced lung injury in rats. Environ. Health Prev. Med. 2015;20:318–324. doi: 10.1007/s12199-015-0469-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gundavarapu S., Wilder J.A., Mishra N.C., Rir-Sima-Ah J., Langley R.J., Singh S.P., Saeed A.I., Jaramillo R.J., Gott K.M., Peña-Philippides J.C., Harrod K.S., McIntosh J.M., Buch S., Sopori M.L. Role of nicotinic receptors and acetylcholine in mucous cell metaplasia, hyperplasia, and airway mucus formation in vitro and in vivo. J. Allergy Clin. Immunol. 2012;130(3):770–780. doi: 10.1016/j.jaci.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekhon H.S., Keller J.A., Benowitz N.L., Spindel E.R. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am. J. Respir. Crit. Care Med. 2001;164:989–994. doi: 10.1164/ajrccm.164.6.2011097. [DOI] [PubMed] [Google Scholar]
- 30.Henricks P.A., Nijkamp F.P. Reactive oxygen species as mediators in asthma. Pulm. Pharmacol. Ther. 2001;14:409–420. doi: 10.1006/pupt.2001.0319. [DOI] [PubMed] [Google Scholar]
- 31.Flouris A.D., Metsios G.S., Carrillo A.E., Jamurtas A.Z., Gourgoulianis K., Kiropoulos T., Tzatzarakis M.N., Tsatsakis A.M., Koutedakis Y. Acute and short-term effects of secondhand smoke on lung function and cytokine production. Am. J. Respir. Crit. Care Med. 2009;179(11):1029–1033. doi: 10.1164/rccm.200812-1920OC. [DOI] [PubMed] [Google Scholar]
- 32.Zemans R.L., Colgan S.P., Downey G.P. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am. J. Respir. Cell. Mol. Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Northway W.H., Rosan R.C., Porter D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease, bronchopulmonary dysplasia. N. Engl. J. Med. 1967;276(7):357–368. doi: 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- 34.Ainsworth S.B. Pathophysiology of neonatal respiratory distress syndrome. Treat. Respir. Med. 2005;4(6):423–437. doi: 10.2165/00151829-200504060-00006. [DOI] [PubMed] [Google Scholar]
- 35.Matthay M.A., Folkesson H.G., Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol. Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 36.Orhon F.S., Ulukol B., Kahya D., Cengiz B., Başkan S., Tezcanet S. The influence of maternal smoking on maternal and newborn oxidant and antioxidant status. Eur. J. Pediatr. 2009;168:975–981. doi: 10.1007/s00431-008-0873-0. [DOI] [PubMed] [Google Scholar]
- 37.Burri P.H. Structural aspects of prenatal and postnatal development and growth of the lung. In: McDonald J.A., editor. Lung Growth and Development. Dekker; New York: 1997. pp. 1–35. [Google Scholar]
- 38.Ferrara N., Bunting S. Vascular endothelial growth factor, a specific regulator of angiogenesis. Curr. Opin. Nephrol. Hypertens. 1996;5:35–44. doi: 10.1097/00041552-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Compernolle V., Brusselmans K., Acker T., Hoet P., Tjwa M., Beck H., Plaisance S., Dor Y., Keshet E., Lupu F., Nemery B., Dewerchin M., Van Veldhoven P., Plate K., Moons L., Collen D., Carmeliet P. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 40.Ambrose J.A., Barua R.S. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J. Am. Coll. Cardiol. 2004;43(10):1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 41.Kanazawa H. Role of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Med. Sci. Monit. 2007;13(11):189–195. [PubMed] [Google Scholar]
- 42.Cochrane C.G. Pulmonary surfactant in allergic inflammation: new insights into the molecular mechanisms of surfactant function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;288(4):608–609. doi: 10.1152/ajplung.00434.2004. [DOI] [PubMed] [Google Scholar]
- 43.Calkovska A. Pulmonary surfactant in the respiratory tract. Cesk Fysiol. 2000;49(3):145–151. [PubMed] [Google Scholar]
- 44.Fu Z.H., Yang Z.C., Hu Z.H., Wu J.P., Liu Z.X. Study on the mechanisms of abnormal SP-a expression after smoke inhalation injury. Med. J. Chin. PLA. 2002;27:518. [Google Scholar]
- 45.Dubick M.A., Carden S.C., Jordan B.S., Langlinais P.C., Mozingo D.W. Indices of antioxidant status in rats subjected to wood smoke inhalation and/or thermal injury. Toxicology. 2002;176:145. doi: 10.1016/s0300-483x(02)00132-4. [DOI] [PubMed] [Google Scholar]
- 46.Fisher A.B. Intermediary metabolism of the lung. Environ. Health Perspect. 1984;55:149–158. doi: 10.1289/ehp.8455149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kordom C., Maritz G.S., De Kock M. Maternal nicotine exposure during pregnancy and lactation: I. Effect on glycolysis in the lungs of the offspring. Exp. Lung Res. 2003;29(2):79–89. doi: 10.1080/01902140303769. [DOI] [PubMed] [Google Scholar]
- 48.Jordan J.A., Gairola C.G. Lung mitochondrial in rat chronically exposed to cigarette smoke. Toxicologist. 1994;14:425. [Google Scholar]
- 49.Beers M.F., Kim C.Y., Dodia C., Fisher A.B. Synthesis of type II cell lamellar body lysozyme-15 kD protein (lbl-15) by perfused by perfused rat lung. Am. J. Cell. Mol. Biol. 1994;11:240–248. doi: 10.1165/ajrcmb.11.2.8049085. [DOI] [PubMed] [Google Scholar]
- 50.Cantin A.M., North S.L., Hubbard R.C., Crystal R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1987;63(1):152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- 51.Willcox J.K., Ash S.L., Catignani G.L. Antioxidants and prevention of chronic disease. Review. Crit. Rev. Food Sci. Nutr. 2004;44:275–295. doi: 10.1080/10408690490468489. [DOI] [PubMed] [Google Scholar]
- 52.Dadhania V.P., Tripathi D.N., Vikram A., Ramarao P., Jena G.B. Intervention of alpha-lipoic acid ameliorates methotrexate-induced oxidative stress and genotoxicity: a study in rat intestine. Chem. Biol. Interact. 2010;183:85–97. doi: 10.1016/j.cbi.2009.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.