

Abstract
Multiple acyl-CoA dehydrogenase deficiency (MADD) is an autosomal recessive disorder of fatty acid, amino acid, and choline metabolism caused by mutations in EFTA, EFTB, or ETFDH. Many MADD patients are responsive to treatment with riboflavin, termed riboflavin-responsive MADD (RR-MADD). Here, we report three novel mutations and one previously reported mutation in ETFDH in four RR-MADD patients who presented at various ages, and characterize the corresponding changes in ETF-QO protein structure. Clinicians should consider MADD in the differential diagnosis when patients present with muscle weakness and biochemical abnormalities. Gene testing plays a critical role in confirming the diagnosis of MADD, and may not only prevent patients from invasive testing, but also allow timely initiation of riboflavin treatment. The novel variants in ETFDH and the corresponding clinical features reported here enrich the allelic heterogeneity of RR-MADD and provide insight into genotype-phenotype relationships.
Keywords: Multiple acyl-CoA dehydrogenase deficiency, Glutaric aciduria II, ETFDH, ETF-QO, Riboflavin
Abbreviations: AST, aspartate aminotransferase; CK, creatine kinase; ETF, electron transfer flavoprotein; ETF-QO, ETF-ubiquinone oxidoreductase; GAII, glutaric aciduria II; LDH, lactate dehydrogenase; MADD, multiple acyl-CoA dehydrogenase deficiency; RR-MADD, riboflavin-responsive MADD; WES, whole exome sequencing
1. Introduction
Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria II (GAII, OMIM #231680), is an autosomal recessive disorder of fatty acid, amino acid, and choline metabolism. MADD is highly clinically heterogeneous, and has been categorized into three types: neonatal onset with congenital abnormalities (Type I), neonatal onset without congenital abnormalities (Type II), and late-onset (Type III) [[1], [2], [3]]. MADD results from a defect in either electron transfer flavoprotein (ETF, encoded by the alpha ETF (ETFA) and beta ETF (ETFB) genes) or ETF-ubiquinone oxidoreductase (ETF-QO, encoded by the ETF dehydrogenase (ETFDH) gene) [4, 5].
Types I and II are severe and typically fatal, characterized by nonketotic hypoglycemia, metabolic acidosis, and accumulation and excretion of metabolites, while Type III is milder and more variable, characterized by recurrent episodes of hypoglycemia, metabolic acidosis, vomiting, and muscle weakness during catabolic stress [6, 7]. Many MADD patients, especially late-onset MADD patients, can be effectively treated with riboflavin, termed riboflavin-responsive MADD (RR-MADD). Previously, it has been reported that ETFDH mutations are the major cause of RR-MADD [8, 9]. Here, we report four additional RR-MADD patients from three unrelated families who presented at various ages and carry three novel mutations and one previously reported mutation in ETFDH. In addition, we predict changes in the ETF-QO protein structure due to these mutations.
2. Materials and methods
2.1. Patients
The four patients are from southern China, and were clinically diagnosed with MADD. Informed consent was obtained, and this study was approved by the local ethics committee.
2.2. Mutation analysis
Genomic DNA was extracted from peripheral blood using standard protocols (QIAGEN, Hilden, Germany). Direct sequencing of ETFA, ETFB, and ETFDH was performed for two patients, and no mutations were identified in ETFA or ETFB. Whole exome sequencing (WES) was performed for the other two patients. Sequence capture, enrichment, and elution were performed using an Illumina TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) according to the manufacturer's protocols, and sequencing was performed on an Illumina HiSeq2000 (Illumina). Sequence alignment and variant calling against the reference human genome (GRCh37) were performed using BWA and the Genome Analysis Toolkit. Variant interpretation of WES data was performed using Ingenuity Variant Analysis pipeline (Ingenuity, Redwood City, CA, USA). SIFT, Polyphen2, and MutationTaster were used for in silico analysis of candidate variants. Deleterious variants were confirmed by Sanger sequencing.
2.3. Structure prediction of ETF:QO
Human and porcine ETF-QO proteins share high amino acid sequence identity (95%), thus the porcine ETF-QO is an excellent structural template. The structural model of the human ETF-QO was made based on the crystallographic structure of porcine ETF-QO [10] (PDB: 2GMH) by homology modeling (Swiss-Model).
3. Results
3.1. Clinical presentations
All four patients were clinically diagnosed with MADD, and changes in blood acylcarnitines, urine organic acids, and muscle enzymes are listed in Table 1.
Table 1.
Blood acylarnitine, urine organic acids, and muscle enzymes in the four MADD patients.
| No. | Sex | Onset | Blood acylcarnitine spectrum (μmol/L) | Urine organic acids spectrum | CK (U/L) | LDH (U/L) | AST (U/L) | Genotype |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 7 yo | ↓: C2 (6.16), C3 (0.47) | ↑: Ethylmalonic acid (8.48), decanedioic acid (54.59) | 193 | 234 | 34 | c.524G>A (p.R175H), c.229G>A (p.G77S) |
| ↑: C4 (1.18), C5 (0.56), C6 (1.06), C8 (1.47), C10 (1.86), C10:1 (0.55), C12 (0.60), C14:1 (0.57) | ||||||||
| 2 | F | 10 do | ↑: C4 (1.03), C5 (0.58), C5DC (0.33), C6 (0.96), C8 (1.50), C8DC (0.23), C10 (2.11), C10:1 (0.34), C12 (2.43), C12:1 (0.45), C14 (2.49), C14:1 (1.53), C16 (6.39), C16:1 (1.47) | ↑: 3-Hydroxyglutaric acid (29.22) | 235 | 331 | 82 | c.524G>A (p.R175H), c.229G>A (p.G77S) |
| 3 | M | 27 yo | ↓: C0(8.38) | ↑: Lactate-2 (34.29), Glycolic acid-2 (14.44), Oxalic acid-2 (4.22), 2-Hydroxybutyric acid-2 (4.58), 3-Hydroxypropionic acid-2 (4.43), Pyruvate-OX-2 (58.29), 2-Hydroxyisovalerate-2 (17.98) | 1318 | 1024 | 557 | c.524G>A (p.R175H), c.1450T>C (p.W484R) |
| ↑: C6 (0.41), C8 (0.63), C10 (1.06) | ||||||||
| 4 | M | 30 do | ↓: C0 (5.37), C3 (0.42) | ↑: Ethylmalonic acid (10.33), 3-Hydroxyglutaric acid (7.03), 4-Hydroxyphenyllactate-2 (28.34) | 976 | 853 | 323 | c.1157G>A (p.G286D), c.1450T>C (p.W484R) |
| ↑: C4 (1.16), C8DC (0.11), C12(0.51), C14 (1.13), C14:1 (1.18), C16 (4.16), C14:1/ C8:1 (32.10), C16/C3 (9.92) | Hypervanillic acid-2 (80.4), Vanillic acid-2 (25.45), Palmitic acid-1 (131.72), Glycerate-3 (14.3), 4-hydroxyphenylacetic acid (303.27) |
↑ above normal level; ↓ below normal level; yo: years old; do: days old.
Upper limit of normal: CK 200 U/L, LDH 245 U/L, and AST 40 U/L.
Patients 1 and 2 are siblings and carry the same mutations but presented at different ages. Patient 2 was diagnosed with newborn screening and presented ill at 10 days old with elevated levels of acylcarnitines, consistent with MADD. Gas chromatography-mass spectrometry revealed a significant increase of 3-hydroxyglutaric acid. In addition, she had elevated levels of creatine kinase (CK), lactate dehydrogenase (LDH), and aspartate aminotransferase (AST). Patient 2 was initially treated with 195 mg riboflavin daily, and her symptoms largely resolved after taking the medication for one month. Then, her parents stopped giving her the medication, and she presented with severe acidosis and convulsions. She was then treated with 300 mg riboflavin daily, which she has continued, and she remains symptom-free. Patient 1 was tested at 7 years old following her sibling's diagnosis. She has had intermittent mild symptoms, which her parents believed were viral upper respiratory infections. Levels of acylcarnitines, ethylmalonic acid, and decanedioic acid were elevated, although her muscle enzymes were within the normal range. Patient 1 was initially treated with 150 mg riboflavin daily. Once improved and lab values normalized, Patient 1 was intermittently treated (her parents stop giving her the medication), and she is now 9 years old and remains symptom-free.
Patient 3, a 27-year old male, was previously healthy with no family history of neuromuscular disease. He presented with pneumonia and reported muscle weakness for 8 months, and was found to have elevated levels of acylcarnitines, urine organic acids, and serum ammonia, as well as fatty liver detected by ultrasound. He was treated with 2 tablets of vitamin B composite (B1: 3.0 mg; riboflavin: 1.5 mg; B6: 0.2 mg) three times a day, and his muscle strength and lab values gradually recovered to normal levels.
Patient 4, a 30-day old male infant, presented with non-ketotic hypoglycemia, metabolic acidosis, electrolyte disturbances, and increased muscle enzymes. His clinical symptoms improved for 20 days after riboflavin treatment (treatment details unknown). At 3 months old, Patient 4 was hospitalized for pneumonia, and passed away due to sepsis. The sister of Patient 4 (genotype unknown) passed away at 3 months old due to encephalitis.
3.2. Mutation analysis
Sequencing studies identified four missense mutations in the three families, three of which are novel (hg19; Chr4:159603400 G>A, c.229G>A, p.Gly77Ser in exon 3; Chr4:159624615 G>A, c.1157G>A, p.Gly386Asp in exon 10; chr4:159627505 T>C, c.1450T>C, p.Trp484Arg in exon 11) (Fig. 1a). Each unaffected parent carries one mutant allele, and the four affected patients are compound heterozygous for two mutant alleles, consistent with an autosomal recessive mode of inheritance (Fig. 1b). Patient 1 (F1.3) and Patient 2 (F1.4) carry p.R175H (v1)/p.G77S (v2), Patient 3 (F2.3) carries p.R175H (v1)/p.W484R (v3), and Patient 4 (F3.4) carries p.W484R (v3)/p.G386D (v4). Thus, the previously reported mutant allele p.R175H (v1, chr4:159606289 G>A, c.524G>A) [11] is shared by Families 1 and 2, and one of the novel mutant alleles, p.W484R (v3), is shared by Families 2 and 3. p.R175H variant is present in only the heterozygous state in gnomAD database with a frequency of 4/246132 chromosomes, and is not reported in the 1000 Genomes Project or ExAC databases. The other three variants are not reported in any of the public databases (Fig. 1c), suggesting that these variants are extremely rare in the general population. All four variants are predicted to be damaging or deleterious by SIFT, PolyPhen2, and MutationTaster (Fig. 1c). The variants were confirmed by Sanger sequencing (red arrows in Fig. 1d) and are highly conserved across species (Fig. 1e).
Fig. 1.

Mutation analysis of ETFDH in three MADD families. a) Schematic of the human ETFDH gene structure with the three novel mutations (c.229G>A, c.1157G>A, c.1450T>C) detected in this study marked with asterisks. b) Segregation of heterozygous missense mutations in Family 1 (F1), Family 2 (F2) and Family 3 (F3). c) Population frequencies and in silico predictions for detected mutations. d) Depiction of the DNA and protein changes, with the mutations marked with red arrows and the corresponding amino acid changes in red. E) The four missense mutations are conserved across species. D: Damaging. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Protein structure analysis
ETF-QO, encoded by ETFDH, is integrated in the inner mitochondrial membrane and its crystal structure is comprised of three domains, as depicted in Fig. 2: FAD domain (FAD shown in green), 4Fe4S cluster domain (4Fe4S shown in magenta), and UQ-binding domain (UQ shown in red). The four variants are shown as cyan spheres (Fig. 2a). In the model, R175, G77, and G386 are located near the FAD binding site, while W484 is located near the UQ binding site. The mutants R175H, G77S, and G386D likely affect the binding affinity of ETFDH to FAD (Fig. 2b–d). The bulky aromatic side chain of W484 forms hydrophobic interactions with Y470 and F474, which are important for UQ10 binding. The mutation W484R disrupts the hydrophobic interactions and likely affects the binding between ETFDH and UQ10 (Fig. 2e). Therefore, these variants may reduce the activity of ETFDH by decreasing the affinity of ETFDH binding to FAD or UQ10.
Fig. 2.
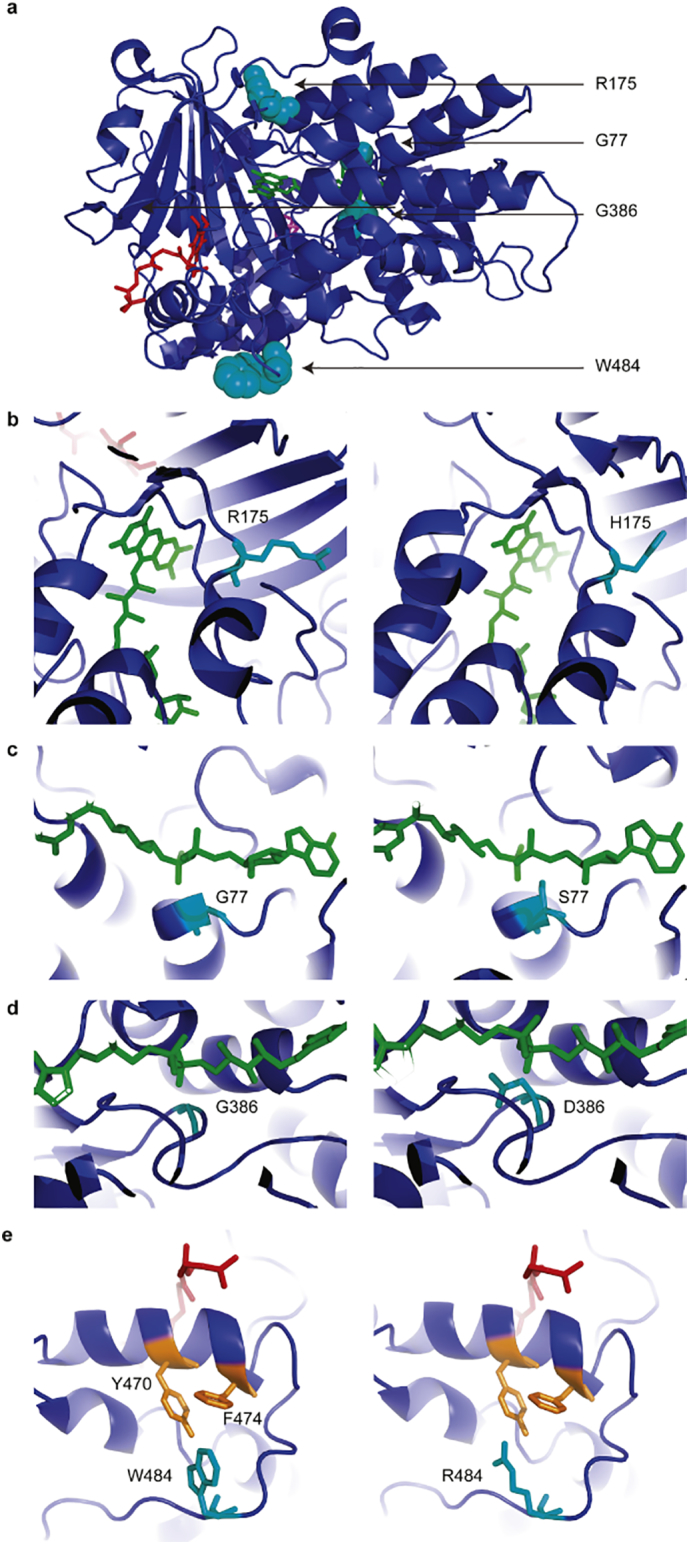
3D protein structure model of ETF-QO based on the crystal structure of porcine ETF-QO reported by Zhang and colleagues [10]. a) ETF-QO is modeled in complex with FAD (green), 4Fe4S (magenta) and UQ (red). The four variants are shown as cyan spheres. b-e) Magnified images of b) R175 and variant H175, c) G77 and variant S77, d) G386 and variant D386, and e) W484 and variant R484. W484 forms hydrophobic interactions with Y470 and F474, while R484 does not.
4. Discussion and conclusions
In this study, we report three novel compound heterozygous mutations in ETFDH in four patients with RR-MADD from three unrelated families. The diagnosis of MADD was primarily based on biochemical data (increased levels of acylcarnitines and urine organic acids), and confirmed by ETFDH mutation analysis. Although RR-MADD is a treatable disease, it is rare and its diagnosis is difficult due to high clinical heterogeneity. MADD patients often present with fluctuating muscle weakness, vomiting, hypoglycemia, metabolic acidosis, encephalopathy, and hepatopathy [[12], [13], [14]]. However, many muscle disorders (such as inflammatory myopathies, metabolic myopathy, and progressive muscular dystrophy) also present with muscle weakness. Thus, MADD may be misdiagnosed as a different type of lipid storage myopathy, a glycogen storage disease, progressive muscular dystrophy, or other muscle disease [15]. Muscle biopsy has traditionally been diagnostically important to differentiate MADD from other muscle diseases, as muscle biopsy showing accumulation of lipid droplets in muscle fibers in combination with the biochemical pattern of acylcarnitines and urine organic acids can confirm the MADD diagnosis [16].
Currently, gene testing plays an important role in MADD diagnosis, and may prevent more invasive diagnostic testing, such as muscle biopsy. Given the current knowledge of the genetic etiology underlying MADD, we suggest a stepwise approach for patients suspected to have MADD involving targeted sequencing of ETFA, ETFB, and ETFDH followed by WES if targeted sequencing is negative. In addition to cost effectiveness, this approach would limit incidental findings such as of variants of unknown significance, which are difficult to interpret and may cause anxiety for the patients and their families. For example, a recent report of a patient with MADD used WES to identify a compound heterozygous mutation in ETFDH as well as a predicted deleterious novel variant in ACOT11, which was of unclear relevance to the phenotype [17].
Early diagnosis is critical because timely riboflavin treatment is effective in many cases. Riboflavin, the precursor of the coenzyme FAD, has been reported to act as a molecular chaperone that promotes the in vitro folding and steady state levels of misfolded ETF-QO proteins in early stages and stabilizes folding intermediates or membrane-inserted proteins in later stages [18, 19]. Riboflavin treatment was given to all four patients in this study, and they showed dramatic improvement in clinical symptoms after riboflavin treatment. Patient 4 was re-hospitalized for pneumonia, and was treated late in his disease course. He suffered from severe liver injury, which was considered to be irreversible, and led to the family's decision to withdraw support.
Although over 80 mutations in ETFDH have been reported worldwide, the same mutation is rarely found in different populations [20]. Hot spot mutations have been identified in Asian populations, with c.250G>A (p.A84T) the most common mutation in southern China [21], and c.770A>G (p.G362R) and c.1227A>C (p.L409F) the most common mutations in northern China [9, [22], [23], [24]]. Indeed, ETFDH mutations may be ethno-specific. In the four RR-MADD patients from southern China in this study, we identified three novel mutations and one previously reported mutation in ETFDH [11]. Of note, Patients 1 and 2 carried the same mutations but presented at different times and with different clinical manifestations, suggesting the role of modifier genes or the environment. Further studies are needed to elucidate these differences.
ETFDH mutations can be classified into two groups: mutations affecting protein folding and assembly or mutations affecting enzymatic activity [25]. Three of the identified mutations in this study are located in the FAD binding domain (p.Arg175His, p.Gly77Ser, and p.Gly386Asp), and one is located in the UQ binding domain (p.Trp484Arg). Most of the previously reported ETFDH mutations are located in the FAD binding domain, suggesting that this region is a sensitive functional hot spot [26]. Riboflavin treatment has been suggested to promote FAD binding to ETF-QO mutants with variants in the FAD domain by affecting the kinetics and/or thermodynamics to stabilize the mutant conformations [27].
In summary, we report four variants including three novel likely pathogenic mutations in ETFDH, enriching the allelic heterogeneity of MADD. A high index of suspicion for MADD is necessary if a patient presents with symptoms including muscle weakness, and it should be included in the differential diagnosis. Gene analysis is critical to confirm the diagnosis of MADD, and may prevent invasive testing, such as muscle biopsy, and allow timely initiation of effective therapy to alleviate symptoms.
Acknowledgments
The authors thank the patients and their families involved in this study. AMD was supported by the NIGMS (grant number T32GM007753). PBA was supported by the NIH/NIAMS (grant number 1R01AR068429-01) and the NICHD/NHGRI/NIH (grant number U19HD077671). The funding sources did not play any role in study design, data collection, analysis, or interpretation, writing of the manuscript, or the decision to submit the manuscript for publication. The authors have no conflicts of interest to report.
Contributor Information
Pankaj B. Agrawal, Email: pagrawal@enders.tch.harvard.edu.
Qifei Li, Email: Qifei.Li@childrens.harvard.edu.
Yiping Shen, Email: yiping.shen@childrens.harvard.edu.
References
- 1.Goodman S.I., Frerman F.E. Glutaric acidaemia type II (multiple acyl-CoA dehydrogenation deficiency) J. Inherit. Metab. Dis. 1984;7(Suppl. 1):33–37. doi: 10.1007/BF03047371. [DOI] [PubMed] [Google Scholar]
- 2.Olsen R.K., Andresen B.S., Christensen E., Bross P., Skovby F., Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- 3.Angelini C. Multiple acyl-CoA dehydrogenase deficiency. Genetic Neuromusc. Disord. 2014:279–284. Springer. [Google Scholar]
- 4.Gregersen N., Kolvraa S., Rasmussen K., Christensen E., Brandt N.J., Ebbesen F., Hansen F.H. Biochemical studies in a patient with defects in the metabolism of acyl-CoA and sarcosine: another possible case of glutaric aciduria type II. J. Inherit. Metab. Dis. 1980;3:67–72. doi: 10.1007/BF02312527. [DOI] [PubMed] [Google Scholar]
- 5.Swigonova Z., Mohsen A.W., Vockley J. Acyl-CoA dehydrogenases: dynamic history of protein family evolution. J. Mol. Evol. 2009;69:176–193. doi: 10.1007/s00239-009-9263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ip W.C., Hammond J.W., Wilcken B. Neonatal multiple acyl-CoA dehydrogenase deficiency: essentially absent fatty acid oxidation activity in proband but normal activity in parental cultured skin fibroblasts. J. Inherit. Metab. Dis. 1996;19:379–380. doi: 10.1007/BF01799277. [DOI] [PubMed] [Google Scholar]
- 7.Frerman F.E., Goodman S.I. Defects of electron transferflavoprotein and electron transfer flavoprotein-ubiquinoneoxidoreductase: glutaric aciduria type II. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Diseases. McGraw-Hill; New York: 2001. pp. 2357–2365. [Google Scholar]
- 8.Olsen R.K., Olpin S.E., Andresen B.S., Miedzybrodzka Z.H., Pourfarzam M., Merinero B., Frerman F.E., Beresford M.W., Dean J.C., Cornelius N., Andersen O., Oldfors A., Holme E., Gregersen N., Turnbull D.M., Morris A.A. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- 9.Wen B., Dai T., Li W., Zhao Y., Liu S., Zhang C., Li H., Wu J., Li D., Yan C. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J. Neurol. Neurosurg. Psychiatry. 2010;81:231–236. doi: 10.1136/jnnp.2009.176404. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J., Frerman F.E., Kim J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. U. S. A. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan M.Y., Fu M.H., Liu Y.F., Huang C.C., Chang Y.Y., Liu J.S., Peng C.H., Chen S.S. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin. Genet. 2010;78:565–569. doi: 10.1111/j.1399-0004.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 12.Dusheiko G., Kew M.C., Joffe B.I., Lewin J.R., Mantagos S., Tanaka K. Recurrent hypoglycemia associated with glutaric aciduria type II in an adult. N. Engl. J. Med. 1979;301:1405–1409. doi: 10.1056/NEJM197912273012601. [DOI] [PubMed] [Google Scholar]
- 13.Gempel K., Topaloglu H., Talim B., Schneiderat P., Schoser B.G., Hans V.H., Palmafy B., Kale G., Tokatli A., Quinzii C., Hirano M., Naini A., DiMauro S., Prokisch H., Lochmuller H., Horvath R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037–2044. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang W.C., Ohkuma A., Hayashi Y.K., Lopez L.C., Hirano M., Nonaka I., Noguchi S., Chen L.H., Jong Y.J., Nishino I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009;19:212–216. doi: 10.1016/j.nmd.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beresford M.W., Pourfarzam M., Turnbull D.M., Davidson J.E. So doctor, what exactly is wrong with my muscles? Glutaric aciduria type II presenting in a teenager. Neuromuscul. Disord. 2006;16:269–273. doi: 10.1016/j.nmd.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Angelini C., Tavian D., Missaglia S. Heterogeneous phenotypes in lipid storage myopathy due to ETFDH gene mutations. JIMD Rep. 2018;38:33–40. doi: 10.1007/8904_2017_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goh L.L., Lee Y., Tan E.S., Lim J.S.C., Lim C.W., Dalan R. Patient with multiple acyl-CoA dehydrogenase deficiency disease and ETFDH mutations benefits from riboflavin therapy: a case report. BMC Med. Genet. 2018;11:37. doi: 10.1186/s12920-018-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelius N., Byron C., Hargreaves I., Guerra P.F., Furdek A.K., Land J., Radford W.W., Frerman F., Corydon T.J., Gregersen N., Olsen R.K. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013;22:3819–3827. doi: 10.1093/hmg/ddt232. [DOI] [PubMed] [Google Scholar]
- 19.Cornelius N., Frerman F.E., Corydon T.J., Palmfeldt J., Bross P., Gregersen N., Olsen R.K. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2012;21:3435–3448. doi: 10.1093/hmg/dds175. [DOI] [PubMed] [Google Scholar]
- 20.Zhuo Z., Jin P., Li F., Li H., Chen X., Wang H. A case of late-onset riboflavin responsive multiple acyl-CoA dehydrogenase deficiency (MADD) with a novel mutation in ETFDH gene. J. Neurol. Sci. 2015;353:84–86. doi: 10.1016/j.jns.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z.Q., Chen X.J., Murong S.X., Wang N., Wu Z.Y. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J. Mol. Med. (Berl) 2011;89:569–576. doi: 10.1007/s00109-011-0725-7. [DOI] [PubMed] [Google Scholar]
- 22.Xi J., Wen B., Lin J., Zhu W., Luo S., Zhao C., Li D., Lin P., Lu J., Yan C. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2014;37:399–404. doi: 10.1007/s10545-013-9671-6. [DOI] [PubMed] [Google Scholar]
- 23.Zhu M., Zhu X., Qi X., Weijiang D., Yu Y., Wan H., Hong D. Riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients. J. Hum. Genet. 2014;59:256–261. doi: 10.1038/jhg.2014.10. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y., Zhao D.H., Hong D.J., Wang Z.X., Yuan Y. Hot spot mutations in electron transfer flavoprotein dehydrogenase gene of riboflavin responsive lipid storage myopathy in 20 Chinese families. Chin. J. Neurol. 2011;44:309–313. [Google Scholar]
- 25.Nagao M., Tanaka K. FAD-dependent regulation of transcription, translation, post-translational processing, and post-processing stability of various mitochondrial acyl-CoA dehydrogenases and of electron transfer flavoprotein and the site of holoenzyme formation. J. Biol. Chem. 1992;267:17925–17932. [PubMed] [Google Scholar]
- 26.Alves E., Henriques B.J., Rodrigues J.V., Prudencio P., Rocha H., Vilarinho L., Martinho R.G., Gomes C.M. Mutations at the flavin binding site of ETF:QO yield a MADD-like severe phenotype in Drosophila. Biochim. Biophys. Acta. 2012;1822:1284–1292. doi: 10.1016/j.bbadis.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Er T.K., Chen C.C., Liu Y.Y., Chang H.C., Chien Y.H., Chang J.G., Hwang J.K., Jong Y.J. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct. Biol. 2011;11:43. doi: 10.1186/1472-6807-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]