

Summary
The extracellular matrix (ECM) is present in all solid tissues and considered a master regulator of cell behaviour and phenotype. The importance of maintaining the correct biochemical and biophysical properties of the ECM, and the subsequent regulation of cell and tissue homeostasis, is illustrated by the simple fact that the ECM is highly dysregulated in many different types of disease, especially cancer. The loss of tissue ECM homeostasis and integrity is seen as one of the hallmarks of cancer and typically defines transitional events in progression and metastasis. The vast majority of cancer studies place an emphasis on exploring the behaviour and intrinsic signalling pathways of tumour cells. Their goal was to identify ways to target intracellular pathways regulating cancer. Cancer progression and metastasis are powerfully influenced by the ECM and thus present a vast, unexplored repository of anticancer targets that we are only just beginning to tap into. Deconstructing the complexity of the tumour ECM landscape and identifying the interactions between the many cell types, soluble factors and extracellular‐matrix proteins have proved challenging. Here, we discuss some of the emerging tools and platforms being used to catalogue and chart the ECM in cancer.
Keywords: cancer, extracellular matrix, imaging, metastasis, proteomics, remodelling
The diverse extracellular‐matrix (ECM) landscape
Composed of hundreds of different building blocks, the ECM makes up the complex, highly cross‐linked, three‐dimensional (3D) network of macromolecules (proteins, glycoproteins and its subgroup of proteoglycans, polysaccharides (glycosaminoglycans), elastins and carbohydrates) that surround cells (Figures 1 and 2). Extracellular matrix molecules assemble into supramolecular structures, each with their own distinct biochemical and biomechanical properties (Pickup et al. 2014). Broadly speaking, the ECM falls into two categories: a) interstitial matrix and b) highly specialized organ or tissue‐specific ECMs such as basement membranes (BM). The ECM is a salient feature of tissues and as such considered one of the most important regulators of cellular and tissue function in the body. That is to say that cells residing in, or passing through discrete tissue and ECM microenvironments activate or deactivate distinct sets of intracellular signalling events, such as clustering of integrin‐based adhesion proteins into focal adhesions (Nicolas et al. 2004), Rho‐ROCK activation/deactivation and downstream cytoskeletal reorganization (Chrzanowska‐Wodnicka & Burridge 1996; Reffay et al. 2014), and nuclear localization of key transcriptional regulators such as YAP/TAZ (Dupont et al. 2011) and SNAIL1 (Zhang et al. 2016; Figure 2). Importantly, these signalling events can elicit both short‐ and long‐term effects (Nasrollahi et al. 2017). Furthermore, the activation of these signalling pathways, such as Rho‐ROCK, typically leads to successive ECM reorganization such as collagen remodelling (Rath et al. 2016). The result is a dynamic reciprocity between cells and the ECM (Bissell & Aggeler 1987; Roskelley et al. 1995; Kass et al. 2007; Alexander & Cukierman 2016; Boyle & Samuel 2016).
Figure 1.
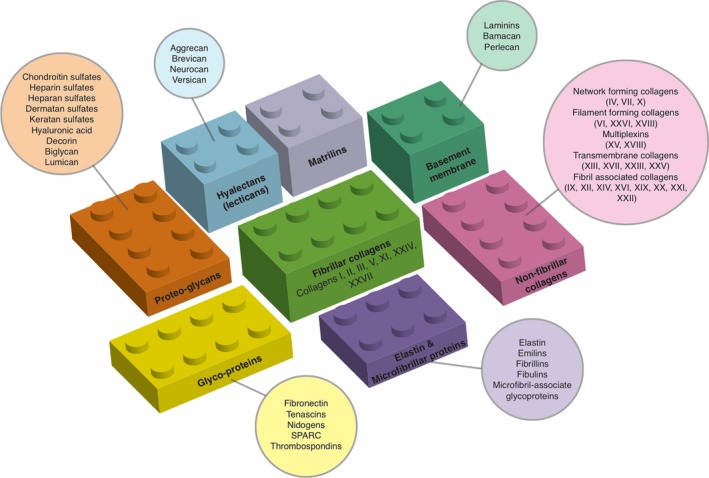
Overview of the basic building blocks of the extracellular matrix that come together to form the highly ordered supramolecular structures that contribute to the various interstitial matrix, microfibrils and the basal laminae within tissues. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Figure 2.
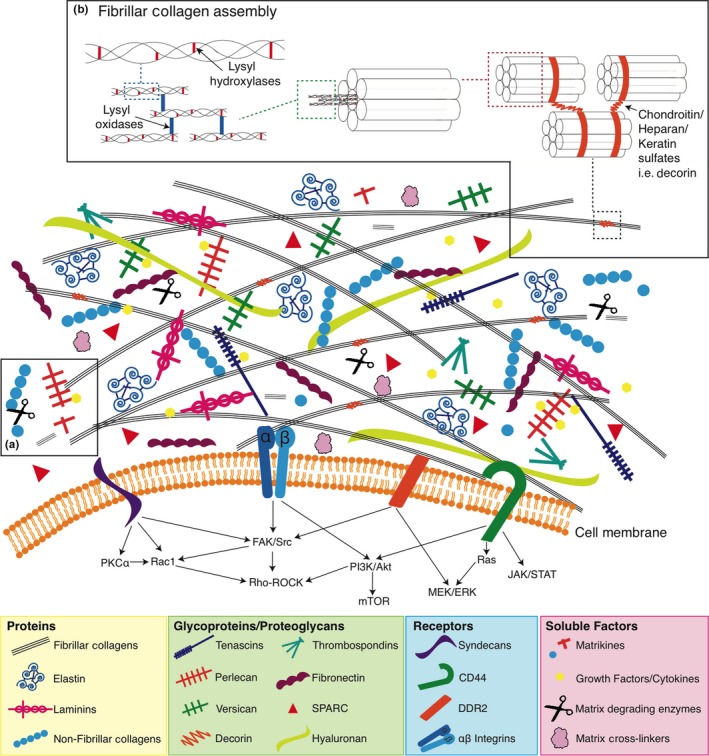
Simplified representation of the extracellular matrix. Made up of many molecules that come together to form the highly ordered supramolecular structures, and the extracellular matrix, through its various cell surface receptors, modulates a number of downstream intracellular signalling pathways to determine cell phenotype and behaviour. [Colour figure can be viewed at http://wileyonlinelibrary.com]
In most tissues, the structure of the ECM is highly dynamic and undergoes constant remodelling controlled by the delicate balance between synthesis and degradation. In doing this, the ECM provides continuously evolving topological, mechanical and biochemical cues to cells (Hynes 2009). Through engagement with various cell surface molecules including integrins, syndecans and discoidin domain receptors (Figure 2), the ECM activates and suppresses intracellular signalling pathways to regulate and fine‐tune diverse cellular programmes including cell proliferation, survival, migration/invasion, differentiation, stemness and plasticity (Hynes 2002, 2009). For example, integrins and their associated receptor tyrosine kinase (RTK) containing focal adhesions (FAs) are critically involved in the cellular response to the ECM through the simultaneous integration of physical and biochemical cues to modulate downstream cellular decisions.
In addition to the canonical structural components, another group of highly disparate, non‐structural matrix proteins exist within the ECM. Termed matricellular proteins (Bornstein & Sage 2002), these are capable of modulating a variety of biological processes within the ECM through altering cell–matrix interactions and cell regulatory functions. Acting via a number of unique mechanisms, matricellular proteins are typically highly context dependent, with many being organ specific. Examples include thrombospondin (TSP) 1 and 2, secreted protein acidic and rich in cysteine (SPARC), tenascin C, osteopontin and CCN family proteins (Figure 2). This group of proteins is dynamically expressed and is highly elevated during embryonic development but shows diminished presence in normal adult tissues. Thus, they are typically associated with tissue renewal and repair programmes and are thought to be important in normalizing the tissue (Chiodoni et al. 2010). For example, SPARC (initially called osteonectin) was originally identified in bone, where it binds collagen and Ca2+, and is key to initiating nucleation during the process of bone mineralization (Termine et al. 1981). However, SPARC is also known to be secreted by non‐epithelial cells in non‐ossifying tissues (Sage et al. 1984) during both development and tissue repair. Here, it plays a role in ECM remodelling and turnover, as well as in mediating cell–ECM interactions, including preventing hypertrophy of adipocytes and hyperplasia of adipocyte progenitors (Engel et al. 1987; Sage et al. 1989; Funk & Sage 1991; Lane & Sage 1994; Murphy‐Ullrich et al. 1995; Chlenski & Cohn 2010). SPARC has also been implicated in several solid cancers with SPARC overexpression being a marker of poor prognosis in cancers such as breast, prostate and melanoma (Podhajcer et al. 2008) and high expression correlating with tumour invasion through extracellular matrix in many aggressive cancers including breast invasive ductal carcinoma, glioblastoma, pancreatic ductal adenocarcinoma, clear‐cell renal cell carcinoma, melanoma and prostate carcinoma (Morrissey et al. 2016). In contrast, it has also been shown to be an inhibitor of cancer cell proliferation in some tumours including pancreatic cancer (Sato et al. 2003). Complicating the story further, SPARC is also known to be a chemoattractant for both breast and prostate cancer cells to home to the bone (Jacob et al. 1999). With this in mind, there is still much to do in order to uncover the precise role and function of matricellular proteins in the different stages of health and disease.
In recent years, there has also been increasing evidence that distinct fragments from specific ECM proteins and glycosaminoglycans (GAGs) are capable of regulating signalling through cell surface receptors and in turn regulating biological functions. Known as matrikines (Figure 2, box a), these ECM fragments are released by partial or limited proteolysis of extracellular‐matrix macromolecules. They are able to regulate cell activities such as proliferation, migration, protease production or apoptosis, leading to the modulation of physiopathological processes including angiogenesis, cancer, fibrosis, inflammation, neurodegenerative diseases and wound healing (Tran et al. 2005; Ricard‐Blum & Salza 2014). Still subject to some debate, matrikines are defined as any ECM fragment which has biological functions distinct from those of the parental macromolecule (Arroyo & Iruela‐Arispe 2010). Conversely, ‘cryptic’ matrikines or matricryptins are defined as enzymatic fragments of the ECM containing exposed cryptic sites (Davis et al. 2000). The term cryptic matrikine is restricted to biologically active fragments from the ECM‐exposing functional cryptic sites that are not normally exposed in the full‐length molecules. For more detailed reviews of matrikines and matricryptins, see Tran et al. (2005); Ricard‐Blum and Salza (2014); Davis et al. (2000).
The biological activity of matrikines is typically mediated through the integrin, heparan sulphate proteoglycan and growth factor receptors. Some examples of matrikines include arresten, canstatin and tumstatin, which are cryptic fragments generated from the NC1 domain of collagen IV α1/α2/α3 chains respectively (Chong et al. 2012). Canstatin, tumstatin, endostatin and tetrastatin bind the αvβ3 integrin (Maeshima et al. 2000; Rehn et al. 2001; Magnon et al. 2005; Faye et al. 2009; Brassart‐Pasco et al. 2012), whereas arrestin and anastellin are examples of matrikines binding β1 integrins (Colorado et al. 2000; Mercurius & Morla 2001; Sudhakar et al. 2005). The physical size of matrikines has been shown to vary from as little as a few amino acids in the case of the classical RGD tripeptide, to over 700 amino acids in endorepellin, a matrikine derived from the C‐terminal fragment of perlecan (Mongiat et al. 2003). Furthermore, the precise biological activity of specific matrikines may also be regulated by their size, an effect which was shown for different size fragments of hyaluronan oligosaccharides in wound healing (Ghazi et al. 2012) and angiogenesis (West et al. 1985; Cui et al. 2009). These findings reinforce the notion that the ECM is a highly dynamic entity that is highly regulated and compartmentalized at the post‐translational level, as well as intracellularly at the transcriptional/translational level.
In addition to engagement with cell surface receptors, there is also emerging evidence that ECM fragments may act as a reservoir for critical nutrients under conditions of nutrient deficiency. Recently, Olivares et al. (2017) showed that in pancreatic cancer models, collagens are catabolized to release ECM fragments that are then taken up as a proline source when other nutrient fuels are limited. The collagen‐derived proline is then metabolized by proline oxidase (PRODH1) to fuel the tricarboxylic acid (TCA) cycle and contributes to pancreatic ductal adenocarcinoma cell survival under restrictive nutrient conditions.
The changing ECM in disease
Remodelling of the ECM during tissue development and homeostasis, and in pathological diseases leads to the dramatic reorganization of biochemical and biomechanical cues presented to the cell. Alterations in the ECM have been linked to various pathologies such as fibrosis and skeletal diseases (Aszódi et al. 2006; Nelson & Bissell 2006; Bateman et al. 2009), and it has long been known that the tumour ECM is biochemically distinct from normal ECM (Cox & Erler 2011). In solid tumours, changes typically include both over‐ and underexpression of ECM molecules; increased and decreased degradation and/or post‐translational modification; and complete loss of, and even de novo synthesis of new ECM molecules. Even small changes can lead to significant alterations in the properties of the tumour ECM. As such, the remodelling of the ECM that accompanies disease onset and progression leads to significant changes in the bioavailability and bioactivity of ECM cues. These cues include altered anchorage points for cells and/or other ECM molecules such as proteoglycans, altered ECM assembly and disassembly dynamics, altered sensitivity to degradation, and altered growth factor sequestration (Bissell et al. 2002).
Despite the rapidly expanding body of literature, there is still an enormous depth and complexity to the heterogeneous 3D ECM landscape that is perhaps currently underappreciated. Apart from the basement membrane, our current level of understanding of cancer ECM structure is very much akin to how the cytoplasm was considered an amorphous ‘soup’ 30 years ago. Just as the understanding of the intracellular space has increased, so the recent advances in matrix biology have begun to shed light on the premise that the tissue ECM is highly compartmentalized and composed of spatially distinct biochemical and biomechanical zones. In its basic sense, this compartmentalization charts distinct differences between basement membrane matrix, pericellular matrix and interstitial matrix, although these represent a gross oversimplification of the ECM organization. Recently, several new approaches have emerged to help us understand and map this spatial complexity and chart the global dynamics of the ECM in health and disease.
We have known for many years, and will discuss in more detail later in this review, how the changes in the tissue ECM of solid tumours is inherently linked to cancer progression from a small isolated tumour to large invasive and/or disseminated metastatic disease. For example, seminal work by Weaver and colleagues showed that post‐translational cross‐linking of the ECM in premalignant stages of breast cancer is required for malignant transformation to occur and will drive breast tumour progression (Paszek et al. 2005; Levental et al. 2009). At the same time, Erler and colleagues have shown that similar ECM remodelling is needed to drive breast and colorectal cancer metastatic dissemination and colonization of secondary organs (Erler et al. 2006, 2009; Baker et al. 2011, 2013a; Cox et al. 2013). However, we still lack a detailed mechanistic understanding of the precise role that the tumour ECM plays at each point of this multistep process. With each transitional step in cancer progression, a number of ECM changes are involved, which modulate the intricate cross‐talk between the cancer cells and their surroundings. Many of these changes are significant in terms of scale or magnitude, but it is now emerging that subtler changes in absolute amounts, relative ratios, and/or spatial organization and topology also lead to profound changes in biochemical and biomechanical properties of the cancer ECM that together promote disease progression. As we can typically only study these ECM changes at defined static endpoints, it makes understanding how the continual evolution of the ECM during disease progression incredibly difficult. However, deconstructing the complexity of the tumour ECM and identifying the interactions between cell populations, soluble factors and ECM proteins is an exciting yet challenging field of research.
Over the last 30 years, researchers have shown that tumour cells not only need to accumulate oncogenic genetic alterations, but also rely on permissive cues from a surrounding ECM for malignant progression (Hanahan & Weinberg 2000, 2011; Egeblad et al. 2010). For example, it has been shown that the ECM is capable of altering level of mRNA and the rates of synthesis and secretion of proteins consistent with the model of ‘dynamic reciprocity’ (Bissell et al. 1982; Bissell & Aggeler 1987). In this model, the ECM exerts an influence on gene expression via transmembrane proteins (including integrins) and cytoskeletal components. The cytoskeleton is closely linked to polyribosomes, which regulate mRNA stability and rates of protein synthesis, and also with the nuclear matrix, which regulates mRNA processing and rates of transcription. Disrupting these ECM cues alone can therefore alter oncogenic programmes to accelerate or delay tumour progression, as was recently shown in pancreatic cancer by Laklai et al. (2016) (Bissell et al. 1982, 2002; Bissell & Aggeler 1987; Nelson & Bissell 2006; Bissell & Hines 2011). Thus, the tumour ECM has rapidly become of great interest to cancer researchers who have begun interrogating the importance of aberrantly regulated ECM proteins produced by tumour‐subverted stroma.
Extracellular matrix molecules such as tenascins (Mori et al. 1996; Hanamura et al. 1997; Yoshida et al. 1997; Pauli et al. 2002; Ilmonen et al. 2004; Brellier et al. 2009; Pedretti et al. 2009; Oskarsson et al. 2011; Saupe et al. 2013; Didem et al. 2014), periostin (Sasaki et al. 2001; Bao et al. 2004; Tilman et al. 2007; Fukushima et al. 2008; Puglisi et al. 2008; Takanami et al. 2008; Contié et al. 2011), hyaluronan (Sironen et al. 2011; Jacobetz et al. 2013) and versican (Ricciardelli et al. 2002, 2007, 2009; Pirinen et al. 2005; Ween et al. 2011; Keire et al. 2014) have all been shown to have important roles in the onset and progression of a multitude of different cancers. Furthermore, some of these individual ECM components have been implicated in controlling disseminated tumour cell dormancy (Barkan et al. 2010; Oskarsson & Massagué 2012).
Defining the ECM in health and disease
Many of the early ECM studies focused on extensively characterizing single ECM components in great detail. Whilst this led to enormous advances in our understanding of the role of these individual ECM components in health and disease, it adopts a reductionist approach to tackling the enormous complexity of the dynamic ECM landscape. However, given the inherently complex heterogeneous 3D nature of the ECM, including the extensive post‐translation modification and cross‐linking of ECM components, it has traditionally been very difficult to study the ECM and its function in vitro and in vivo at the global level. Much of our understanding of the 3D nature of the ECM has come from in vivo mouse models which leverage gene‐targeting approaches, transgene expression and spontaneous mutations of ECM proteins (Aszódi et al. 2006). This has significantly contributed to, and accelerated our mechanistic understanding of the structure and function of the different ECM molecules in tissue homeostasis and disease. In particular, they have been critical in refining our understanding that not all ECM components contribute purely scaffolding roles to the matrix as basic structural elements, but many, if not all, act as organizers/modulators of the structural network and bioactive signalling cues.
The development of high‐throughput ‘omics’‐based approaches has significantly accelerated the widespread global cataloguing of proteomes in almost every living organism. Thanks to the completion of the genomes of many species and to previous studies, it is now clear that vertebrate genomes contain hundreds of genes that encode, or potentially encode ECM proteins. Unbiased global approaches to mapping the proteome of tissue and organ ECMs are giving us new information into the make‐up of tissues and how they are altered in the disease setting. In most tissues, it has been shown that the primary ECM scaffold is typically made up of organ‐ and tissue‐specific blends of collagen fibrils or networks. Collagens are seen as the principal load‐bearing elements in all tissues from skin to bone. The tissue‐specific collagen networks typically stiffen under mechanical loads to prevent excessive and potentially damaging deformation of the tissues. To date, there are 16 different types of collagen, with collagen types I, II and III comprising approximately 75‐80% of all of the collagen within our body (Figure 1). These collagens are packed together to form long thin, mainly structural fibrils. In contrast, collagen type IV forms a two‐dimensional reticulum and is core to all basal laminae. Several of the other collagen types such as VI and XXVI then form filament networks associated with these fibrillar collagens and act to link them to one another or to other matrix components (Figures 1 and 2, box B). These different collagens and their resulting networks, along with the various other ECM components, undergo constant remodelling to provide required biochemical properties such as tensile strength and elasticity.
Importantly, the ratios of individual collagens and other ECM components to one another are critical in the subsequent provision of assembly points or sequestration of various proteoglycans and glycoproteins in both a spatial and temporal manner. The result is the formation of highly ordered supramolecular structures that contribute to the various biochemical and biomechanical requirements of the tissue. For example, the specific make‐up of glycosaminoglycans on proteoglycans will alter the hydration of the matrix (Figure 2), which in turn alters bulk tissue stiffness, interstitial pressure of tissues as well as rates of diffusion and perfusion within the tissues. At the same time, proteoglycans play a major role in signal transduction with regulatory functions in various cellular processes (Schaefer & Schaefer 2010). Therefore, the biomechanical properties of the ECM can synergize with and antagonize the biochemical cues to drive single‐cell and collective cell behaviour resulting in correct tissue and organ function. Recently, there have also emerged exciting reports that tissue stiffness may even regulate cellular circadian clocks, with stiff extracellular matrix dampening the oscillations of the molecular clock within cells (Yang et al. 2017; Williams et al. 2018). As such, disruption of any single ECM component can, and often does, lead to an enormous array of downstream changes that typically force the establishment of a new dynamic equilibrium and underpin the gross changes in tissue and organ architecture and function typically associated with cancer progression (Mayorca‐Guiliani et al. 2017).
For example, it was initially thought that increased mammographic density was caused purely by increased deposition of collagens within the mammary tissue. However, recent studies using approaches such as second harmonic generation (SHG) imaging, picrosirius red (PSR) and polarizing light microscopy have demonstrated that raised mammographic density is not associated with merely increased collagen deposition, but the specific spatial organization and topology of collagen fibrils (Huo et al. 2015; McConnell et al. 2016; Sherratt et al. 2016). This organization and topology are, in part, controlled by altered proteoglycan composition, and as a result, several laboratories are investigating how proteoglycans may underpin the physical properties of mammographic density and ultimately contribute to the associated increase in breast cancer risk. Despite numerous studies investigating proteoglycans in relation to mammographic density and breast cancer, the reported results are sometimes discordant. For a comprehensive review of the role of proteoglycans in mammographic density and breast cancer, see Shawky et al. (2015). At present, it is not known how localized ECM changes in composition and organization contribute to globally raised mammographic density and subsequently to increased breast cancer progression and metastasis at the mechanistic level. Likewise, there is still a knowledge gap in the specific changes in ECM composition and properties during tumour progression. Understanding how the changing ECM landscape is linked to alterations in mammographic density and subsequently to the onset and progression of breast cancer as well as survival is critically important. High mammographic density is not uncommon in the community, yet the biological basis of the ECM in high mammographic density, and how it raises breast cancer risk remains elusive.
There are many reports detailing ECM changes which have been shown to be involved in cancer progression, poor prognosis and malignant cell invasion at both primary and secondary sites. For example, elevated levels of fibronectin have been shown to promote ovarian cancer invasion and metastasis through α5β1 integrin and activating downstream c‐Met/FAK/Src‐dependent signalling (Kenny et al. 2014). Increased levels of versican, fibronectin, collagen I and decreased levels of vitronectin and tenascin C have been shown in the premetastatic liver in models of pancreatic cancer (Costa‐Silva et al. 2015). It is becoming increasingly clear that ECM remodelling in tumour progression does not only occur at the primary site of tumour cells, but can also be driven systemically through the release of tumour‐secreted factors and exosomes to form so‐called premetastatic niches. These premetastatic niches exhibit altered expression of ECM molecules as well as aberrant post‐translational modification, which leads to the generation of a growth permissive milieu that acts to support tumour cell colonization of secondary organs. Premetastatic remodelling of the ECM has been shown to occur in several cancers including breast cancer (Erler et al. 2009; Hiratsuka et al. 2013; Cox et al. 2015), lung, pancreatic cancer and melanoma (Kaplan et al. 2005; Peinado et al. 2012) leading to increases in the resulting metastatic burden in these secondary organs.
Prolonged exposure to the extracellular proteoglycan, lumican, has been shown to restrain pancreatic adenocarcinoma growth (Li et al. 2017). In contrast, versican and tenascin have both been shown to be increased in the peri‐tumoral stroma of breast cancer and predictive of poor relapse‐free and overall survival respectively, in women with lymph node‐negative breast cancer. In fact, versican is localized within the peri‐tumoral stromal tissue of many solid cancers and is secreted into the ECM by both host tissue fibroblasts, or expressed by the cancer cells themselves. Several mechanisms have been proposed for regulating versican levels in the tumour ECM. For example, TGF‐β‐driven secretion of versican in the ovarian tumour microenvironment activates NF‐κB signalling and upregulates expression of CD44, matrix metalloproteinase‐9 and the hyaluronan‐mediated motility receptor in the tumour cells to enhance aggressiveness (Yeung et al. 2013; Figure 2). Therefore, inhibiting versican synthesis may be a potential mechanism for reducing versican levels in cancer tissues. Treatment with the tyrosine kinase inhibitor, genistein, has been shown to inhibit versican synthesis induced by growth factors in malignant mesothelioma cell lines (Syrokou et al. 1999) and vascular SMCs (Schönherr et al. 1997). The biological role and regulation of versican in cancer are discussed in more detail in Ricciardelli et al. (2009).
Proteomic cataloguing of the ECM
Previous proteomic analyses of tumours have typically not focused on the ECM, although ECM proteins are usually detected among the tumour proteins analysed (Zanivan et al. 2008; Sutton et al. 2010; Shaheed et al. 2013). This is predominantly as a result of the physical and chemical properties of the ECM. ECM components are heavily cross‐linked, highly insoluble and often strongly charged which poses significant technical challenges to mass spectrometry pipelines and have, until recently, prevented effective and routine analysis. However, new proteomic protocols and pipelines have been developed that selectively enrich for ECM proteins based on their relative insolubility compared to intracellular proteins. Quantitative proteomics is now seen as the gold standard method for comprehensively profiling the dynamic changes in the composition of the ECM that occur during disease progression. Selective enrichment for ECM components is primarily based on removal of soluble intracellular proteins from tissue samples in solution (Naba et al. 2012, 2015, 2017a) or more recently in situ using protocols such as ISDoT (Mayorca‐Guiliani et al. 2017; Figure 3). In addition, the depletion of highly abundant ECM elements such as collagens, through treatment with collagenases, can enhance the proteomic coverage of low‐abundance proteins in decellularized tissues (Kuljanin et al. 2017).
Figure 3.
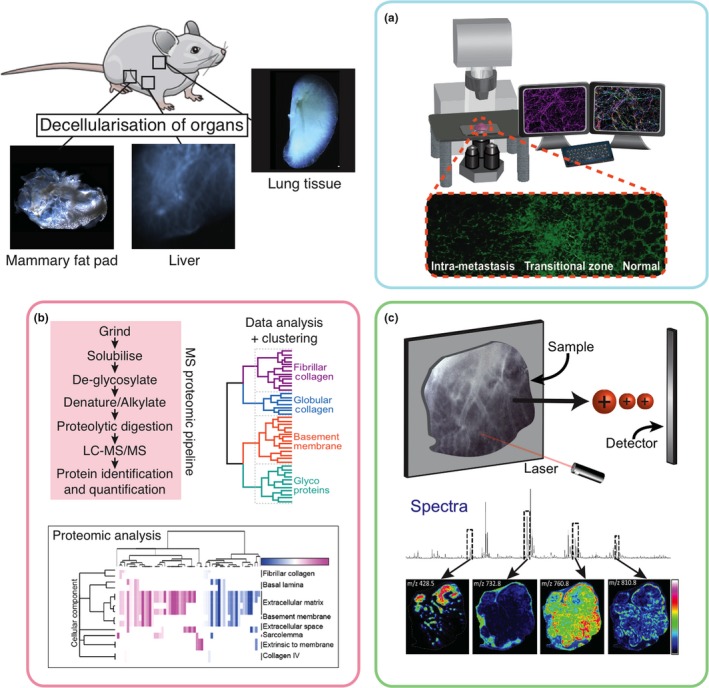
Multiplexing new platforms to study the extracellular matrix in health and disease. Approaches such as tissue decellularization significantly boost our ability to, and resolution at which we can study the extracellular matrix in downstream applications such as; (a) confocal and light sheet microscopy; (b) proteomic cataloguing using liquid chromatography‐mass spectrometry and computational analysis; (c) MALDI‐Mass Spec Imaging (MALDI‐MSI) and spatial mapping of ECM components. [Colour figure can be viewed at http://wileyonlinelibrary.com]
In addition to proteomics‐based cataloguing, Naba and colleagues have also pioneered efforts to combine proteomics‐based methods with bioinformatic pipelines to reshape our definition of the so‐called matrisome. The matrisome is classified as both the ECM and ECM‐associated proteins which make up the protein composition of the tissue extracellular matrix (Naba et al. 2012). The establishment of the ‘Matrisome Project’ and the generation of reference matrisomes for different organisms (MatrisomeDB; http://matrisomeproject.mit.edu/) include proteins predicted (based on genomic sequence) to encode matrix proteins because they contain characteristic InterPro (https://www.ebi.ac.uk/interpro/) domain‐based structures (Hohenester & Engel 2002), a classical hallmark of ECM proteins. Their inclusion criterion consists of 55 diagnostic InterPro domains commonly found in ECM proteins. The challenge now is to map expression patterns spatially to tissues and temporally to organ development and disease. For a comprehensive review of some of the new bioinformatic tools and experimental strategies for cataloguing the ECM in health and disease and how they can be exploited to provide novel insights in our understanding of ECM biology, see Naba et al. (2016).
Building on this, the recent establishment of the ECM Atlas (http://matrisomeproject.mit.edu/ecm-atlas/; Naba et al. 2016) also provides a rapidly expanding resource for matrix biologists. The ECM atlas is built through the compilation of proteomic data sets of ECM composition from multiple different tissues and disease types. The next release scheduled for later in 2018 will dramatically expand the ECM Atlas to incorporate new tissues and diseases and mark significant collaborative efforts in the matrix biology field to deepen our understanding of the ECM. Whilst it is becoming commonplace for journals to require that mass spectrometry data sets are made available upon publication, they are typically unwieldy and provide little information to non‐experts. Web interfaces such as the ECM Atlas are now providing a common point of reference to easily access and extract useful information from this vast and rapidly expanding reservoir of data.
Moving forward, the next step is to determine the mechanisms leading to this presence of lower or higher abundance of specific ECM proteins in the microenvironment during normal development or disease. In particular, determining whether these changes are the result of transcriptional or translational control mechanisms, or altered secretion, assembly or stability of ECM proteins in the tissue microenvironment. We and others have previously identified novel ECM proteins and ECM modulators in tissue samples from tumour progression models that have previously not been associated with tumour progression (Mayorca‐Guiliani et al. 2017; Naba et al. 2017a; Figure 3, box B). Refining our understanding of the build‐up and breakdown of the ECM both in normal tissue and in disease will be critical to understanding how the dynamic ECM regulates tissue homeostasis and disease progression. Furthermore, the ability to define ECM signatures (Figure 3, box B) based on proteomic analysis will be incredibly useful in disease staging. Using label‐based quantitative proteomics, Naba et al. (2017a) have achieved this and reported the characterization of the matrisome in pancreatic islets and identified a 35‐ECM–protein signature that is characteristic of, and correlated with insulinoma progression, whilst in high‐grade serous ovarian cancer (HGSOC), Pearce et al. (2017) reported a matrix index that could be used to predict poor prognosis. More recently, researchers have identified a nine‐gene ECM signature (based on mining of gene transcriptional data sets), which strongly predicts outcome across multiple cancer types, although it still requires validation in prospective cancer cohorts to confirm its prognostic ability (Yuzhalin et al. 2018). Just as we now currently use genomic signatures to identify high‐risk patients and predict outcome across a wide array of cancer types, it will not be long before we can use ECM‐based proteomic signatures in a similar manner.
Seeing is believing: charting the matrix
Cataloguing the in vivo ECM composition of normal and diseased tissues using selective ECM enrichment has accelerated the pace of ECM cataloguing in disease (for a full review, see Byron et al. 2013). Whilst the information generated is invaluable in understanding changing ECM composition in disease, we are still lacking a matching repertoire of tools facilitating high‐definition interrogation of global 3D topology of the ECM and the subsequent study of cell interactions with this native ECM. In addition to cataloguing the ECM, a better characterization of native ECM composition and its spatial and temporal distribution and organization is needed to provide a deeper understanding of its role in disease settings such as cancer.
Whilst several groups, including our own, have investigated ECM changes in 2D patient tissue sections, until very recently no 3D studies have thoroughly characterized or linked the spatial topology of the tumour ECM to disease progression. As a result, we have little insight into the global pathophysiology of the ECM in cancer, where ECM remodelling is a fundamental driver of cancer cell survival, proliferation and metastasis (Cox & Erler 2011). Currently, there are several approaches designed to facilitate high‐resolution 3D microscopy of native tissues. These have almost exclusively focused on tissue fixation/clearing approaches and advances in microscopy to improve optical clarity of large volumes of tissue. These approaches include PACT, CLARITY, Scale/ScaleA2, SeeDB, PARS, ClearT/ClearT2, 3DISCO, RIMS, FocusClear, CUBIC, LUMOS, DBE and BABB (Dodt et al. 2007; Tseng et al. 2009; Hama et al. 2011; Becker et al. 2012; Chung & Deisseroth 2013; Ertürk & Bradke 2013; Ke et al. 2013; Kuwajima et al. 2013; Moy et al. 2013; Susaki et al. 2014; Tainaka et al. 2014; Tomer et al. 2014). Some are specifically tailored to organs of interest such as the brain, whilst others such as 3DISCO can be applied more widely. Indeed, it was recently shown that RI‐CUBIC (refractive index‐optimized clear unobstructed brain/body imaging cocktails and computational analysis) protocols enable whole‐body examination of cancer models at the single‐cell resolution (Kubota et al. 2017). The focus of these methodologies, however, is to increase imaging resolution whilst preserving cellular structure. These approaches are unfortunately not optimized for improving imaging resolution of the ECM architecture. Similarly, many of these approaches involve tissue fixation and swelling, which may affect tissue integrity and/or biomolecular structures, introduce artefacts, distort native ECM architecture and even irreversibly denature biomolecules.
In vivo the precise assembly of collagens into different fibrils and fibre organizations is also typically tissue dependent. For example, collagens can form thin fibrils in the region of 30 nm in diameter within in the cornea of the eye. However, they will also form much thicker fibres, in some cases several orders of magnitude larger, at several microns, in tissues such as tendons which experience very high tensile strains (Raspanti et al. 2018). Therefore, the preservation of the delicate 3D structure of the ECM is critical when attempting to dissect the processes underlying its spatial organization and construction, and the resulting effects on cell behaviour and phenotype. The overall organization of the ECM is largely determined by the positioning and orientation of the collagen fibrils to one another. The resulting ECM pore size, fibre alignment and visco‐elasticity, as well as biomolecular composition in turn provide important cues controlling cell behaviour. The organization, alignment and spacing of collagen fibrils are determined, in addition to their abundance, by proteoglycans. This in turn is critical to controlling elements such as the mechanical properties of the tissue. The precise spacing between collagen fibrils is controlled by dermatan/chondroitin sulphate or keratan sulphate chains bound to the protein cores of small leucine‐rich proteoglycans (Figure 2, box B). These proteoglycans bind to the specific binding domains of collagen fibrils (approximately every 65 nm), and their sulphated side chains bridge and control the interfibrillar space (Scott et al. 1998). Disrupting these proteoglycans significantly alters collagen fibre assembly and the organization of ECM architecture. For example, decorin, a small leucine‐rich proteoglycan abundant in normal breast tissue, has been shown to coat the shaft of mature collagen fibrils preventing fibril fusion (Ishiba et al. 2014). Decorin knockout (KO) mouse models still show high levels of collagen fibrils within tissues such as the skin, similar to that shown in wild‐type mice. However, in KO mice, these collagen fibrils exhibit an abnormal morphology which leads to altered tensile strength and dramatically increases skin fragility (Danielson et al. 1997).
In terms of disease, it has previously been shown that the precise organization and orientation of collagen fibres in the tumour microenvironment are also tightly correlated with breast tumour progression (Provenzano et al. 2006). Collagen fibre alignment perpendicular to and out from the tumour may be an enabling feature for breast cancer cells to migrate persistently along these fibres away from the primary site (Riching et al. 2014). It has recently been shown that increased confinement of MV3 melanoma and HT1080 fibrosarcoma cells, due to increasing collagen concentrations, can lead to ‘cell jamming’ and acts to drive a switch from single‐ to collective cell migration (Haeger et al. 2014). The importance of mechanoreciprocity, ECM organization and remodelling in determining cell migration and invasion and the concept of cell jamming are discussed in more detail in van Helvert et al. (2018), and a comprehensive review of cellular mechanotransduction at the cell–matrix interface is discussed in Jansen et al. (2017).
Decellularization of tissues to produce native ECMs is a powerful tool to study the matrix (Figure 3). Decellularized ECM (assuming the use of correct decellularization treatments) is identical to the composition of native ECM and is generally considered to possess native mechanical properties (e.g. stiffness) and microstructure. In the tissue regeneration and bioengineering field, decellularized tissues and organs are often used as scaffolds to facilitate the repair and reconstruction of tissues. Recently, researchers have also begun investigating the way that decellularization approaches affect the integrity of the ECM including the preservation of glycosaminoglycans (GAG), collagens and cytokines (Fischer et al. 2017). A clear benefit of tissue decellularization is that it improves the optical clarity of large volumes of tissue for studying the ECM using conventional approaches such as confocal and multiphoton microscopy as well as emerging techniques such as light‐sheet microscopy. As such, tissue decellularization for matrix biology bridges the gap between in vivo whole‐organ imaging and standard 2D histology.
We recently developed a fast and efficient approach to enhance the study of ECM composition and 3D structure of normal and diseased tissues in exquisite detail (Figure 3). Termed In situ decellularization of tissues (ISDoT), it allows tissues to be decellularized without collapse to leave the delicate native ECM architecture intact. The optically clear 3D ISDoT decellularized tissues can be studied using high‐resolution confocal and state‐of‐the‐art multiphoton microscopy without the need for extensive preparation, tissue fixation or sectioning. Furthermore, ISDoT acts to enrich ECM molecules making it perfectly suited to quantitative proteomic interrogation of the ECM to accurately catalogue global changes during cancer progression (workflow shown in Figure 3). Using ISDoT, we have shown that this enrichment facilitates an approximate order of magnitude increase in proteomic coverage compared to non‐decellularized tissues (Mayorca‐Guiliani et al. 2017). This therefore allows reliable identification and quantification of ECM molecules otherwise masked in the presence of cellular content. Specifically, it allows the accurate detection of subtle changes in ratiometric levels of ECM components. As discussed above, the changing ECM in cancer is not only characterized by increased ECM deposition. It is also important to distinguish between increased absolute amounts and increased concentrations of ECM components relative to one another during disease progression (Cox & Erler 2011). This can only be carried out with global ‘omics’ approaches and is not immediately possible when looking at single molecules in isolation. Using matched ISDoT samples, we can both comprehensively catalogue the ECM and map the spatial distribution of components in 3D in high resolution in the normal vs. tumour setting. As such, this work was the first to provide detailed 3D characterization of the metastatic niche in breast cancer progression and identified several ECM components, which have not previously been implicated in cancer onset and progression (Mayorca‐Guiliani et al. 2017). Recently, complementary decellularization approaches to study the ECM in development of whole murine embryos has also begun to visually map and interrogate proteoglycan‐rich ECMs in multiple developing tissues, including the forelimb, eye and spinal cord (Acuna et al. 2018).
This work is part of the emerging trend of applying cutting‐edge technologies to ECM research and is set to revolutionize the way in which we study the ECM in disease. For decades, the ECM was seen as ‘the stuff outside cells’, but we have shown that there is a highly ordered compartmentalization and localization of specific matrix molecules within the tumour microenvironment (Figure 3a, note the observed presence of the transition zone at the tumour interface). This compartmentalization is a fundamental feature of the ECM landscape and underpins the very essence of tissue and organ complexity. Each compartment constitutes a unique and specific context of biochemical and biomechanical cues based on its protein composition and spatial organization/topology. As such, the ECM is pivotal in contributing to both inter and intratumour heterogeneity. The ability to spatially map ECM heterogeneity will be critical to understanding how it modulates cancer cell phenotype, invasive/migratory behaviour, proliferative capacity and even stemness and plasticity (Bissell & Hines 2011). Similarly, by comparing and contrasting different ECM compositions and topologies across cancer types, we may be able to stratify and treat patient tumours using ECM‐based signatures rather than the organ from which they originate.
Using ‘omics to see the matrix – MALDI‐MSI
Matrix‐assisted laser desorption/ionization mass spectrometry imaging (MALDI‐MSI) has emerged as a powerful analytical technique that combines the specificity and sensitivity of mass spectrometry with spatial information to map the tissue‐wide distribution of multiple analytes including proteins (Caprioli et al. 1997), peptides (DeKeyser et al. 2007), lipids (Woods & Jackson 2006; Jackson et al. 2007) and small molecules (Reyzer et al. 2003; Cornett et al. 2008; Dekker et al. 2009) directly from a single tissue section (Figure 3, box C). Current approaches can accurately identify and quantify hundreds of native (or modified) analytes simultaneously in a single experiment, with a typical spatial resolution of between 10 to 200 microns. MALDI‐MSI allows direct label‐free measurement of analytes from tissue sections, and spatial ‘molecular histology’ images can be reconstituted for each observed signal. Whilst the approach is conducted on 2D sections of tissue, it creates a digital MALDI image for each ECM component, which combined with serial sectioning and image registration and integration has the potential to generate 3D detailed reconstructions of the ECM topology and architecture (Figure 3, box C).
Cancer samples are highly heterogeneous with many different histological ECM structures on a small scale. As MALDI‐MSI preserves the spatial distribution and the histology of the samples, it is perfectly suited for the analysis of these highly heterogeneous cancer samples. As such, MALDI‐MSI is a highly flexible and powerful imaging technology and is not only proving to be a uniquely useful discovery tool for mapping tissue‐wide distributions of analytes, but can also be used to look for specific molecules from pre‐generated targeted lists. Moreover, subtle molecular ECM signals or patterns can be identified and correlated with tumour subtypes or other parameters such as outcome or response to treatment.
Recently, MALDI‐MSI has been successfully combined with decellularization approaches to significantly improve identification rates and the resolution of spatial mapping of ECM components in tissues (Gessel et al. 2015; Figure 3). However, there are limitations to MALDI‐MSI when it comes to high molecular weight (100's of kDa) components such as glycoproteins abundant in some tissues, because they are currently beyond the practical mass range for protein imaging experiments (Gessel et al. 2015). Whilst the spatial resolution of MALDI‐MSI is typically less than that obtained by confocal, multiphoton and light‐sheet microscopy, it does not rely on the availability of validated antibodies that may hamper other visualization approaches. It can in some cases also be used to map post‐translational modifications such as protease cleavage, cross‐linking, citrullination, nitrosylation, glycosylation and isomerization of ECM molecules.
Post‐translational modification and regulation of the ECM
The natural ageing process of the ECM and various pathologies correlate closely with alterations in the post‐translational modification of ECM molecules. These modifications affect the structural and mechanical integrity of the tissues, altering among other things their susceptibility to degradation. Classic examples of this are skin wrinkling, bone and cartilage deterioration, as well as cardiovascular and respiratory malfunctions. Typically, specific post‐translational modifications are the result of local physiological or pathobiological processes. As such, the identification of specific post‐translational modifications could be used as disease‐specific markers for staging or ageing of the tumour. For a review on how post‐translational ECM modifications are seen as key events in cancer progression, see Leeming et al. (2011).
Excessive post‐translational modification of the ECM is associated with many solid tumours. In particular, the post‐translational modification of fibrillar collagens has become an area of intense interest given its importance in tumour‐associated desmoplasia. Normal collagen synthesis requires several post‐translational modifications, such as hydroxylation of proline and lysine residues, propeptide cleavage and the covalent cross‐linking by lysyl oxidases (Figure 2, box B; Gerstenfeld et al. 1993; Myllyharju & Kivirikko 2004; Mäki 2009). The overexpression of the collagen cross‐linking lysyl oxidases has been shown by us and others to be key players in the progression of several solid cancers including breast, pancreatic, colorectal, head and neck and more (Payne et al. 2005; Erler et al. 2006, 2009; Laczko et al. 2007; Bondareva et al. 2009; Levental et al. 2009; Sakai et al. 2009; Baker et al. 2011, 2013a,b; Taylor et al. 2011; Chu et al. 2012; El‐Haibi et al. 2012; Cox et al. 2013, 2015; Pickup et al. 2013; Miller et al. 2015; Chang et al. 2017; Rachman‐Tzemah et al. 2017; Reynaud et al. 2017). For a comprehensive full review of the role of the lysyl oxidase family in cancer see (Barker et al. 2012). Importantly, targeting the lysyl oxidase family has shown promising preclinical results and is seen to offer significant promise in targeting the remodelling of the ECM in cancer.
In addition to altered deposition, turnover or reorganization of the ECM, there are a large number of long‐lived proteins that are prone to the accumulation of damage with both age and disease (Sherratt 2009). In fact, many structural ECM proteins exhibit a remarkable longevity in vivo, often measured in years as opposed to hours for intracellular proteins. This longevity also appears to be tissue specific and likely comes down to fibril organization and structure (discussed above). For example, types I and II collagen in human skin, articular cartilage and intervertebral disc exhibit 15‐, 95‐ and 117‐year half‐lives respectively (Verzijl et al. 2000; Sivan et al. 2008). When we consider these timescales, events such as glycation‐mediated collagen cross‐linking, the normal and slow accumulation of cross‐links that form advanced glycation end products (AGEs) become important and are thought to play a key role in many age‐associated diseases, including degenerative eye disease, pulmonary fibrosis, arterial stiffening and cardiovascular disease, and neurodegeneration. Furthermore, these manifestations are increasingly accelerated in diabetes patients who exhibit chronically elevated blood glucose levels (Bunn et al. 1978; Frank 1991; Vitek et al. 1994; Sasaki et al. 1998; Glenn & Stitt 2009). The aberrant glycation of proteins leading to AGEs has been shown to be associated with increased tissue stiffness in tissues such as the prostate (Rodriguez‐Teja et al. 2015), and such changes in tissue stiffness are also closely associated with cancer onset and progression (Bailey 2001; Paszek et al. 2005; Sharaf et al. 2015).
If we apply this to the cancer setting, it could be argued that the development of cancer in an aged individual may be more likely to be accompanied by accelerated progression and enhanced propensity for metastasis, than if the same cancer was to develop at a younger stage of life. A hypothesis of this nature would centre around a central theme that a slowing down of active ECM remodelling and the gradual replacement with passive cross‐linked, and/or post‐translationally modified ECM components would ultimately lead to a more aggressive tumour. In support of this, work by Maller et al. (2013) has attempted to address why clinically, there is a reduction in breast cancer risk in postpregnancy women. Following pregnancy, during involution, a vast developmental programme remodels the breast tissue following cessation of milk production. In their study, the authors reported an approximately 50% increase in abundance of collagen I in postparous rat mammary tissue and also in clinical samples. However, more importantly, they noted a significant decrease in the level of collagen I linearization as measured by second harmonic generation (SHG) imaging compared to nulliparous mice. Thus, it is clear that collagen I organization and not merely abundance plays an important role in parity‐induced protection. These findings support the hypothesis that the active reorganization brought about by pregnancy‐induced remodelling of the mammary gland may act to reset the accumulation of detrimental and/or protumorigenic post‐translational modifications which slowly accumulate over time (Cox & Erler 2011).
The therapeutic repertoire of the matrix in disease
The vast majority of cancer studies place an emphasis on studying the behaviour and intrinsic signalling pathways of tumour cells. Their goal is to identify ways to target intracellular pathways regulating cancer. Indeed, most, if not all, of the chemotherapeutic regimens for cancer are directed at the tumour cells and not the ECM. We and others have shown that cancer progression and metastasis are strongly influenced by the microenvironment and in particular the ECM. As such, we argue that the ECM presents a vast, and at present, unexplored repository of anticancer targets and will briefly summarize some current and emerging approaches to targeting it in cancer.
One approach to therapeutically treating diseases such as solid tumours via their aberrant ECM is to exploit the presence of unique, or overexpressed ECM molecules as targeting beacons. For example, Ishihara and colleagues recently showed that they could conjugate immune checkpoint antibodies to the heparin binding domain (HBD) of placental growth factor‐2 (PIGF‐2123‐144) which shows exceptionally high affinity for multiple ECM proteins (Martino et al. 2014). Peri‐tumoral injection of PIGF‐2123‐144–αPD‐L1 led to higher retention within tumour tissue. This in turn significantly reduced systemic toxicity and elicited a highly effective antitumour response in the B16F10 melanoma model (Ishihara et al. 2017). Similarly, collagen I targeting of cetuximab, an anti‐EGFR monoclonal antibody, showed good therapeutic efficacy in the A431 epidermoid cancer model. By targeting cetuximab to the collagen‐rich tumour ECM, it led to a longer retention time in tumours with no loss in potency (Liang et al. 2016). However, despite this encouraging data, it may prove more powerful to target the less ubiquitous molecules, such as those that are expressed exclusively in specific tumours.
A recent success of note in targeting ECM molecules directly in cancer to improve outcome is highlighted by the phase II HALO 202 clinical trial (NCT01839487). Metastatic pancreatic ductal adenocarcinoma is typically characterized by accumulation of excessive hyaluronan (HA), a large, linear polysaccharide within the tumour microenvironment. The increased HA leads to elevated interstitial pressure and as a consequence is thought to impair perfusion and chemotherapy delivery to tumours. In preclinical studies, pegvorhyaluronidase alfa (PEGPH20), a pegylated recombinant human hyaluronidase, was shown to successfully degrade intratumoral HA and remodel the tumour stroma to improve perfusion and drug delivery (Thompson et al. 2010; Provenzano et al. 2012; Jacobetz et al. 2013). Based on this, the recent phase II clinical trial in patients with previously untreated metastatic pancreatic ductal adenocarcinoma showed that in those with high HA tumours, treatment with PEGPH20 plus standard‐of‐care chemotherapy (gemcitabine + Nab‐paclitaxel) significantly improved progression‐free survival (Hingorani et al. 2017), highlighting how combinatorial approaches which target the ECM as well as cancer cells offer enormous promise.
The tightly controlled homeostasis of the ECM is sensitive to altered expression of matrix‐degrading enzymes such as matrix metalloproteinases (MMPs; Figure 2), which are typically altered for prolonged periods of time in fibrotic diseases and cancer. Unfortunately, a range of matrix metalloproteinase (MMP) inhibitors have to date proven largely unsuccessful in the clinic. This is likely due (among other reasons) to their lack of specificity and ubiquitous nature of expression throughout multiple tissues in the body (Coussens et al. 2002). For a full and comprehensive review on MMP‐targeting successes and failures, see Cathcart et al. (2015).
A recent example of the difficulties associated with targeting post‐translational cross‐linking in the matrix has been highlighted with the lysyl oxidase‐like 2 (LOXL2) antibody, simtuzumab, which showed no significant improvement in overall survival in a phase II trial in late‐stage metastatic pancreatic cancer (Benson et al. 2017), or in a phase II trial in metastatic KRAS mutant colorectal adenocarcinoma (Hecht et al. 2017). However, it should be pointed out here that in these trials, the late‐stage terminal nature of disease in these patients was unlikely to yield significant results, and as such, earlier treatment will likely lead to more promising results in line with preclinical studies using lysyl oxidase inhibitors in breast (Erler et al. 2009; Cox et al. 2013, 2015; Chang et al. 2017), colorectal (Baker et al. 2011, 2013a,b) and pancreatic cancers (Miller et al. 2015).
In addition to targeting or exploiting ECM elements directly, other approaches have focussed on targeting the downstream cellular response to altered ECM in diseases such as cancer, multiple sclerosis and inflammatory bowel disease. The most prominent target is the major matrix engagers, the integrins and their downstream signalling machinery. However, success has been painfully limited on this front, and of the 24 known human integrins, three are currently targeted therapeutically by monoclonal antibodies, peptides or small molecules. To date, targeting the platelet αIIbβ3 integrin is used to prevent thrombotic complications after percutaneous coronary interventions, and compounds targeting the lymphocyte α4β1 and α4β7 integrins are showing promising indications in multiple sclerosis and inflammatory bowel disease. Building on this, new antibodies and small molecules targeting β7 integrins (α4β7 and αEβ7 integrins) as well as their ligands are currently in clinical development for the treatment of inflammatory bowel diseases. Generally, integrin‐based therapeutics have shown clinically significant benefits in many patients, and as a result, this has led to the continued interest in further developing novel integrin inhibitors. Of note is that almost all integrin antagonists in use or in late‐stage clinical trials target either the ligand‐binding site or the ligand itself. As we begin to expand our repertoire of integrin‐based therapies, it will be essential to co‐develop companion biomarkers (discussed below) to ensure appropriate administration to maximize patient benefit. For a comprehensive review, including recent clinical trials, see Ley et al. (2016).
Downstream of integrin–ECM engagement is the molecular machinery, comprised of large complexes of multiple kinases that action change within the cells in response to exogenous ECM cues. As such, the use of small molecule kinase inhibitors that target these signalling pathways regulated by the ECM such as focal adhesion kinase (FAK; Lightfoot et al. 2004; Golubovskaya 2014; Jean et al. 2014; Lee et al. 2015) 89,90 and Rho‐associated protein kinase (ROCK; Kular et al. 2015; Rath et al. 2016; Guerra et al. 2017; Vennin et al. 2017b; Whatcott et al. 2017) have proven to be effective in targeting the accompanying desmoplasia in cancer. Whilst these approaches show promise, without the addition of adjuvant cytotoxic therapy or radiotherapy, they do little to physically debulk and regress tumours. For a detailed discussion on the clinical potential of targeting ECM stiffness through downstream cellular signalling to combat diseases, see Lampi and Reinhart‐King (2018). Finally, as mentioned above targeting intracellular signalling to prevent ECM secretion and deposition, such as treatment with the tyrosine kinase inhibitor, genistein, to inhibit versican synthesis, has also showed promise in preclinical models of malignant mesothelioma (Syrokou et al. 1999).
The call for better companion biomarkers to stratify patients for therapeutic intervention is clear. In our own studies in preclinical models of breast cancer, we have shown that during cancer progression and metastatic colonization of secondary sites, there are changes in expression of both unusual and unreported ECM proteins within the tumour and metastatic microenvironment (Mayorca‐Guiliani et al. 2017), identifying them as potentially new diagnostic and/or prognostic markers for ECM‐based therapeutics. The above‐mentioned HALO 202 phase II clinical trial only reported benefit of administration of pegvorhyaluronidase alfa (PEGPH20) in HA‐high pancreatic tumours showing little positive effect in HA‐low pancreatic tumours (Hingorani et al. 2017). In high‐grade serous ovarian cancer (HGSOC), Pearce et al. (2017) recently reported a matrix index that could be used to predict poor prognosis, extending earlier work by Naba et al. (2017b) on the proteomic characterization of ECM changes in triple‐negative breast cancer and adjacent mammary tissue, and omental metastasis from high‐grade serous ovarian cancer and normal omentum. Importantly, these studies collectively illustrate that despite high levels of genetic heterogeneity within tumours, the changes in the ECM and the underlying mechanisms behind these changes, as well as the pathways they subsequently activate, may be more consistent and as a result represent more robust therapeutic targets.
Finally, the significant ECM remodelling that accompanies cancer progression has also been shown to be critical to facilitating metastatic colonization (Cox et al. 2013, 2015) and as such may represent markers for patients with already‐disseminated disease. By leveraging this information, we can better stratify cancer patients, as well as identify and deploy new combination treatments that co‐target the ECM and ECM remodelling alongside standard‐of‐care cancer therapies (Miller et al. 2015; Vennin et al. 2017a) to improve patient outcome.
Concluding remarks
The composition of the ECM and the distribution of its components define tissue architecture and modulate cellular responses to a plethora of exogenous cues. The complexity of the ECM makes it difficult to identify the precise functional repertoire of ECM proteins by biochemical or cell culture experiments alone. Hence, mouse models and human biopsies are critical to analysing the biological significance of ECM components in a wide variety of in vivo processes.
For the most part, we have developed sensitive methods to generate a list of building materials and can look at a final snapshot of the matrix composition and architecture. However, we must still conjecture the assembly dynamics. It would be the same as looking at a completed building and the materials list and piecing together how it was constructed. We are missing the process, and at present, we lack the tools required to watch in real time the assembly process of the ECM taking place in vivo. However, new approaches and platforms are continually being developed and it should hopefully not be long before we can watch ECM assembly and dynamics in vivo in 3D in real time.
From a therapeutic perspective, further studies are necessary to more clearly delineate deleterious and beneficial aspects of the ECM in the biology of cancer, but will likely offer a reservoir of new therapeutic avenues. Furthermore, rather than just identifying the presence of tumours in routine screening approaches such as biopsy, the ability to predict, based on a tumour's ECM landscape and genetic status, whether it may or may not have metastasized, and to where, and more importantly whether it may be refractive or responsive to a particular therapy will be of great significance in the clinical setting. This will significantly contribute to refining and personalizing the treatment regimens of patients and bring us closer to evidence‐based, personalized medicine and improved patient care.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We apologize to authors whose work could not be discussed here because of space limitations. TRC is supported by a Susan G. Komen Career Catalyst award and the Cancer Institute of New South Wales (CINSW). JLC and ECF are supported by the National Health and Medical Research Council (NHMRC) of Australia. TRC was the recipient of the 2016 British Society for Matrix Biology (BSMB) John Scott Young Investigator Award.
References
- Acuna A., Drakopoulos M.A., Leng Y., Goergen C.J. & Calve S. (2018) Three‐dimensional visualization of extracellular matrix networks during murine development. Dev. Biol. 435, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J. & Cukierman E. (2016) Stromal dynamic reciprocity in cancer: intricacies of fibroblastic‐ECM interactions. Curr. Opin. Cell Biol. 42, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo A.G. & Iruela‐Arispe M.L. (2010) Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc. Res. 86, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszódi A., Legate K.R., Nakchbandi I. & Fässler R. (2006) What mouse mutants teach us about extracellular matrix function. Annu. Rev. Cell Dev. Biol. 22, 591–621. [DOI] [PubMed] [Google Scholar]
- Bailey A.J. (2001) Molecular mechanisms of ageing in connective tissues. Mech. Ageing Dev. 122, 735–755. [DOI] [PubMed] [Google Scholar]
- Baker A.‐M., Cox T.R., Bird D. et al (2011) The role of lysyl oxidase in SRC‐dependent proliferation and metastasis of colorectal cancer. J. Natl Cancer Inst. 103, 407–424. [DOI] [PubMed] [Google Scholar]
- Baker A.M., Bird D., Lang G., Cox T.R. & Erler J.T. (2013a) Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 32, 1863–1868. [DOI] [PubMed] [Google Scholar]
- Baker A.‐M., Bird D., Welti J.C. et al (2013b) Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Can. Res. 73, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S., Ouyang G., Bai X. et al (2004) Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 5, 329–339. [DOI] [PubMed] [Google Scholar]
- Barkan D., Green J.E. & Chambers A.F. (2010) Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer 46, 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker H.E., Cox T.R. & Erler J.T. (2012) The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 12, 540–552. [DOI] [PubMed] [Google Scholar]
- Bateman J.F., Boot‐Handford R.P. & Lamandé S.R. (2009) Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 10, 173–183. [DOI] [PubMed] [Google Scholar]
- Becker K., Jährling N., Saghafi S., Weiler R. & Dodt H.‐U. (2012) Chemical clearing and dehydration of GFP expressing mouse brains. PLoS ONE 7, e33916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A.B., Wainberg Z.A., Hecht J.R. et al (2017) A phase II randomized, double‐blind, placebo‐controlled study of simtuzumab or placebo in combination with gemcitabine for the first‐line treatment of pancreatic adenocarcinoma. Oncologist 22, 241–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell M.J. & Aggeler J. (1987) Dynamic reciprocity: how do extracellular matrix and hormones direct gene expression? Prog. Clin. Biol. Res. 249, 251–262. [PubMed] [Google Scholar]
- Bissell M.J. & Hines W.C. (2011) Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell M.J., Hall H.G. & Parry G. (1982) How does the extracellular matrix direct gene expression? J. Theor. Biol. 99, 31–68. [DOI] [PubMed] [Google Scholar]
- Bissell M.J., Radisky D.C., Rizki A., Weaver V.M. & Petersen O.W. (2002) The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareva A., Downey C.M., Ayres F. et al (2009) The lysyl oxidase inhibitor, beta‐aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS ONE 4, e5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P. & Sage E.H. (2002) Matricellular proteins: extracellular modulators of cell function. Curr. Opin. Cell Biol. 14, 608–616. [DOI] [PubMed] [Google Scholar]
- Boyle S.T. & Samuel M.S. (2016) Mechano‐reciprocity is maintained between physiological boundaries by tuning signal flux through the Rho‐associated protein kinase. Small GTPases 7, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brassart‐Pasco S., Sénéchal K., Thevenard J. et al (2012) Tetrastatin, the NC1 domain of the α4(IV) collagen chain: a novel potent anti‐tumor matrikine. PLoS ONE 7, e29587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brellier F., Tucker R.P. & Chiquet‐Ehrismann R. (2009) Tenascins and their implications in diseases and tissue mechanics. Scand. J. Med. Sci. Sports 19, 511–519. [DOI] [PubMed] [Google Scholar]
- Bunn H.F., Gabbay K.H. & Gallop P.M. (1978) The glycosylation of hemoglobin: relevance to diabetes mellitus. Science 200, 21–27. [DOI] [PubMed] [Google Scholar]
- Byron A., Humphries J.D. & Humphries M.J. (2013) Defining the extracellular matrix using proteomics. Int. J. Exp. Pathol. 94, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli R.M., Farmer T.B. & Gile J. (1997) Molecular imaging of biological samples: localization of peptides and proteins using MALDI‐TOF MS. Anal. Chem. 69, 4751–4760. [DOI] [PubMed] [Google Scholar]
- Cathcart J., Pulkoski‐Gross A. & Cao J. (2015) Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Lucas M.C., Leonte L.E. et al (2017) Pre‐clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 8, 26066–26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodoni C., Colombo M.P. & Sangaletti S. (2010) Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 29, 295–307. [DOI] [PubMed] [Google Scholar]
- Chlenski A. & Cohn S.L. (2010) Modulation of matrix remodeling by SPARC in neoplastic progression. Semin. Cell Dev. Biol. 21, 55–65. [DOI] [PubMed] [Google Scholar]
- Chong H.C., Tan C.K., Huang R.‐L. & Tan N.S. (2012) Matricellular proteins: a sticky affair with cancers. J. Oncol. 2012, 351089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska‐Wodnicka M. & Burridge K. (1996) Rho‐stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu I.M., Michalowski A.M., Hoenerhoff M. et al (2012) GATA3 inhibits lysyl oxidase‐mediated metastases of human basal triple‐negative breast cancer cells. Oncogene 31, 2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K. & Deisseroth K. (2013) CLARITY for mapping the nervous system. Nat. Methods 10, 508–513. [DOI] [PubMed] [Google Scholar]
- Colorado P.C., Torre A., Kamphaus G. et al (2000) Anti‐angiogenic cues from vascular basement membrane collagen. Can. Res. 60, 2520–2526. [PubMed] [Google Scholar]
- Contié S., Voorzanger‐Rousselot N., Litvin J., Clézardin P. & Garnero P. (2011) Increased expression and serum levels of the stromal cell‐secreted protein periostin in breast cancer bone metastases. Int. J. Cancer 128, 352–360. [DOI] [PubMed] [Google Scholar]
- Cornett D.S., Frappier S.L. & Caprioli R.M. (2008) MALDI‐FTICR imaging mass spectrometry of drugs and metabolites in tissue. Anal. Chem. 80, 5648–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa‐Silva B., Aiello N.M., Ocean A.J. et al (2015) Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat. Cell Biol. 17, 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens L.M., Fingleton B. & Matrisian L.M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392. [DOI] [PubMed] [Google Scholar]
- Cox T.R. & Erler J.T. (2011) Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model Mech 4, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T.R., Bird D., Baker A.‐M. et al (2013) LOX‐mediated collagen crosslinking is responsible for fibrosis‐enhanced metastasis. Can. Res. 73, 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T.R., Rumney R.M.H., Schoof E.M. et al (2015) The hypoxic cancer secretome induces pre‐metastatic bone lesions through lysyl oxidase. Nature 522, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cui X., Xu H., Zhou S. et al (2009) Evaluation of angiogenic activities of hyaluronan oligosaccharides of defined minimum size. Life Sci. 85, 573–577. [DOI] [PubMed] [Google Scholar]
- Danielson K.G., Baribault H., Holmes D.F., Graham H., Kadler K.E. & Iozzo R.V. (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 136, 729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis G.E., Bayless K.J., Davis M.J. & Meininger G.A. (2000) Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 156, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKeyser S.S., Kutz‐Naber K.K., Schmidt J.J., Barrett‐Wilt G.A. & Li L. (2007) Imaging mass spectrometry of neuropeptides in decapod crustacean neuronal tissues. J. Proteome Res. 6, 1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker L.J.M., van Kampen J.J.A., Reedijk M.L. et al (2009) A mass spectrometry based imaging method developed for the intracellular detection of HIV protease inhibitors. Rapid Commun. Mass Spectrom. 23, 1183–1188. [DOI] [PubMed] [Google Scholar]
- Didem T., Faruk T., Senem K. et al (2014) Clinical significance of serum tenascin‐c levels in epithelial ovarian cancer. Tumour Biol. 35, 6777–6782. [DOI] [PubMed] [Google Scholar]
- Dodt H.‐U., Leischner U., Schierloh A. et al (2007) Ultramicroscopy: three‐dimensional visualization of neuronal networks in the whole mouse brain. Nat. Methods 4, 331–336. [DOI] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M. et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Egeblad M., Nakasone E.S. & Werb Z. (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell 18, 884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Haibi C.P., Bell G.W., Zhang J. et al (2012) Critical role for lysyl oxidase in mesenchymal stem cell‐driven breast cancer malignancy. Proc. Natl Acad. Sci. USA 109, 17460–17465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Taylor W., Paulsson M., Sage H. & Hogan B. (1987) Calcium binding domains and calcium‐induced conformational transition of SPARC/BM‐40/osteonectin, an extracellular glycoprotein expressed in mineralized and nonmineralized tissues. Biochemistry 26, 6958–6965. [DOI] [PubMed] [Google Scholar]
- Erler J.T., Bennewith K.L., Nicolau M. et al (2006) Lysyl oxidase is essential for hypoxia‐induced metastasis. Nature 440, 1222–1226. [DOI] [PubMed] [Google Scholar]
- Erler J.T., Bennewith K.L., Cox T.R. et al (2009) Hypoxia‐induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A. & Bradke F. (2013) High‐resolution imaging of entire organs by 3‐dimensional imaging of solvent cleared organs (3DISCO). Exp. Neurol. 242, 57–64. [DOI] [PubMed] [Google Scholar]
- Faye C., Moreau C., Chautard E. et al (2009) Molecular interplay between endostatin, integrins, and heparan sulfate. J. Biol. Chem. 284, 22029–22040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer I., Westphal M., Rossbach B. et al (2017) Comparative characterization of decellularized renal scaffolds for tissue engineering. Biomed. Mater. 12, 045005. [DOI] [PubMed] [Google Scholar]
- Frank R.N. (1991) On the pathogenesis of diabetic retinopathy. A 1990 update. Ophthalmology 98, 586–593. [DOI] [PubMed] [Google Scholar]
- Fukushima N., Kikuchi Y., Nishiyama T., Kudo A. & Fukayama M. (2008) Periostin deposition in the stroma of invasive and intraductal neoplasms of the pancreas. Mod. Pathol. 21, 1044–1053. [DOI] [PubMed] [Google Scholar]
- Funk S.E. & Sage E.H. (1991) The Ca2(+)‐binding glycoprotein SPARC modulates cell cycle progression in bovine aortic endothelial cells. Proc. Natl Acad. Sci. USA 88, 2648–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstenfeld L.C., Riva A., Hodgens K., Eyre D.R. & Landis W.J. (1993) Post‐translational control of collagen fibrillogenesis in mineralizing cultures of chick osteoblasts. J. Bone Miner. Res. 8, 1031–1043. [DOI] [PubMed] [Google Scholar]
- Gessel M., Spraggins J.M., Voziyan P., Hudson B.G. & Caprioli R.M. (2015) Decellularization of intact tissue enables MALDI imaging mass spectrometry analysis of the extracellular matrix. J. Mass Spectrom. 50, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi K., Deng‐Pichon U., Warnet J.‐M. & Rat P. (2012) Hyaluronan fragments improve wound healing on in vitro cutaneous model through P2X7 purinoreceptor basal activation: role of molecular weight. PLoS ONE 7, e48351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn J.V. & Stitt A.W. (2009) The role of advanced glycation end products in retinal ageing and disease. Biochem. Biophys. Acta. 1790, 1109–1116. [DOI] [PubMed] [Google Scholar]
- Golubovskaya V.M. (2014) Targeting FAK in human cancer: from finding to first clinical trials. Front. Biosci. (Landmark Ed) 19, 687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra F.S., de Oliveira R.G., Fraga C.A.M., Mermelstein C.D.S. & Fernandes P.D. (2017) ROCK inhibition with Fasudil induces beta‐catenin nuclear translocation and inhibits cell migration of MDA‐MB 231 human breast cancer cells. Sci. Rep. 7, 13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeger A., Krause M., Wolf K. & Friedl P. (2014) Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochem. Biophys. Acta. 1840, 2386–2395. [DOI] [PubMed] [Google Scholar]
- Hama H., Kurokawa H., Kawano H. et al (2011) Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat. Neurosci. 14, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R.A. (2000) The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R.A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanamura N., Yoshida T., Matsumoto E., Kawarada Y. & Sakakura T. (1997) Expression of fibronectin and tenascin‐C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int. J. Cancer 73, 10–15. [DOI] [PubMed] [Google Scholar]
- Hecht J.R., Benson A.B., Vyushkov D., Yang Y., Bendell J. & Verma U. (2017) A phase II, randomized, double‐blind, placebo‐controlled study of simtuzumab in combination with FOLFIRI for the second‐line treatment of metastatic KRAS mutant colorectal adenocarcinoma. Oncologist 22, 243–e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvert S., Storm C. & Friedl P. (2018) Mechanoreciprocity in cell migration. Nat. Cell Biol. 20, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Zheng L., Bullock A.J. et al (2017) HALO 202: randomized phase II study of PEGPH20 plus nab‐paclitaxel/gemcitabine versus nab‐paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 36, 359–366. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S., Ishibashi S., Tomita T. et al (2013) Primary tumours modulate innate immune signalling to create pre‐metastatic vascular hyperpermeability foci. Nat. Commun. 4, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenester E. & Engel J. (2002) Domain structure and organisation in extracellular matrix proteins. Matrix Biol. 21, 115–128. [DOI] [PubMed] [Google Scholar]
- Huo C.W., Chew G., Hill P. et al (2015) High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 17, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R.O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. (2009) The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilmonen S., Jahkola T., Turunen J.P., Muhonen T. & Asko‐Seljavaara S. (2004) Tenascin‐C in primary malignant melanoma of the skin. Histopathology 45, 405–411. [DOI] [PubMed] [Google Scholar]
- Ishiba T., Nagahara M., Nakagawa T. et al (2014) Periostin suppression induces decorin secretion leading to reduced breast cancer cell motility and invasion. Sci. Rep. 4, 7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara J., Fukunaga K., Ishihara A. et al (2017) Matrix‐binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 9, pii: eaan0401. [DOI] [PubMed] [Google Scholar]
- Jackson S.N., Wang H.‐Y.J. & Woods A.S. (2007) In situ structural characterization of glycerophospholipids and sulfatides in brain tissue using MALDI‐MS/MS. J. Am. Soc. Mass Spectrom. 18, 17–26. [DOI] [PubMed] [Google Scholar]
- Jacob K., Webber M., Benayahu D. & Kleinman H.K. (1999) Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Can. Res. 59, 4453–4457. [PubMed] [Google Scholar]
- Jacobetz M.A., Chan D.S., Neesse A. et al (2013) Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen K.A., Atherton P. & Ballestrem C. (2017) Mechanotransduction at the cell‐matrix interface. Semin. Cell Dev. Biol. 71, 75–83. [DOI] [PubMed] [Google Scholar]
- Jean C., Chen X.L., Nam J.‐O. et al (2014) Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 204, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan R.N., Riba R.D., Zacharoulis S. et al (2005) VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 438, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass L., Erler J.T., Dembo M. & Weaver V.M. (2007) Mammary epithelial cell: influence of extracellular matrix composition and organization during development and tumorigenesis. Int. J. Biochem. Cell Biol. 39, 1987–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M.‐T., Fujimoto S. & Imai T. (2013) SeeDB: a simple and morphology‐preserving optical clearing agent for neuronal circuit reconstruction. Nat. Neurosci. 16, 1154–1161. [DOI] [PubMed] [Google Scholar]
- Keire P.A., Bressler S.L., Lemire J.M. et al (2014) A role for versican in the development of leiomyosarcoma. J. Biol. Chem. 289, 34089–34103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny H.A., Chiang C.‐Y., White E.A. et al (2014) Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 124, 4614–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S.I., Takahashi K., Nishida J. et al (2017) Whole‐body profiling of cancer metastasis with single‐cell resolution. Cell Rep. 20, 236–250. [DOI] [PubMed] [Google Scholar]
- Kular J., Scheer K.G., Pyne N.T. et al (2015) A negative regulatory mechanism involving 14‐3‐3ζ limits signaling downstream of ROCK to regulate tissue stiffness in epidermal homeostasis. Dev. Cell 35, 759–774. [DOI] [PubMed] [Google Scholar]
- Kuljanin M., Brown C.F.C., Raleigh M.J., Lajoie G.A. & Flynn L.E. (2017) Collagenase treatment enhances proteomic coverage of low‐abundance proteins in decellularized matrix bioscaffolds. Biomaterials 144, 130–143. [DOI] [PubMed] [Google Scholar]
- Kuwajima T., Sitko A.A., Bhansali P., Jurgens C., Guido W. & Mason C. (2013) ClearT: a detergent‐ and solvent‐free clearing method for neuronal and non‐neuronal tissue. Development 140, 1364–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczko R., Szauter K.M., Jansen M.K. et al (2007) Active lysyl oxidase (LOX) correlates with focal adhesion kinase (FAK)/paxillin activation and migration in invasive astrocytes. Neuropathol. Appl. Neurobiol. 33, 631–643. [DOI] [PubMed] [Google Scholar]
- Laklai H., Miroshnikova Y.A., Pickup M.W. et al (2016) Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 22, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampi M.C. & Reinhart‐King C.A. (2018) Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci. Transl. Med. 10, eaao0475. [DOI] [PubMed] [Google Scholar]
- Lane T.F. & Sage E.H. (1994) The biology of SPARC, a protein that modulates cell‐matrix interactions. FASEB J. 8, 163–173. [PubMed] [Google Scholar]
- Lee B.Y., Timpson P., Horvath L.G. & Daly R.J. (2015) FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 146, 132–149. [DOI] [PubMed] [Google Scholar]
- Leeming D.J., Bay‐Jensen A.C., Vassiliadis E., Larsen M.R., Henriksen K. & Karsdal M.A. (2011) Post‐translational modifications of the extracellular matrix are key events in cancer progression: opportunities for biochemical marker development. Biomarkers 16, 193–205. [DOI] [PubMed] [Google Scholar]
- Levental K.R., Yu H., Kass L. et al (2009) Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K., Rivera‐Nieves J., Sandborn W.J. & Shattil S. (2016) Integrin‐based therapeutics: biological basis, clinical use and new drugs. Nat. Rev. Drug Discovery 15, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Kang Y., Roife D. et al (2017) Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene 36, 5432–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H., Li X., Wang B. et al (2016) A collagen‐binding EGFR antibody fragment targeting tumors with a collagen‐rich extracellular matrix. Sci. Rep. 6, 18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot H.M., Lark A., Livasy C.A. et al (2004) Upregulation of focal adhesion kinase (FAK) expression in ductal carcinoma in situ (DCIS) is an early event in breast tumorigenesis. Breast Cancer Res. Treat. 88, 109–116. [DOI] [PubMed] [Google Scholar]
- Maeshima Y., Colorado P.C. & Kalluri R. (2000) Two RGD‐independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti‐tumor properties. J. Biol. Chem. 275, 23745–23750. [DOI] [PubMed] [Google Scholar]
- Magnon C., Galaup A., Mullan B. et al (2005) Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Can. Res. 65, 4353–4361. [DOI] [PubMed] [Google Scholar]
- Mäki J.M. (2009) Lysyl oxidases in mammalian development and certain pathological conditions. Histol. Histopathol. 24, 651–660. [DOI] [PubMed] [Google Scholar]
- Maller O., Hansen K.C., Lyons T.R. et al (2013) Collagen architecture in pregnancy‐induced protection from breast cancer. J. Cell Sci. 126(Pt 18), 4108–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino M.M., Briquez P.S., Güç E. et al (2014) Growth factors engineered for super‐affinity to the extracellular matrix enhance tissue healing. Science 343, 885–888. [DOI] [PubMed] [Google Scholar]
- Mayorca‐Guiliani A.E., Madsen C.D., Cox T.R., Horton E.R., Venning F.A. & Erler J.T. (2017) ISDoT: in situ decellularization of tissues for high‐resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 23, 890–898. [DOI] [PubMed] [Google Scholar]
- McConnell J.C., O'Connell O.V., Brennan K. et al (2016) Increased peri‐ductal collagen micro‐organization may contribute to raised mammographic density. Breast Cancer Res. 18, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurius K.O. & Morla A.O. (2001) Cell adhesion and signaling on the fibronectin 1st type III repeat; requisite roles for cell surface proteoglycans and integrins. BMC Cell Biol. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.W., Morton J.P., Pinese M. et al (2015) Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 7, 1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongiat M., Sweeney S.M., San Antonio J.D., Fu J. & Iozzo R.V. (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 278, 4238–4249. [DOI] [PubMed] [Google Scholar]
- Mori M., Muramatsu Y., Yamada K., Shrestha P. & Takai Y. (1996) Intracellular localization of tenascin in squamous cell carcinoma of oral cavity: an immunohistochemical study. Anticancer Res. 16(5B), 3075–3079. [PubMed] [Google Scholar]
- Morrissey M.A., Jayadev R., Miley G.R. et al (2016) SPARC promotes cell invasion in vivo by decreasing type IV collagen levels in the basement membrane. PLoS Genet. 12, e1005905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy A.J., Wiersma M.P. & Choi B. (2013) Optical histology: a method to visualize microvasculature in thick tissue sections of mouse brain. PLoS ONE 8, e53753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy‐Ullrich J.E., Lane T.F., Pallero M.A. & Sage E.H. (1995) SPARC mediates focal adhesion disassembly in endothelial cells through a follistatin‐like region and the Ca(2 + )‐binding EF‐hand. J. Cell. Biochem. 57, 341–350. [DOI] [PubMed] [Google Scholar]
- Myllyharju J. & Kivirikko K.I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43. [DOI] [PubMed] [Google Scholar]
- Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A. & Hynes R.O. (2012) The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 11, M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A., Clauser K.R. & Hynes R.O. (2015) Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. J. Vis. Exp. 101, e53057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A. & Hynes R.O. (2016) The extracellular matrix: tools and insights for the “omics” era. Matrix Biol. 49, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A., Clauser K.R., Mani D.R., Carr S.A. & Hynes R.O. (2017a) Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci. Rep. 7, 40495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A., Pearce O.M.T., Del Rosario A. et al (2017b) Characterization of the extracellular matrix of normal and diseased tissues using proteomics. J. Proteome Res. 16, 3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrollahi S., Walter C., Loza A.J., Schimizzi G.V., Longmore G.D. & Pathak A. (2017) Past matrix stiffness primes epithelial cells and regulates their future collective migration through a mechanical memory. Biomaterials 146, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C.M. & Bissell M.J. (2006) Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 22, 287–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A., Geiger B. & Safran S.A. (2004) Cell mechanosensitivity controls the anisotropy of focal adhesions. Proc. Natl Acad. Sci. USA 101, 12520–12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares O., Mayers J.R., Gouirand V. et al (2017) Collagen‐derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 8, 16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T. & Massagué J. (2012) Extracellular matrix players in metastatic niches. EMBO J. 31, 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T., Acharyya S., Zhang X.H.‐F. et al (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M.J., Zahir N., Johnson K.R. et al (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Pauli C., Stieber P., Schmitt U.M., Andratschke M., Hoffmann K. & Wollenberg B. (2002) The significance of Tenascin‐C serum level as tumor marker in squamous cell carcinoma of the head and neck. Anticancer Res. 22, 3093–3097. [PubMed] [Google Scholar]
- Payne S.L., Fogelgren B., Hess A.R. et al (2005) Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide‐mediated mechanism. Can. Res. 65, 11429–11436. [DOI] [PubMed] [Google Scholar]
- Pearce O.M.T., Delaine‐Smith R., Maniati E. et al (2017) Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov. 8, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedretti M., Soltermann A., Arni S., Weder W., Neri D. & Hillinger S. (2009) Comparative immunohistochemistry of L19 and F16 in non‐small cell lung cancer and mesothelioma: two human antibodies investigated in clinical trials in patients with cancer. Lung Cancer 64, 28–33. [DOI] [PubMed] [Google Scholar]
- Peinado H., Alečković M., Lavotshkin S. et al (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nat. Med. 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M.W., Laklai H., Acerbi I. et al (2013) Stromally derived lysyl oxidase promotes metastasis of transforming growth factor‐β‐deficient mouse mammary carcinomas. Can. Res. 73, 5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M.W., Mouw J.K. & Weaver V.M. (2014) The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen R., Leinonen T., Böhm J. et al (2005) Versican in nonsmall cell lung cancer: relation to hyaluronan, clinicopathologic factors, and prognosis. Hum. Pathol. 36, 44–50. [DOI] [PubMed] [Google Scholar]
- Podhajcer O.L., Benedetti L., Girotti M.R., Prada F., Salvatierra E. & Llera A.S. (2008) The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 27, 523–537. [DOI] [PubMed] [Google Scholar]
- Provenzano P.P., Eliceiri K.W., Campbell J.M., Inman D.R., White J.G. & Keely P.J. (2006) Collagen reorganization at the tumor‐stromal interface facilitates local invasion. BMC Med. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Cuevas C., Chang A.E., Goel V.K., Von Hoff D.D. & Hingorani S.R. (2012) Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglisi F., Puppin C., Pegolo E. et al (2008) Expression of periostin in human breast cancer. J. Clin. Pathol. 61, 494–498. [DOI] [PubMed] [Google Scholar]
- Rachman‐Tzemah C., Zaffryar‐Eilot S., Grossman M. et al (2017) Blocking surgically induced lysyl oxidase activity reduces the risk of lung metastases. Cell Rep. 19, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspanti M., Reguzzoni M., Protasoni M. & Basso P. (2018) Not only tendons: the other architecture of collagen fibrils. Int. J. Biol. Macromol. 107(Pt B), 1668–1674. [DOI] [PubMed] [Google Scholar]
- Rath N., Morton J.P., Julian L. et al (2016) ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 9, 198–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reffay M., Parrini M.C., Cochet‐Escartin O. et al (2014) Interplay of RhoA and mechanical forces in collective cell migration driven by leader cells. Nat. Cell Biol. 16, 217–223. [DOI] [PubMed] [Google Scholar]
- Rehn M., Veikkola T., Kukk‐Valdre E. et al (2001) Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl Acad. Sci. USA 98, 1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud C., Ferreras L., Di Mauro P. et al (2017) Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Can. Res. 77, 268–278. [DOI] [PubMed] [Google Scholar]
- Reyzer M.L., Hsieh Y., Ng K., Korfmacher W.A. & Caprioli R.M. (2003) Direct analysis of drug candidates in tissue by matrix‐assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 38, 1081–1092. [DOI] [PubMed] [Google Scholar]
- Ricard‐Blum S. & Salza R. (2014) Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp. Dermatol. 23, 457–463. [DOI] [PubMed] [Google Scholar]
- Ricciardelli C., Brooks J.H., Suwiwat S. et al (2002) Regulation of stromal versican expression by breast cancer cells and importance to relapse‐free survival in patients with node‐negative primary breast cancer. Clin. Cancer Res. 8, 1054–1060. [PubMed] [Google Scholar]
- Ricciardelli C., Russell D.L., Ween M.P. et al (2007) Formation of hyaluronan‐ and versican‐rich pericellular matrix by prostate cancer cells promotes cell motility. J. Biol. Chem. 282, 10814–10825. [DOI] [PubMed] [Google Scholar]
- Ricciardelli C., Sakko A.J., Ween M.P., Russell D.L. & Horsfall D.J. (2009) The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev. 28, 233–245. [DOI] [PubMed] [Google Scholar]
- Riching K.M., Cox B.L., Salick M.R. et al (2014) 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J. 107, 2546–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Teja M., Gronau J.H., Breit C. et al (2015) AGE‐modified basement membrane cooperates with Endo180 to promote epithelial cell invasiveness and decrease prostate cancer survival. J. Pathol. 235, 581–592. [DOI] [PubMed] [Google Scholar]
- Roskelley C.D., Srebrow A. & Bissell M.J. (1995) A hierarchy of ECM‐mediated signalling regulates tissue‐specific gene expression. Curr. Opin. Cell Biol. 7, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage H., Johnson C. & Bornstein P. (1984) Characterization of a novel serum albumin‐binding glycoprotein secreted by endothelial cells in culture. J. Biol. Chem. 259, 3993–4007. [PubMed] [Google Scholar]
- Sage H., Vernon R.B., Funk S.E., Everitt E.A. & Angello J. (1989) SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2‐dependent binding to the extracellular matrix. J. Cell Biol. 109, 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M., Kato H., Sano A. et al (2009) Expression of lysyl oxidase is correlated with lymph node metastasis and poor prognosis in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 16, 2494–2501. [DOI] [PubMed] [Google Scholar]
- Sasaki N., Fukatsu R., Tsuzuki K. et al (1998) Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am. J. Pathol. 153, 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H., Dai M., Auclair D. et al (2001) Serum level of the periostin, a homologue of an insect cell adhesion molecule, as a prognostic marker in nonsmall cell lung carcinomas. Cancer 92, 843–848. [DOI] [PubMed] [Google Scholar]
- Sato N., Fukushima N., Maehara N. et al (2003) SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor‐stromal interactions. Oncogene 22, 5021–5030. [DOI] [PubMed] [Google Scholar]
- Saupe F., Schwenzer A., Jia Y. et al (2013) Tenascin‐C downregulates wnt inhibitor dickkopf‐1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep. 5, 482–492. [DOI] [PubMed] [Google Scholar]
- Schaefer L. & Schaefer R.M. (2010) Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 339, 237–246. [DOI] [PubMed] [Google Scholar]
- Schönherr E., Kinsella M.G. & Wight T.N. (1997) Genistein selectively inhibits platelet‐derived growth factor‐stimulated versican biosynthesis in monkey arterial smooth muscle cells. Arch. Biochem. Biophys. 339, 353–361. [DOI] [PubMed] [Google Scholar]
- Scott J.E., Dyne K.M., Thomlinson A.M. et al (1998) Human cells unable to express decoron produced disorganized extracellular matrix lacking “shape modules” (interfibrillar proteoglycan bridges). Exp. Cell Res. 243, 59–66. [DOI] [PubMed] [Google Scholar]
- Shaheed S., Rustogi N., Scally A. et al (2013) Identification of stage‐specific breast markers using quantitative proteomics. J. Proteome Res. 12, 5696–5708. [DOI] [PubMed] [Google Scholar]
- Sharaf H., Matou‐Nasri S., Wang Q. et al (2015) Advanced glycation endproducts increase proliferation, migration and invasion of the breast cancer cell line MDA‐MB‐231. Biochem. Biophys. Acta. 1852, 429–441. [DOI] [PubMed] [Google Scholar]
- Shawky M.S., Ricciardelli C., Lord M. et al (2015) Proteoglycans: potential agents in mammographic density and the associated breast cancer risk. J. Mammary Gland Biol. Neoplasia 20, 121–131. [DOI] [PubMed] [Google Scholar]
- Sherratt M.J. (2009) Tissue elasticity and the ageing elastic fibre. Age 31, 305–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherratt M.J., McConnell J.C. & Streuli C.H. (2016) Raised mammographic density: causative mechanisms and biological consequences. Breast Cancer Res. 18, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironen R.K., Tammi M., Tammi R., Auvinen P.K., Anttila M. & Kosma V.M. (2011) Hyaluronan in human malignancies. Exp. Cell Res. 317, 383–391. [DOI] [PubMed] [Google Scholar]
- Sivan S.‐S., Wachtel E., Tsitron E. et al (2008) Collagen turnover in normal and degenerate human intervertebral discs as determined by the racemization of aspartic acid. J. Biol. Chem. 283, 8796–8801. [DOI] [PubMed] [Google Scholar]
- Sudhakar A., Nyberg P., Keshamouni V.G. et al (2005) Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J. Clin. Investig. 115, 2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Susaki E.A., Tainaka K., Perrin D. et al (2014) Whole‐brain imaging with single‐cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739. [DOI] [PubMed] [Google Scholar]
- Sutton C.W., Rustogi N., Gurkan C. et al (2010) Quantitative proteomic profiling of matched normal and tumor breast tissues. J. Proteome Res. 9, 3891–3902. [DOI] [PubMed] [Google Scholar]
- Syrokou A., Tzanakakis G.N., Hjerpe A. & Karamanos N.K. (1999) Proteoglycans in human malignant mesothelioma. Stimulation of their synthesis induced by epidermal, insulin and platelet‐derived growth factors involves receptors with tyrosine kinase activity. Biochimie 81, 733–744. [DOI] [PubMed] [Google Scholar]
- Tainaka K., Kubota S.I., Suyama T.Q. et al (2014) Whole‐body imaging with single‐cell resolution by tissue decolorization. Cell 159, 911–924. [DOI] [PubMed] [Google Scholar]
- Takanami I., Abiko T. & Koizumi S. (2008) Expression of periostin in patients with non‐small cell lung cancer: correlation with angiogenesis and lymphangiogenesis. Int. J. Biol. Markers 23, 182–186. [DOI] [PubMed] [Google Scholar]
- Taylor M.A., Amin J.D., Kirschmann D.A. & Schiemann W.P. (2011) Lysyl oxidase contributes to mechanotransduction‐mediated regulation of transforming growth factor‐β signaling in breast cancer cells. Neoplasia 13, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termine J.D., Belcourt A.B., Conn K.M. & Kleinman H.K. (1981) Mineral and collagen‐binding proteins of fetal calf bone. J. Biol. Chem. 256, 10403–10408. [PubMed] [Google Scholar]
- Thompson C.B., Shepard H.M., O'Connor P.M. et al (2010) Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 9, 3052–3064. [DOI] [PubMed] [Google Scholar]
- Tilman G., Mattiussi M., Brasseur F., van Baren N. & Decottignies A. (2007) Human periostin gene expression in normal tissues, tumors and melanoma: evidences for periostin production by both stromal and melanoma cells. Mol. Cancer. 6, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer R., Ye L., Hsueh B. & Deisseroth K. (2014) Advanced CLARITY for rapid and high‐resolution imaging of intact tissues. Nat. Protoc. 9, 1682–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K.T., Lamb P. & Deng J.‐S. (2005) Matrikines and matricryptins: implications for cutaneous cancers and skin repair. J. Dermatol. Sci. 40, 11–20. [DOI] [PubMed] [Google Scholar]
- Tseng S.‐J., Lee Y.‐H., Chen Z.‐H., Lin H.‐H., Lin C.‐Y. & Tang S.‐C. (2009) Integration of optical clearing and optical sectioning microscopy for three‐dimensional imaging of natural biomaterial scaffolds in thin sections. J. Biomed. Optics 14, 044004. [DOI] [PubMed] [Google Scholar]
- Vennin C., Chin V.T., Warren S.C. et al (2017a) Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 9, pii: eaai8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennin C., Rath N., Pajic M., Olson M.F. & Timpson P. (2017b) Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2017 Oct 3: 1–8 doi: 10.1080/21541248.2017.1345712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijl N., DeGroot J., Thorpe S.R. et al (2000) Effect of collagen turnover on the accumulation of advanced glycation end products. J. Biol. Chem. 275, 39027–39031. [DOI] [PubMed] [Google Scholar]
- Vitek M.P., Bhattacharya K., Glendening J.M. et al (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl Acad. Sci. USA 91, 4766–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ween M.P., Hummitzsch K., Rodgers R.J., Oehler M.K. & Ricciardelli C. (2011) Versican induces a pro‐metastatic ovarian cancer cell behavior which can be inhibited by small hyaluronan oligosaccharides. Clin. Exp. Metas. 28, 113–125. [DOI] [PubMed] [Google Scholar]
- West D.C., Hampson I.N., Arnold F. & Kumar S. (1985) Angiogenesis induced by degradation products of hyaluronic acid. Science 228, 1324–1326. [DOI] [PubMed] [Google Scholar]
- Whatcott C.J., Ng S., Barrett M.T., Hostetter G., Hoff D.D.Von & Han H. (2017) Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 12, e0183871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J., Yang N., Wood A., Zindy E., Meng Q.‐J. & Streuli C.H. (2018) Epithelial and stromal circadian clocks are inversely regulated by their mechano‐matrix environment. J. Cell Sci. 131, pii: jcs208223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A.S. & Jackson S.N. (2006) Brain tissue lipidomics: direct probing using matrix‐assisted laser desorption/ionization mass spectrometry. AAPS J 8, E391–E395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Williams J., Pekovic‐Vaughan V. et al (2017) Cellular mechano‐environment regulates the mammary circadian clock. Nat. Commun. 8, 14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T.‐L., Leung C.S., Wong K.‐K. et al (2013) TGF‐β modulates ovarian cancer invasion by upregulating CAF‐derived versican in the tumor microenvironment. Can. Res. 73, 5016–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Matsumoto E., Hanamura N. et al (1997) Co‐expression of tenascin and fibronectin in epithelial and stromal cells of benign lesions and ductal carcinomas in the human breast. J. Pathol. 182, 421–428. [DOI] [PubMed] [Google Scholar]
- Yuzhalin A.E., Urbonas T., Silva M.A., Muschel R.J. & Gordon‐Weeks A.N. (2018) A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 118, 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanivan S., Gnad F., Wickström S.A. et al (2008) Solid tumor proteome and phosphoproteome analysis by high resolution mass spectrometry. J. Proteome Res. 7, 5314–5326. [DOI] [PubMed] [Google Scholar]
- Zhang K., Grither W.R., Van Hove S. et al (2016) Mechanical signals regulate and activate SNAIL1 protein to control the fibrogenic response of cancer‐associated fibroblasts. J. Cell Sci. 129, 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]