

Abstract
Background and Purpose
Agonists for the liver X receptor (LXR) are considered promising therapeutic moieties in cholesterol‐driven diseases by promoting cellular cholesterol efflux pathways. However, current clinical application of these agents is hampered by concomitant LXR‐induced activation of a lipogenic transcriptional network, leading to hepatic steatosis. Recent studies have suggested that protein arginine methyltransferase 3 (PRMT3) may act as a selective co‐activator of LXR activity. Here, we verified the hypothesis that PRMT3 inhibition selectively disrupts the ability of LXR to stimulate lipogenesis while maintaining its capacity to modulate macrophage cholesterol homeostasis.
Experimental Approach
A combination of the LXR agonist T0901317 and palm oil was administered to C57BL/6 mice to maximally stimulate LXR and PRMT3 activity. PRMT3 activity was inhibited using the allosteric inhibitor SGC707.
Key Results
Treatment with SGC707 did not negatively influence the T0901317/palm oil‐induced up‐regulation of the cholesterol efflux ATP‐binding cassette transporter genes, ABCA1 and ABCG1, in peritoneal cells. In contrast, SGC707 treatment was associated with a significant decrease in the hepatic expression of the lipogenic gene fatty acid synthase (−64%). A similar trend was observed for stearoyl‐coenzyme A desaturase and acetyl CoA carboxylase expression (−43%; −56%). This obstruction of lipogenic gene transcription coincided with a significant 2.3‐fold decrease in liver triglyceride content as compared with the T0901317 and palm oil‐treated control group.
Conclusion and Implications
We showed that inhibition of PRMT3 activity by SGC707 treatment selectively impairs LXR‐driven transcription of hepatic lipogenic genes, while the positive effect of LXR stimulation on macrophage cholesterol efflux pathways is maintained.
Abbreviations
- ABCA1
ATP‐binding cassette transporter A1
- ABCG1
ATP‐binding cassette transporter G1
- ACC
acetyl CoA carboxylase
- FAS
fatty acid synthase
- LXR
Liver X receptor
- PRMT3
protein arginine methyltransferase 3
- SCD‐1
stearoyl‐coenzyme A desaturase
- SREBP1c
sterol regulatory element‐binding protein 1c
- TG
triglyceride
Introduction
The http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=602 (LXR) is a nuclear oxysterol receptor, which functions as a transcriptional regulator of genes involved in lipid metabolism (Repa et al., 2000). Active LXR regulates cholesterol absorption, transport and catabolism (Venkateswaran et al., 2000; Yu et al., 2003). Because of its cholesterol efflux and reverse cholesterol transport stimulating properties via up‐regulation of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=756 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=155#791, LXR agonists are widely investigated as potential therapeutic agents for the treatment of cholesterol‐driven diseases such as atherosclerosis (Joseph et al., 2002b; Naik et al., 2006; Viennois et al., 2012; Komati et al., 2017). However, the LXR also regulates de novo hepatic lipogenesis via activation of the transcription factor sterol regulatory element‐binding protein 1c (SREBP1c) and direct interaction with hepatic lipogenic genes (Joseph et al., 2002a; Talukdar and Hillgartner, 2006; Cha and Repa, 2007). As a result, LXR activation by the use of LXR agonists leads to unwanted hepatic steatosis (Schultz et al., 2000; Grefhorst et al., 2002). This steatotic effect of LXR agonism has hampered the development of LXR agonists for clinical use in humans. Hence, preventing the induction of hepatic lipogenesis by LXR agonists, while maintaining its cholesterol efflux‐stimulating properties, is essential for their future therapeutic development.
LXR‐dependent transcription of target genes occurs via the recruitment of co‐repressors and co‐activators (Perissi and Rosenfeld, 2005; Komati et al., 2017). The recruitment of these cofactors is cell type‐ and gene‐dependent. Modifying cofactor availability or functionality could therefore provide a tool to selectively control LXR‐mediated transcription. In the absence of an LXR agonist, LXR‐dependent transcription is inhibited by the recruitment of co‐repressors (Hu et al., 2003). LXR activation by ligands results in the dissociation of the co‐repressor and moderate activation of target gene transcription. Upon subsequent recruitment of co‐activators, the transcription of target genes is highly increased (Herzog et al., 2007). A few of these selective LXR cofactors have been identified and are being investigated as therapeutic agents (Viennois et al., 2012). TRAP80 has been identified as a co‐activator of LXR, stimulating the transcription of lipogenesis genes in the liver but not of genes controlling cholesterol metabolism (Kim et al., 2015b). Based on this, 20(S)‐protopanaxatriol was shown to hamper the recruitment of TRAP80 to the target gene SREBP1c, thereby reducing the effect of this lipogenic co‐activator upon LXR stimulation (Oh et al., 2015). Recently, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1254 (PRMT3) was identified as a novel co‐activator for LXR‐driven lipogenic gene expression in vitro (Kim et al., 2015a). In the current study, the in vivo relevance of PRMT3 as a specific co‐activator of LXR‐mediated hepatic lipogenesis is investigated, and the overall effect of PRMT3 inhibition on the therapeutic potential of LXR agonists is evaluated.
Methods
Experimental mice
Animal experiments were performed at the Gorlaeus Laboratories of the Leiden Academic Centre for Drug Research in Leiden, The Netherlands. Male C57BL/6 mice, 13–19 weeks old, were bred in‐house at the Gorlaeus Laboratories. A total of 27 mice (6 DMSO; 7 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2755 + palm oil; 9 T0901317 + palm oil + http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8552 10 mg·kg−1 and 5 T0901317 + palm oil + SGC707 30 mg·kg−1) were used in this experiment. Mice were randomized at the start of the experiment based on age and body weight. Group size was determined based on the paper by Grefhorst et al. (2002) showing that n = 4 per group was the minimum number of mice to show effects on gene expression (our primary readout). Since the SGC707 compound has not been used in this context before, we slightly increased the group size while maintaining minimal amounts of mice needed. Due to the expected smaller effect size using a lower dose of SGC707 compound, the number of mice was increased to secure sufficient power. Mice were housed in a temperature and light cycle (12 h light/12 h dark) controlled environment. Food and water were supplied ad libitum. All animal experiments were performed in accordance with the principles of laboratory animal care and regulations of Dutch law on animal welfare and the BJP guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All experimental protocols were approved by the Animal Ethics Committee of Leiden University.
Treatment procedure
C57BL/6 mice received normal chow diet and were treated with a combination of T0901317 and palm oil to induce hepatic steatosis or an equal amount of DMSO as a negative control. Body weight was monitored daily. For four continuous days, T0901317 (10 mg·kg−1 in 10 μL DMSO) was injected i.p., and 200 μL palm oil (Sigma‐Aldrich Corp., St. Louis, MO, USA) was administered with an orogastric tube in the hepatic steatosis experimental groups. Mice were injected i.p. with SGC707 (10 or 30 mg·kg−1 in 10 μL DMSO) three times in this 4 day time span (day 1 9.00 am, day 2 4.00 pm and day 4 9.00 am). Food was removed at 8.00 am on day 5. After a 4 h fasting period, mice were anaesthetised with an s.c. injection using a mixture of ketamine (100 mg·kg−1), xylazine (12.5 mg·kg−1) and atropine (125 μg·kg−1) and monitored by checking the withdrawal reflex. Under deep anaesthesia, mice were killed by exsanguination. Blood was collected by orbital bleeding in EDTA‐coated tubes (Greiner Bio One, Kremsmünster, Austria). Plasma was collected after 10 min centrifugation at 2415 × g. Peritoneal leukocytes were obtained by lavaging the peritoneal cavity of the mice with 10 mL of ice‐cold PBS. The percentage of macrophages in the total leukocyte population was analysed using an automated Sysmex XT‐2000iV Veterinary Hematology analyzer (Sysmex Corporation, Etten‐Leur, The Netherlands). Subsequently, in preparation for harvesting the vital organs and to confirm the mice were dead, the diaphragm was severed, and the organs were perfused with PBS. Organs were harvested, and parts of the organs were fixed for 24 h in formalin and stored in 0.1% NaAzide/PBS until histological analysis. The remaining parts were snap‐frozen in liquid nitrogen and stored at −20°C until further analysis.
Immunohistochemistry analysis
Formalin‐fixed liver specimens were embedded in Tissue‐Tek® O.C.T. Compound (Sakura Finetek Europe, Alphen an den Rijn, NL), and cryosections (8 μm) were prepared on a Leica CM3050‐S cryostat (Leica Ltd., Cambridge, UK). Liver cryosections were stained for neutral lipids with Oil red O. Images were obtained with a Leica image analysis system, which consisted of a Leica DMRE microscope coupled to a video camera and Leica Qwin Imaging software (Leica Ltd., Cambridge, UK). For each liver, three images were obtained from three different liver sections per mouse. Images were blinded, and each image was assigned a score between 0 and 3 by an unbiased observer for the amount of red staining in the liver. Representative photomicrographs were taken for each group using the same system with 40× magnification.
Liver lipid extraction and quantification
Triglycerides (TGs) were extracted from liver tissue using Nonidet™ P 40 Substitute (Sigma‐Aldrich Corp.). In brief, approximately 50 mg of liver tissue was homogenized with 500 μL of Nonidet™ P 40 Substitute, heated until 90°C and chilled on ice. The heating and cooling procedure was repeated once more to solubilize all TGs. All insoluble material was removed by centrifuging at 13 148 × g. The concentration of TGs was determined by the use of an enzymatic colorimetric assay. Cholesterol was extracted from liver tissue ing the Folch extraction method (Folch et al., 1953). The concentration of free cholesterol and cholesteryl esters was determined by the use of enzymatic colorimetric assays as described by Out et al. (2006). The concentration of TGs, free cholesterol and cholesteryl esters was corrected for the total protein concentration, determined using a Pierce™ BCA Protein Assay Kit (ThermoFisher Diagnostics, Waltham, MA, USA).
Gene expression by quantitative real‐time PCR
Total RNA was extracted from the liver and peritoneal leukocytes using the guanidinium thiocyanate/chloroform/phenol extraction method (Chomczynski and Sacchi, 1987). RNA concentrations were determined using a Nanodrop Spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) after which cDNA was synthesized from 1 μg of RNA using RevertAid™ reverse transcriptase (ThermoFisher Diagnostics, Waltham, MA, USA). mRNA levels were measured using real‐time SYBR Green technology (Eurogentec, Seraing, Belgium) with PCR primers designed with Primer Express 1.5 Software (Applied Biosystems, Carlsbad, CA, USA). Primer sequences are available on request. β‐Actin, hypoxanthine‐guanine phosphoribosyltransferase, glyceraldehyde‐3‐phosphate dehydrogenase, ribosomal protein L27, ribosomal protein 36B4, hydroxymethylbilane synthase and peptidylprolyl isomerase A were used as standard housekeeping genes. No significant difference in the average expression of the six housekeeping genes was measured between treatment groups.
Plasma lipid analysis
The concentration of TGs, free cholesterol and cholesteryl esters was determined in plasma, obtained after orbital bleeding, by the use of enzymatic colorimetric assays as described by Out et al. (2006) (Roche Diagnostics, Mannheim, Germany).
Statistical analysis
All values are expressed as means ± SEM. Statistical analyses were performed using GraphPad Prism Software (GraphPad Software, La Jolla, California, USA) on the raw data. A Gibbs's outlier test and a Levene's test or Bartlett's test to check for equal variances between groups were performed on the raw data. Significance was calculated using Student's two‐tailed t‐test or ANOVA with multiple comparisons. Welch's and Bonferroni's post hoc tests were conducted only if F was significant, and there was no variance inhomogeneity. Probability values of <0.05 were considered significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Results
PRMT3 inhibition using recommended dosage of SGC707 impairs the metabolic state of mice
In order to investigate the effect of PRMT3 inhibition by the allosteric inhibitor SGC707 on LXR‐mediated lipogenesis, a T0901317‐induced hepatic steatosis C57Bl/6 mouse model was used as previously described by Grefhorst et al. (2002). The mice received palm oil to further induce hepatic steatosis and ensure optimal stimulation of PRMT3 translocation to the nucleus (Chisholm et al., 2003; Go et al., 2015; Kim et al., 2015a). To determine the effect of PRMT3 inhibition under these steatosis‐inducing conditions, mice were treated for 4 days with a total of three injections of SGC707 (30 mg·kg−1), which was the dose recommended for in vivo usage (Kaniskan et al., 2015). An additional experimental group was included to assess the effectiveness of a lower dosage of 10 mg·kg−1 of SGC707. T0901317 and palm oil treatment significantly increased plasma cholesteryl esters as compared with the DMSO negative control group (69% increase; Figure 1A). Free cholesterol levels followed the same trend but just failed to reach significance (Figure 1B). These values were similar to the plasma lipid levels of T0901317‐treated mice described by Grefhorst et al. (2002). Subsequent treatment with SGC707 did not affect plasma cholesterol levels at the dose of 10 mg·kg−1, while the group treated with 30 mg·kg−1 SGC707 showed an additional significant increase in cholesteryl esters (1.9‐fold increase) and free cholesterol levels (2.3‐fold increase) as compared with the T0901317 + palm oil‐treated group (Figure 1A and B). Similarly, the 30 mg·kg−1 SGC707 treatment group showed a significant increase in plasma TG levels (2.7‐fold increase), an effect which was not seen in the 10 mg·kg−1 group (Figure 1C). Importantly, we noticed a significant decrease in body weight in the 30 mg·kg−1 SGC707 treatment group even after treatment for only 4 days (12% decrease) albeit without any other visible signs of discomfort (Figure 1D). Combined, the effects observed after treatment with 30 mg·kg−1 SGC707 suggest that this dosage severely impairs the metabolic state of the mice. Therefore, we considered this experimental group unreliable for the current mechanistic study. For this reason, this experimental group was excluded from further analyses.
Figure 1.
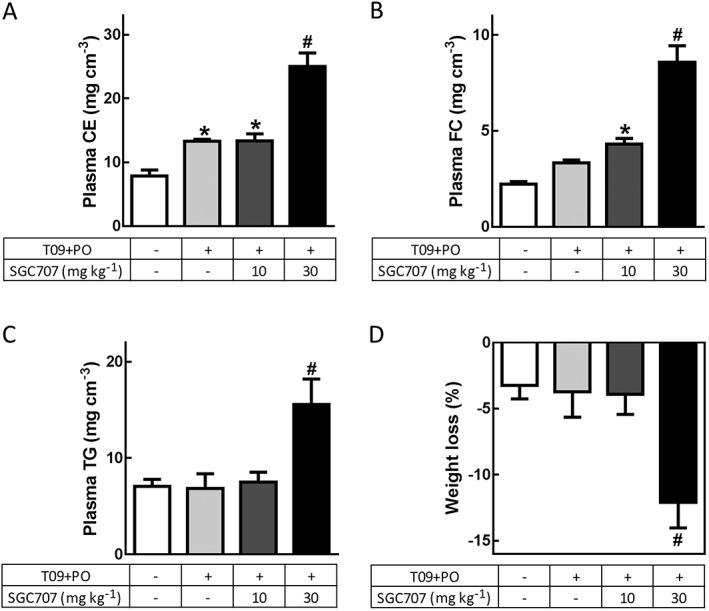
PRMT3 inhibition by a low dose of SGC707 does not impair body weight or plasma lipid levels. (A and B) Only the 30 mg·kg−1 SGC707 treatment showed an additional significant increase in both cholesteryl ester (CE) and free cholesterol (FC) levels as compared with all other groups. (C) Plasma TG levels were significantly higher only in the 30 mg·kg−1 SGC707 treatment group as compared with the other groups. (D) The recommended dosage of 30 mg·kg−1 SGC707 induces an unexpected highly significant change in body weight in comparison with all other experimental groups. T09 + PO, T0901317 and palm oil. DMSO, n = 6; T09 + PO, n = 7; T09 + PO + SGC707 10 mg·kg−1, n = 9; T09 + PO + SGC707 30 mg·kg−1, n = 5. Data represent mean ± SEM. #Significant as compared with all other groups; *significant as compared with DMSO group.
PRMT3 inhibition does not impair expression of LXR‐induced genes involved in macrophage cholesterol metabolism
For therapeutic application, LXR agonist‐induced up‐regulation of the cholesterol efflux receptors ABCA1 and ABCG1 is essential. To determine if in vivo PRMT3 inhibition preserves the LXR‐mediated induction of macrophage cholesterol efflux genes, the mRNA expression of ABCA1 and ABCG1 in peritoneal leukocytes of the SGC707‐treated mice was analysed. As expected based on literature (Zhang et al., 2008), the average percentage of macrophages in the freshly isolated peritoneal leukocytes was 41.5% and did not differ between the experimental groups (data not shown). The relative gene expression levels of PRMT3 did not differ between the groups (Figure 2B). LXRα gene expression levels showed a trend towards increase in the SGC707‐treated mice as compared with the other groups; however, this failed to reach significance (Figure 2A). Importantly, treatment with SGC707 did not affect the LXR‐induced expression of ABCA1 and ABCG1 (Figure 2C).
Figure 2.
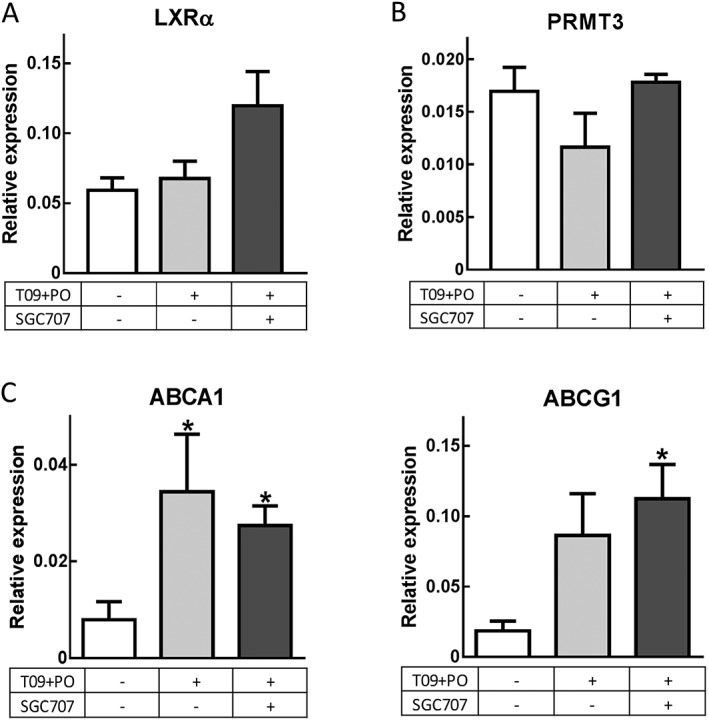
PRMT3 inhibition does not decrease LXR‐mediated cholesterol transporters in macrophages. (A and B) T0901317 and palm oil and the additional treatment with SGC707 did not affect the relative gene expression of LXR or PRMT3. (C) PRMT3 inhibition does not affect the LXR‐regulated expression of reverse cholesterol transport genes ABCA1 and ABCG1 by peritoneal macrophages. T09 + PO T0901317 and palm oil. DMSO, n = 6; T09 + PO, n = 7; T09 + PO + SGC707, n = 9. Data represent mean ± SEM. *Significant as compared with DMSO group.
PRMT3 inhibition impairs the ability of LXR to induce lipogenesis in the liver
To determine the effect of PRMT3 inhibition on hepatic lipogenic gene expression, the LXR‐induced hepatic expression of genes involved in the conversion of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3038 to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5219 by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=255#1263 (ACC), the conversion of malonyl‐CoA to saturated fatty acids by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2608 and the conversion of saturated fatty acids to monounsaturated fatty acids by stearoyl‐coenzyme A desaturase (SCD‐1) were assessed. These are the key processes leading to the production of TGs that are known to be regulated by LXR (Schultz et al., 2000) (Figure 3A). As previously described by Chisholm et al. (2003), the expression of LXRα in the liver decreased significantly upon treatment with T091317 and palm oil, while the expression of lipogenesis genes increased as compared with the DMSO control‐treated mice (Figure 3B and D). Interestingly, the increase in hepatic expression of the lipogenic LXR target genes was severely impaired upon PRMT3 inhibition in T091317 and palm oil‐treated mice. The hepatic mRNA expressions of ACC, FAS and SCD‐1 were lowered 56, 64 and 43%, respectively, upon treatment with SGC707 (Figure 3B). Although the effects on ACC and SCD‐1 expression just failed to reach significance, a similar change in expression was observed compared with the expression pattern of FAS (Figure 3B). The expression of the lipogenic transcription factor SREBP1c follows a similar pattern, though not as pronounced as its target genes (Figure 3B). A highly significant positive correlation between FAS expression and SCD‐1 expression and between ACC expression and SCD‐1 expression was observed (Figure 3C). Finally, also the correlation between FAS expression and ACC expression was highly significant (Figure 3C). These observations indicate that the effect of PRMT3 inhibition is similar for the LXR‐mediated transcription of all these three downstream LXR targets. The effect on the hepatic expression of these lipogenic genes could not be explained by a change in LXRα or PRMT3 gene expression as treatment with the PRMT3 inhibitor SGC707 did not affect the expression of these genes in the liver (Figure 3D and E).
Figure 3.
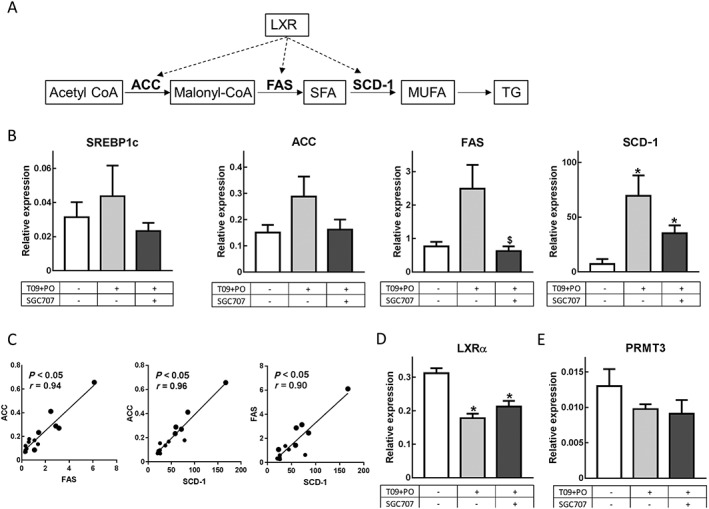
PRMT3 inhibition decreases hepatic expression of LXR target genes involved in lipogenesis. (A) Schematic summary of the LXR‐regulated lipogenic processes involving ACC, FAS and SCD‐1. SFA, saturated fatty acids; MUFA, monounsaturated fatty acids. (B) PRMT3 inhibition with SGC707 results in decreased hepatic expression of SREBP1c, ACC, FAS and SCD‐1. (C) There is a highly significant positive correlation for ACC versus FAS, ACC versus SCD‐1 and FAS versus SCD‐1. (D and E) The effects of SC707 on lipogenic genes are not caused by an effect on gene expression of LXRα or PRMT3. T09 + PO, T0901317 and palm oil. DMSO, n = 6; T09 + PO, n = 7; T09 + PO + SGC707, n = 9. Data represent mean ± SEM. $Significant as compared with T09 + PO group; *significant as compared with DMSO group.
PRMT3 inhibition by SGC707 decreases hepatic lipid accumulation
Next, we investigated whether the effects of PRMT3 inhibition on lipogenic gene expression resulted in phenotypic changes in the livers of SGC707‐treated mice. As expected based on the observations of Grefhorst et al. (2002) and Chisholm et al. (2003), the T0901317 and palm oil treatment resulted in an increased liver weight as compared with the DMSO control group (Figure 4A). SGC707 treatment had no significant effect on relative liver weight as compared with T0901317 + palm oil‐treated controls (Figure 4A). Formalin‐fixed liver cryosections were stained with Oil red O for neutral lipids. Treatment with T0901317 + palm oil resulted in a vast increase in the presence of neutral lipids in the liver as compared with the livers of the DMSO‐treated mice (Figure 4B), similar to data shown by Grefhorst et al. (2002). In line with the reduced expression of lipogenic genes, Oil red O staining showed that treatment with 10 mg·kg−1 SGC707 resulted in a striking decrease in liver lipids in comparison with the livers of the experimental group that received T0901317 + palm oil only (Figure 4B). The stained sections were assigned an Oil red O score between 0 and 3 for increased presence of stained lipids by a blinded observer. Treatment with 10 mg·kg−1 SGC707 was associated with a significantly lower Oil red O score (1.0 ± 0.5), indicating a significant decrease in stained liver lipids as compared with the T0901317 + palm oil control group (3.0 ± 0.2; Figure 4C).
Figure 4.
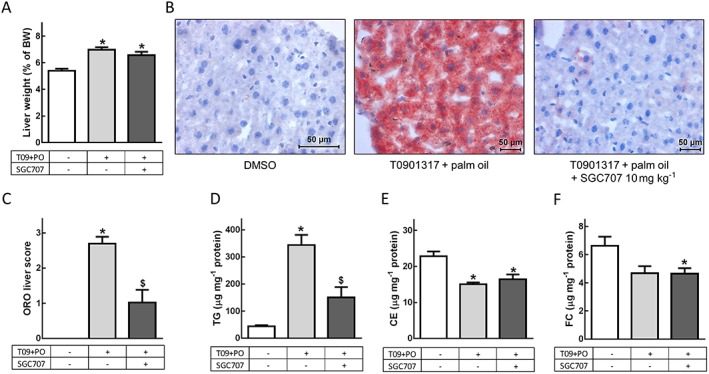
PRMT3 inhibition decreases hepatic lipid accumulation. (A) PRMT3 treatment does not affect liver weight. (B) PRMT3 inhibition decreases hepatic lipid content, as visualized with representative Oil red O (ORO)‐stained (red) liver sections. Magnification: 40×. (C) ORO‐based hepatic lipid score: 0, no red present in the liver section; 3, liver section is totally red. Treatment with SGC707 decreases liver lipid accumulation. (D) Liver TG content is significantly decreased upon PRMT3 inhibition. (E and F) Liver cholesterol levels are not affected by PRMT3 inhibition. T09 + PO, T0901317 and palm oil. DMSO, n = 6; T09 + PO, n = 7; T09 + PO + SGC707, n = 9. Data represent mean ± SEM. $Significant as compared with T09 + PO group; *significant as compared with DMSO group.
To analyse the effect of PRMT3 inhibition on TG and cholesterol content of the liver, lipids were extracted from the liver and quantified. As expected based on literature (Grefhorst et al., 2002), T0901317 and palm oil increased liver TG content 7.9‐fold, while total cholesterol was reduced, mainly driven by a significant 34% decrease in cholesteryl esters (Figures 4D–F). In accordance with the Oil red O scores, additional treatment with 10 mg·kg−1 SGC707 resulted in a remarkable and significant 2.3‐fold decrease in liver TG content as compared with T0901317 + palm oil treatment alone, indicating that PRMT3 protects against the palm oil and T0901317‐induced hepatic steatosis (Figure 4D). Importantly, SGC707 treatment did not affect hepatic cholesterol levels (Figure 4E and F). These results suggest that PRMT3 selectively acts as a cofactor for LXR‐induced lipogenesis without affecting LXR‐regulated cholesterol homeostasis.
Discussion
The development of LXR agonists as therapeutic agents for cholesterol‐driven diseases is currently limited by the increased hepatic lipogenesis that occurs alongside the positive stimulatory effects on macrophage cholesterol efflux (Grefhorst et al., 2002). In this study, we investigated the role of PRMT3 as specific co‐activator for LXR‐mediated lipogenesis in vivo. We aimed to explore PRMT3 inhibition as a tool to uncouple the beneficial effects of LXR activation in macrophages and the adverse effects on hepatic lipogenesis as basis for a novel therapeutic strategy.
LXR is a nuclear receptor that functions as a transcription factor. LXR activity can be regulated at many different levels: transcription or protein levels in the cell but also via the interaction with cofactors at the site of transcription. Selective LXR modulators are a group of molecules that, by specific interaction with LXR, can modulate its transcriptional activity. These selective modulators are increasingly recognized for their therapeutic potential, although for most modulators the mechanisms through which they act remain to be elucidated (Viennois et al., 2012). Kim et al. (2015a) suggested that PRMT3 functions as a selective LXR modulator in vitro. They have shown a specific induction of lipogenic genes via LXR in a PRMT3 overexpression model and reversal of these effects upon PRMT3 silencing and deletion. Our study follows up on those experiments, validating the relevance in an in vivo setting.
We show that PRMT3 inhibition using the allosteric PRMT3 inhibitor SGC707 significantly impairs the up‐regulation of the LXR target genes involved in lipogenesis, thereby hampering LXR‐dependent hepatic accumulation of TGs in vivo. Importantly, PRMT3 inhibition did not affect the LXR‐regulated expression of atheroprotective genes involved in cholesterol efflux in macrophages. Thus, by combined treatment with the LXR agonist T0901317 and the PRMT3 inhibitor SGC707, the ultimate therapeutic goal can be achieved.
Our results show that the effects of SGC707 treatment on LXR‐mediated lipogenesis in vivo are equally pronounced as the effects on lipogenesis after in vitro knockdown of PRMT3. SGC707 thus is a highly potent inhibitor of PRMT3 in vivo and therefore confirms previous studies investigating the effect of SGC707 on PRMT3 activity (Kaniskan et al., 2015). Further studies should be conducted to investigate the molecular mechanisms underlying the PRMT3–LXR interaction and selective effects on gene expression in detail. Kim et al. (2015a,b) showed in vitro that PRMT3 and LXR co‐localize in the nucleus and that silencing of PRMT3 reduces both the expression of LXR and the binding of LXR to its target sequences. We show in our murine in vivo model that despite the obvious changes in hepatic lipogenesis, PRMT3 inhibition using SGC707 does not affect the cellular mRNA levels of LXR. Park et al. (2015) have shown that blocking the migration of PRMT3 to the nucleus using the myokine irisin also decreased the induction of lipogenesis via LXR. We therefore anticipate that in our in vivo setting, PRMT3 does not directly modulate LXR transcription or translation efficiency but rather serves like a selective LXR modulator that controls the transcription of a selected cluster of lipogenic LXR target genes.
In conclusion, we have shown that PRMT3 acts as a specific co‐activator for LXR‐induced hepatic lipogenesis in vivo. Our results indicate that short‐term combination treatment with the LXR agonist T0901317 and the PRMT3 inhibitor SGC707 is associated with a beneficial gene expression profile, that is, increased macrophage cholesterol efflux transcription with only limited hepatic lipogenesis gene induction. As such, co‐treatment appears to be able to overcome hepatic steatosis as the major constraint for using the LXR agonist T0901317 as a therapeutic tool in atherosclerosis. Further studies in established atherosclerosis models are warranted to elucidate whether long term co‐treatment indeed serves as a valuable cardiovascular therapeutic strategy.
Author contributions
J.E.N. performed experiments, analysed and interpreted data and wrote the manuscript. C.G. performed experiments, analysed and interpreted data and contributed to the manuscript. M.H. designed experiments, analysed and interpreted data and aided in the preparation of the manuscript. J.J.G. interpreted data and aided in the preparation of the manuscript. M.V.E. supervised the study and provided financial support. All authors revised the manuscript and approved its final version.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank M.J. Kröner, R.J. van der Sluis and K.E. Dzobo for their practical support during the experiments. We acknowledge the support from the Netherlands CardioVascular Research Initiative: ‘the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences’ for the GENIUS project ‘Generating the best evidence‐based pharmaceutical targets for atherosclerosis’ (CVON2011‐19 to M.V.E). This study was supported by the Netherlands Organization for Scientific Research (VICI grant 91813603 to M.V.E) and Dutch Heart Foundation grant 2012T080 awarded to M.H; M.V.E is an established investigator of the Netherlands Heart Foundation (grant 2007T056).
Nahon, J. E. , Groeneveldt, C. , Geerling, J. J. , van Eck, M. , and Hoekstra, M. (2018) Inhibition of protein arginine methyltransferase 3 activity selectively impairs liver X receptor‐driven transcription of hepatic lipogenic genes in vivo . British Journal of Pharmacology, 175: 3175–3183. https://doi.org/10.1111/bph.14361.
References
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JY, Repa JJ (2007). The liver X receptor (LXR) and hepatic lipogenesis: the carbohydrate‐response element‐binding protein is a target gene of LXR. J Biol Chem 282: 743–751. [DOI] [PubMed] [Google Scholar]
- Chisholm JW, Hong J, Mills SA, Lawn RM (2003). The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44: 2039–2048. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987). Single‐step method of RNA isolation by acid guanidinium extraction. Anal Biochem 162: 156–159. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1953). A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509. [PubMed] [Google Scholar]
- Go R‐E, Hwang K‐A, Kim Y‐S, Kim S‐H, Nam K‐H, Choi K‐C (2015). Effects of palm and sunflower oils on serum cholesterol and fatty liver in rats. J Med Food 18: 363–369. [DOI] [PubMed] [Google Scholar]
- Grefhorst A, Elzinga BM, Voshol PJ, Plösch T, Kok T, Bloks VW et al (2002). Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride‐rich very low density lipoprotein particles. J Biol Chem 277: 34182–34190. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog B, Hallberg M, Seth A, Woods A, White R, Parker G (2007). The nuclear receptor cofactor RIP140 is required for the regulation of hepatic lipid and glucose metabolism by LXR. Mol Endocrinol 21: 2687–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li S, Wu J, Xia C, Lala DS (2003). Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol 17: 1019–1026. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R et al (2002a). Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem 277: 11019–11025. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Mckilligin E, Pei L, Watson MA, Collins AR, Laffitte BA et al (2002b). Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A 99: 7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan HÜ, Szewczyk MM, Yu Z, Eram MS, Yang X, Schmidt K et al (2015). A potent, selective and cell‐active allosteric inhibitor of protein arginine methyltransferase 3. Angew Chem Int Ed Engl 54: 5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Park M, Lim S, Park J, Yoon K, Han H et al (2015a). PRMT3 regulates hepatic lipogenesis through direct interaction with LXRα. Diabetes 64: 60–71. [DOI] [PubMed] [Google Scholar]
- Kim GH, Oh G, Yoon J, Lee GG, Lee K, Kim S (2015b). Hepatic TRAP80 selectively regulates lipogenic activity of liver X receptor. 125: 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komati R, Spadoni D, Zheng S, Sridhar J, Riley KE, Wang G (2017). Ligands of therapeutic utility for the liver X receptors. Molecules 22: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP et al (2006). Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation : 90–98. [DOI] [PubMed] [Google Scholar]
- Oh G, Yoon J, Gu G, Keun W, Kim S (2015). 20(S)‐protopanaxatriol inhibits liver X receptor α‐mediated expression of lipogenic genes in hepatocytes. J Pharmacol Sci 128: 71–77. [DOI] [PubMed] [Google Scholar]
- Out R, Hoekstra M, Hildebrand RB, Kruit JK, Meurs I, Li Z et al (2006). Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor‐deficient mice. Arterioscler Thromb Vasc Biol 26: 2295 LP–2300. [DOI] [PubMed] [Google Scholar]
- Park M, Kim D, Choi J, Heo Y, Park S (2015). New role of irisin in hepatocytes: the protective effect of hepatic steatosis in vitro. Cell Signal 27: 1831–1839. [DOI] [PubMed] [Google Scholar]
- Perissi V, Rosenfeld MG (2005). Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6: 542–554. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JA, Shimomura I et al (2000). Regulation of mouse sterol regulatory by oxysterol receptors, LXRα and LXRβ. Genes Dev 14: 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L et al (2000). Role of LXRs in control of lipogenesis. Genes Dev 14: 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Hillgartner FB (2006). The mechanism mediating the activation of acetyl‐coenzyme A carboxylase‐α gene transcription by the liver X receptor agonist T0‐901317. J Hepatol 47; 2451–2461. [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA et al (2000). Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc Natl Acad Sci U S A 97: 12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viennois E, Mouzat K, Dufour J, Morel L, Lobaccaro J, Baron S (2012). Molecular and cellular endocrinology selective liver X receptor modulators (SLiMs): what use in human health? Mol Cell Endocrinol 351: 129–141. [DOI] [PubMed] [Google Scholar]
- Yu L, York J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2003). Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP‐binding cassette transporters G5 and G8. J Biol Chem 278: 15565–15570. [DOI] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, and Mosser DM (2008). The isolation and characterization of murine macrophages. Curr Protoc Immunol/Ed. by John E. Coligan ...[et Al.] CHAPTER: Unit‐14. [DOI] [PMC free article] [PubMed]