

Abstract
Background and Purpose
PEP06, a polypeptide modified from endostatin, was investigated for its antitumour effects on colorectal cancer (CRC) and the possible mechanisms of this antitumour activity were examined in in vitro and in vivo models.
Experimental Approach
After PEP06 treatment, cell proliferation and migration assays were performed in CRC cells. Epithelial–mesenchymal transition (EMT) progression was determined by Western blotting, immunofluorescent staining and immunohistochemistry in vitro and in a residual xenograft model. MiRNAs regulated by PEP06 were identified by miRNA microarray and verified by in situ hybridization and quantitative real‐time PCR. The interactions between PEP06 and integrin αvβ3 were determined with Biacore SA biochips. The cellular function of miR‐146b‐5p was validated by gain‐of‐function and loss‐of‐function approaches. A mouse model of lung metastasis was used to determine the effect of PEP06 on metastatic growth.
Key Results
PEP06 did not affect cell viability but reduced migration and EMT in SW620 and HCT116 cells. PEP06 significantly repressed the expression of miR‐146b‐5p in these two cell lines through binding to integrin αvβ3. MiR‐146b‐5p was shown to increase EMT by targeting Smad4, and the miR‐146b‐5p‐Smad4 cascade regulated EMT in CRC. PEP06 also suppressed CRC pulmonary metastasis, increased survival of mice and hampered residual tumour growth by inhibiting EMT through down‐regulating miR‐146b‐5p.
Conclusions and Implications
PEP06 is a polypeptide that inhibits the growth and metastasis of colon cancer through its RGD motif binding to integrin αvβ3, thereby down‐regulating miR‐146b‐5p to inhibit EMT in vitro and in vivo. It might have potential as a therapeutic for CRC.
Abbreviations
- 5‐FU
5‐fluorouracil
- ATO
arsenic trioxide
- BLI
bioluminescence imaging
- CRC
colorectal cancer
- EMT
epithelial–mesenchymal transition
- FC2
flow cells 2
- NC
negative control
- SPR
surface plasmon resonance
- TMA
tissue microarray
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer in developed countries and a serious social and economic burden (Torre et al., 2015; Paolillo et al., 2016). According to the Cancer Statistics of China in 2015, CRC is the fifth and fourth most commonly diagnosed cancer in men and women, respectively, in the Chinese population (Chen et al., 2016b). Undoubtedly, suppressing metastasis is a highly and urgently desirable approach for treating patients with CRC. Metastatic progression is regulated by changes in the micro‐environment, such as angiogenesis, inflammation and cancerized stroma and intravasation, which involve epithelial–mesenchymal transition (EMT) (Gupta and Massague, 2006). EMT in tumour progression allows a polarized epithelial cell to acquire a mesenchymal cell phenotype (Kalluri and Weinberg, 2009). It also facilitates intravasation of tumour cells into blood or lymph vessels and subsequent formation of distant metastasis (Radisky, 2005). EMT has also been proposed to be involved in the pathogenesis of CRC (Zhu et al., 2013).
Endostatin, a 20 kDa peptide fragment generated from collagen XVIII, is known to be an inhibitor of both angiogenesis (O'Reilly et al., 1997) and endothelial cell proliferation and/or migration (Dkhissi et al., 2003). This peptide has the potential to suppress colon tumourigenesis (Sudhakar et al., 2003) and cancer metastasis. However, the full‐length endostatin protein cannot be produced as a therapeutic for colon cancer because of certain unfavourable issues, such as difficulties in its isolation, purification and delivery (Harris, 1998; Marshall, 2002; Folkman, 2006). It has been reported that the folded endostatin structure may influence the antitumour activity of Escherichia coli‐derived insoluble endostatin (Marshall, 2002). To solve this issue, a short peptide was synthesized. The antitumour properties of this NH2‐terminal 27 amino‐acid peptide of endostatin was found to be comparable with the full‐length endostatin (Tjin Tham Sjin et al., 2005). Likewise, we have designed a number of peptides consisting of amino acids 1–30 of endostatin with amino acids 25–31 mutated from RGIRGAD to RGDRGD (peptide 30) and investigated their ability to inhibit tumour growth (Li et al., 2011). To produce cost‐effective and stable peptides, we have tested a number of different polypeptides modified from endostatin and found PEP06 to be the best with strong anti‐CRC effects and a high production yield.
The RGD sequence, arginine–glycine–aspartate, is believed to be the site of cell attachment for adhesive proteins, such as cell surface proteins (Pierschbacher and Ruoslahti, 1984; Ruoslahti and Pierschbacher, 1987). A cluster of known integrins, the heterodimeric cell surface proteins highly expressed in malignant cells, can recognize the RGD sequence within their adhesion protein ligands (Ruoslahti, 1996). RGD has anti‐metastasic properties in both in vitro and in vivo models (Saiki et al., 1990; Li et al., 2011). When a peptide/protein is conjugated or fused with RGD, it acquires a new feature and can be targeted to tumour tissues (Rubtsov et al., 2016). Therefore, we attempted to fuse RGD sequences with the active fragments of endostatin to form a new clinical‐stage polypeptide drug that possesses stronger antitumour effects.
MiRNAs can act as either carcinogenic genes or tumour suppressor genes to facilitate or retard EMT; they are critically involved in tumour metastasis and have the potential to act as biomarkers for cancer diagnosis, treatment and prognosis (Tang et al., 2016). The EMT‐regulating miRNAs can also interact with some key transcription factors to regulate the function of epithelial or mesenchymal cells (Bracken et al., 2015). For instance, miR‐146b‐5p has been shown to induce EMT via targeting ZNRF3 in thyroid cancer (Deng et al., 2015). In colon cancer, aberrant regulation of a subset of miRNAs has been demonstrated to be involved in cancer progression (Valeri et al., 2014; Cantini et al., 2015). Yet the potential pathophysiological role of miR‐146b‐5p in EMT of CRC has not been investigated.
In this study, we examined the antitumour effects of an endostatin‐RGDRGD 30‐amino‐acid fusion polypeptide (PEP06) both in vitro and in vivo. We found that PEP06 attenuated CRC metastasis without affecting cell viability or generating cytotoxicity. We further determined that the molecular mechanism of its antitumour activity involves down‐regulating miR‐146b‐5p to derepress its target gene http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=303.
Methods
Cell lines, culture conditions and reagents
The human SW620 colorectal adenocarcinoma [American Type Culture Collection (ATCC)® CCL‐227™], HCT116 colorectal carcinoma (ATCC CCL‐247™), LOVO colorectal adenocarcinoma (ATCC CCL‐229™) and HUVEC (ATCC PCS‐100‐010™) cell lines were purchased from the ATCC (Manassas, VA, USA). With the exception of SW620, the cell lines were maintained in RPMI‐1640 medium (SH30027.01; Hyclone, Logan, UT, USA), supplemented with 10% FBS and 1% antibiotic–antimycotic solution (Beyotime, Shanghai, China) at 37°C in 5% CO2. The SW620 cells were cultured in L‐15 medium (SH30525.01; Hyclone) containing 2 mM glutamine, 10% FBS and 1% antibiotic–antimycotic solution in free gas exchange with atmospheric air.
PEP06 (Arg‐Gly‐Asp‐Arg‐Gly‐Asp‐Met‐His‐Ser‐His‐Arg‐Asp‐Phe‐Gln‐Pro‐Val‐Leu‐His‐Leu‐Val‐Ala‐Leu‐Asn‐Ser‐Pro‐Leu‐Ser‐Gly‐Gly‐Met) was dissolved in glucose, which was synthesized by Chengdu Shengnuo Tech Co., Ltd. (Chengdu, China). The cyclic RGD peptide http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6597 was also dissolved in glucose (Aladdin, Shanghai, China). An artificial 24 amino acid with the RGD motif deleted was synthesized by ChinaPeptides (Shanghai, China) and dissolved in glucose. These three solutions were all filtered through a microporous membrane (0.22 μm; Millipore, Billerica, MA, USA). A similar positive peptide drug ENDOSTAR® was purchased from Simcere Pharmaceutical Group (Shandong, China). The oligonucleotide fragments were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China), including miR‐146b‐5p mimic, a negative control (NC) duplex, miR‐146b‐5p inhibitor and miRNA inhibitor NC. The sequences are listed in Supporting Information Table S1.
Cell viability by MTT assay
SW620, HCT116, LOVO cells or HUVECs were plated in 96‐well plates at a density of 4–8 × 103 cells per well. After the cells had reached about 80% confluency, they were starved overnight. The cells were then exposed to different concentrations of PEP06 (50, 100 and 200 μg·mL−1) for 24 and 48 h respectively. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4789 or arsenic trioxide (ATO) was added as a positive control and 5% glucose solution as a negative control . After incubation, MTT (20 μL per well) solution was added, and the plates were incubated for 6 h. The medium in the plate was discarded, and 150 μL DMSO was added to each well. The absorbance was then measured at 570 nm using a PowerWave HT microplate spectrophotometer (BioTek, Winooski, VT, USA). The data were normalized to the NC in each group.
TUNEL assay
SW620 cells cultured in the presence of PEP06 (50, 100 and 200 μg·mL−1) for 24 h were analysed for apoptosis with TUNEL assay by using the In Situ Cell Death Detection Kit (cat. no. 11684817910; Roche, Mannheim, Germany) according to the manufacturer's instructions. Cells were fixed with 4% paraformaldehyde, infused with 0.1% Triton X‐100 and incubated with TUNEL Reaction Mixture for 1 h at 37°C in the dark. After DAPI counterstain for 10 min at room temperature, cells were photographed with a fluorescence microscope. The assay was repeated in five times (Zeiss, Jena, Germany).
Cell‐cycle analysis
After treatment with PEP06 for 24 h, the cells were harvested and stained with the Cycletest Plus DNA Reagent Kit (BD Biosciences, Franklin Lakes, NJ, USA) following the instructions of the manufacturer. Flow cytometry (LSRFortessa; BD Biosciences, San Jose, CA, USA) was used to determine the changes in cell cycle. The results were analysed using FlowJo 7.6.1 (BD Biosciences, Franklin Lakes, NJ, USA).
Tube formation
The tube formation assay was conducted as described previously (Kim et al., 2013). Briefly, 24‐well plates were coated with growth factor‐reduced Matrigel (250 µL per well; Corning, New York, NY, USA). HUVECs pretreated with 200 µg·mL−1 were plated in 200 µL EBM‐2 medium at a cell density of 5 × 104 cells/well, and incubated with 5% CO2 at 37°C for 12 h to allowed for tube formation. HUVECs were harvested, and 1.5 × 105 cells were resuspended in EBM‐2 medium containing 0.5% FBS. Cells were plated onto the Matrigel layer, then PEP06 was added at a final concentration of 200 μg·mL−1. After 6 h incubation at 37°C, images showing the formation of capillary‐like structures were obtained with an inverted microscope (Olympus, Tokyo, Japan) at 100 × magnification. Tubular structures were assessed by measuring the number of branches using Image‐Pro Plus software. Tube formation was quantified by counting the number of branching nodes and the tube length in three randomly selected fields five times.
Wound‐healing assay
HCT116 and SW620 cells were seeded onto a six‐well plate at a density of 5 × 105 cells mL−1 until they reached full confluence. The cells were cultured for 24 h in the presence of varying concentrations of PEP06. Then they were starved overnight and scratched by using a 200 μL pipette tube the next day. Glucose solution (5%) was used as a negative control. In another experiment, SW620 cells transfected with miR‐146b‐5p lentiviral expression vector and control vector were also seeded. Photographs were captured by a digital single lens reflex camera (Canon, Tokyo, Japan) at 0 and 24 h respectively. The migration gap area of the cells was measured by ImageJ software (https://imagej.nih.gov/ij/; Center for Information Technology, National Institute of Health, Bethesda, MA, USA). Each measurement was repeated five times.
Transwell migration assay
SW620 and HCT116 cells (4–5 × 104 cells per well) were seeded on upper chambers (6.5 mm Transwell® with 8.0 μm Pore Polycarbonate Membrane Insert; Corning, NY, USA) in serum‐free medium with 0.1% BSA. Other transwell assays using miR‐146b‐5p lentiviral expression vector or the control lentiviral vector transfected SW620 cells were carried out using the same procedure. A total of 600 μL medium containing 10% FBS was added to the lower chambers. After incubation for 24 h, non‐migrating and non‐invading cells were gently removed by a cotton swab. The cells were then fixed with methanol for 30 min and stained with 0.1% crystal violet for another 20 min. The area of dyed pores was then calculated under a microscope at a magnification of ×400. Five views were selected randomly for photography and analysis. Each measurement was repeated five times.
Western blot analysis
Whole‐cell lysates were extracted by RIPA (Beyotime) with protease (Roche, Mannheim, Germany). Phosphatase inhibitors (Roche) were added for primary antibodies against phosphorylated Smad2/3 (pSmad2/3). Protein samples of 70 μg per lane were loaded onto 10% SDS‐PAGE and transferred onto nitrocellulose membranes (PALL, New York, NY) for 90 min. For immunodetection, the membranes were incubated with primary antibodies at 4°C overnight: E‐cadherin (ab76055; Abcam, Cambridge, UK) at 1:1000 dilution, α‐catenin (ab52227; Abcam) at 1:10000 dilution, vimentin (HPA001762; Sigma‐Aldrich, St. Louis, MO, USA) at 1:250 dilution, N‐cadherin (C3865; Sigma‐Aldrich) at 1:500 dilution, Smad4 (sc‐7966; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at 1:200 dilution, p‐Smad2/3 (sc‐11769, Santa Cruz Biotechnology, Inc.) at 1:200 dilution, or Smad2/3 (sc‐133098; Santa Cruz Biotechnology, Inc.) at 1:200 dilution or http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=963 (19003‐1‐AP; Proteintech, Wuhan, China) at 1:300 dilution. After incubation with secondary antibody with gentle shaking at room temperature for 30–60 min, the membranes were scanned by Odyssey IR Imaging System (Li‐COR, Lincoln, NE, USA). All experiments were done at least five times. The values of protein band densities were normalized to those of the solution or blank control group.
Immunofluorescent staining for E‐cadherin and vimentin
SW620 and HCT116 cells were plated onto a six‐well plate and treated with PEP06 at 200 μg·mL−1 per well for 24 h or transfected with miRNA. The cells were washed with cold sterilized PBS five times, fixed in 4% paraformaldehyde for 1 h, infused with 0.5% Triton X‐100 (Sigma‐Aldrich) for 15 min and blocked with 0.1% BSA for 1 h. The cells were incubated with E‐cadherin (ab76055; Abcam) at 1:200 dilution or vimentin (HPA001762; Sigma‐Aldrich) at 1:200 dilution at 4°C overnight. The samples were incubated with https://www.thermofisher.com/order/genome-database/details/antibody/A-11034?ICID=search-product (Invitrogen™; Thermo Fisher Scientific, Waltham, MA, USA), or goat anti‐rabbit IgG (H + L), a highly cross‐adsorbed secondary antibody, Alexa Fluor 594 (Invitrogen™; Thermo Fisher Scientific) at 1:200 dilution for 1 h. Nuclei were stained with DAPI (Beyotime) at room temperature for 5 min. The samples were visualized under a fluorescence microscope (Nikon 80i; Nikon, Tokyo, Japan).
MiRNA microarray analysis
The miRNA profiling analysis was conducted by Shanghai KangCheng Biosciences using the Exiqon miRCURY™ LNA Array system, which contained 2043 capture probes covering all human miRNAs (Exiqon, Vedbæk, Denmark). PEP06 was added to SW620 or HCT116 cells for 24 h, and the addition of 5% glucose served as a control. Total RNA was extracted from pretreated cells with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and miRNeasy mini kit (Qiagen, Shenzhen, China). After quality control, the miRCURY™ Hy3™/Hy5™ Power labelling kit (Exiqon) was used according to the manufacturer's guideline for miRNA labelling. Afterwards, the Hy3™‐labelled samples were hybridized on the miRCURY™ LNA Array (v.19.0; Exiqon) according to the array manual. Then images of the chip were scanned using an Axon GenePix 4000B microarray scanner (Axon Instruments, Foster City, CA, USA). The spot density‐derived raw data were extracted from non‐coding RNA profiling by array software and deposited on the Gene Expression Omnibus database (accession number GSE111493). MiRNAs with intensities >50 were used to calculate the normalization reference. Expression data were analysed using the median normalization, and then differentially expressed miRNAs between the two groups were identified through fold change (FDNAs w and P‐value, P < 0.05).
Quantitative real‐time PCR
Total RNA was extracted from pretreated cells with TRIzol reagent (Invitrogen). The cDNAs were produced from the RNA samples with Reverse Transcription Kit (Toyobo, Shanghai, China) and miRNA specific Bulge‐loop™ miRNA RT primers (Invitrogen). Human U6 snRNA was used as an endogenous control for data normalization. Real‐time PCR was performed on the ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using SYBR Green I Real‐Time PCR kit (Roche Diagnostics, Mannheim, Germany). Relative miRNA expression was determined using the Ct method. All experiments were performed at least five times. The primer sequences are listed in Supporting Information Table S1.
Surface plasmon resonance
PEP06‐binding affinities to integrin http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=760#2583 at 25°C were evaluated by surface plasmon resonance (SPR) technology using a Biacore T200 instrument (GE Healthcare, Piscataway, NJ, USA) equipped with an SA chip under different acid conditions. Biotinylated PEP06 (Chengdu Shengnuo Tech Co., Ltd., Chengdu, China) was conjugated to an SA Biacore chip (BR‐10531; GE Healthcare) by the capture coupling method. The PEP06–biotin conjugate (50 nM) was diluted in deionized water (ddH2O) in 1 × HBS‐EP buffer (10 mM HEPES, 150 mM sodium chloride, 3 mM EDTA and 0.005% polysorbate surfactant P20 pH 7.4 buffer) (BR1006‐69; GE Healthcare). The solution at a volume of 100 μL was injected over flow cells 2 (FC2) of the SA chip at a flow rate of 10 μL·min−1. Human recombinant protein integrin αvβ3 was purchased from R&D Systems (3050‐AV; Millipore). Integrin αvβ3, as a series of increasing concentrations (0, 2, 4, 8, 16 and 32 nM), as an analyte was applied to two channels (FC1 and FC2) at a flow rate of 30 μL·min−1 for 120 s of association phase followed by a 240 s dissociation phase and a 30 s MgCl2 regeneration phase. The control flow cell (FC1) was prepared by a 1 min injection of saturated biotin. Data were analysed using the Biacore T200 Control Software for analysing binding affinity between PEP06 and integrin αvβ3. Biotinylated 24aa (0, 2, 4, 8, 16 and 32 nM) was immobilized under the same conditions as a negative control. Response unit values at each concentration were measured during the equilibration phase for steady‐state affinity fittings using either Biacore T200 evaluation software 2.0.3. Kinetic rate constants were determined by fitting globally to the 1:1 Langmuir (one‐to‐one binding) model embedded in the Biacore T200 evaluation software v3.0.
Transfection of miRNA
MiR‐146b‐5p mimic (miR‐146b), miR‐146b‐5p inhibitor (AMO‐146b) and miR‐146b‐5p NC were synthesized by Shanghai GenePharma Co., Ltd. SW620 cells were plated onto a six‐well plate at 5 × 104 cells in 2 mL culture medium overnight until the desired density of 50–70% confluency was achieved. Then they were transfected with the mimic, NC or the mixture of mimic and inhibitor by using X‐tremeGENE siRNA Transfection Reagent (Roche) following the manufacturer's instructions.
Establishment of SW620 cells stably overexpressing miR‐146b‐5p
Stable transfectants overexpressing miR‐146b‐5p were generated by lentiviral transduction using LV‐3 (pGLVH1/GFP + Puro) plasmid harbouring miR‐146b‐5p‐coding sequence. MiR‐146b‐5p‐expressing lentiviruses (miR‐146b‐OE) and its negative control (miR‐Ctl) containing the GFP were packaged by Shanghai GenePharma Co., Ltd. SW620 cells (5 × 104) were seeded in six‐well plates and cultured to about 90% confluence. The cells were transfected with the miR‐146b‐5p or negative control lentiviral plasmids using X‐tremeGENE siRNA Transfection Reagent and incubated in fresh medium containing 1.0 μg·mL−1 puromycin (Sigma‐Aldrich) 6 h post‐transfection. The presence of GFP was detected through the fluorescence microscopy at 24 h after infection. Drug‐resistant cell colonies were individually picked up using tips, seeded into a 24‐well plate, cultured and passed with selection pressure. The expression level of miR‐146b‐5p was determined with quantitative real‐time PCR (qRT‐PCR).
Smad4 overexpression using lentiviral expression system
Lentiviral vectors of Smad4 and a negative control were purchased from GeneChem (Shanghai, China). Stably expressed miR‐146b‐5p (miR‐146b‐OE) and the matched control lines (miR‐Ctl) SW620 cells (6 × 105) were plated in the six‐well plates and cultured to about 90% confluence. Cells were then infected by adding the Smad4 or negative control lentiviral vectors in 2 mL of L‐15 medium at multiplicity of infection of 35 plaque‐forming units per cell, followed by addition of 10 times volume of virus enhanced infection solution. Mock infection (Ctl) was performed by treating cells with vehicle (medium) only. After an 8 h incubation, the medium was replaced with 2 mL of fresh L‐15 medium containing 10% FBS and 1% antibiotic–antimycotic. Cells were trypsinized after 72 h and prepared for Western blot analysis.
In vivo animal experiments
Our protocol for animal use was approved by the Institution Animal Care and Use Committee of Harbin Medical University, and all animal experiments were carried out according to the Guide for the Care and Use of Laboratory Animals, and in strict accordance with the People's Republic of China Legislation Regarding the Use and Care of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Postsurgical residual tumour xenograft models and antitumour effect assay
Adult female athymic BALB/c nude mice (15–20 g), 4 weeks‐old, were purchased from Nanjing Biomedical Research Institute of University [certificate no. SCXK (Su) 2015‐0001]. The animals were housed in a controlled environment at 23 ± 2°C under a 12 h dark/light cycle with free access to irradiated food and sterile water. We established a murine model with a postsurgical residual tumour to mimic the recurrence of solid tumour as described previously (An et al., 2014). The LOVO tumour cells (over 5 × 107 mL−1) were harvested and washed with sodium chloride. Cell suspension of 1 × 106 in 0.2 mL was injected s.c. into the right axillary of mice to establish the traditional tumour xenograft nude mouse model. When the tumours reached an average volume of 1000–1500 mm3, they were removed from the mice aseptically and diced into small cubes (2.0 × 2.0 × 2.0 mm). These tumour chunks were injected s.c. into the right axillary of the mice to establish a murine model with a postsurgical residual tumour. When the tumours had reached an average volume of 300–350 mm3, the mice were anaesthetized by an i.v. injection of 1.2% avertin (2,2,2‐tribromoethanol) solution (0.2 mL per 10 g mouse weight). A small incision was made to remove tumour tissues around the tumour edge aseptically, leaving a residual tumour volume of 60–100 mm3. Twenty‐four hours after the surgery, the mice were sorted into three groups (n = 8) on the basis of tumour volume. Body weight was measured twice a week. The mice in the PEP06 treatment groups were administered PEP06 by i.v. injection at a low dosage of 10 mg·kg−1·day−1 or a high dosage of 20 mg·kg−1·day−1 for 17 days. The control mice were treated with 5% glucose solution in the same manner. Tumours were measured by calipers in two dimensions every 2–3 days. Tumour volume was calculated according to the formula: volume (mm3) = (width)2 × (length)2/2. The mice were killed 17 days after the first injection of PEP06. At the end of the experiments, tumours were excised, weighed and fixed in 4% paraformaldehyde at room temperature before paraffin sections were obtained. The inhibitory rate of tumour volume was calculated as inhibitory rate (%) = (Vt − V0) ∕ V0, where V 0 is the tumour volume for the control mice and V t is the tumour volume for the PEP06 groups. The inhibitory rate of tumour weight was calculated as inhibitory rate (%) = (Wt − W0) ∕ Wt × 100%, where W t and W 0 represent the tumour weights for the treatment and control groups respectively.
Mouse model of colon cancer pulmonary metastasis
Adult female athymic BALB/c nude mice (18–20 g, 4 weeks‐old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. [certificate no. SCXK (Jing) 2016‐0011 (no. 11400700161156)]. The animals were housed in a controlled environment at 23 ± 2°C, 40–70% humidity under a 12 h dark/light cycle with free access to irradiated food and sterile water. They were housed in individually ventilated cages: five per cage, with 4–6 mm corncob bedding after 60Co radiation disinfection.
The CRC cell line, CLY, derived from liver metastasis of a 64‐year‐old Chinese CRC patient was established and kindly donated by Professor Shoujun Yuan (Beijing Institute of Radiation Medicine, Beijing, China) (Li et al., 2007). CLY cells were cultured in DMEM medium (Gibco®; Thermo Fisher Scientific) supplemented with 10% FBS (Beijing YuanHeng ShengMa Biology Technology Research Institute, Beijing, China) and 1% antibiotic–antimycotic solution (Chinese Academy of Medical Science and Peking Union Medical College Institute of Biomedical Engineering, Tianjin, China) at 37°C in 5% CO2.
CLY cells were transduced by self‐inactivating lentiviral vector pCDH‐luc2‐GFP and positive cell clones selected by 1 μg·mL−1 puromycine (InvivoGen, San Diego, CA, USA) to create a stable luciferase 2 (luc2) and GFP expressing CLY‐Luc2‐GFP cell line. A total of 1.5 × 106 of CLY‐Luc2‐GFP tumour cells suspended in 200 μL of physiological saline (0.9% NaCl) was slowly injected (i.v.) into the tail vein. Five minutes after injection, mice were anaesthetized using 2% isoflurane and injected i.p. with 150 mg·kg−1 d‐luciferin and then imaged using IVIS Spectrum CT platform (PerkinElmer, Waltham, MA, USA). Living image 4.3.1 software was used for data analysis (PerkinElmer). The mice were weighed every 5 days. After the experiments had been terminated, the lungs were removed to calculate the lung/body weight ratio. During the course of the treatment, the percentage of tumour growth was determined using the formula: %tumour growth = (total flux of photons of the control group − total flux of photons of PEP06 groups) ∕ total flux of photons of the control group × 100%.
To determine the pulmonary metastasis of the CRC tumour model and the efficacy of PEP06, 5 min after injection of CLY tumour cells, mice (n = 30) were randomly divided into three groups, and one of them was used as the normal group with regular food and drink. According to bioluminescence imaging (BLI) signals, the remaining mice (n = 20) were randomly divided into two groups: control (n = 10) and PEP06 (n = 10). The animals received an i.v. injection of 5% glucose solution (control) or 20 mg·kg−1 PEP06 once a day for 45 days. The mice were killed on day 45, and the main internal organs (lung, brain, colon, spleen, kidney, heart, stomach and small intestine) were harvested for ex vivo bioluminescent imaging and macroscopic weighting.
In another colon cancer metastasis xenograft model used to evaluate the effects of miR‐146b‐5p, SW620‐miR‐Ctl or SW620‐miR‐146b cells (2 × 106) were suspended in 200 μL PBS and then injected into the tail vein of the 8‐week‐old BALB/c nude mice (n = 8 for each group). Forty days post‐implantation, the animals were killed, and the tumours, livers and lungs were dissected, fixed in 4% formalin overnight and embedded in paraffin. To evaluate metastasis, serial sections from the lungs with primary tumours were stained with haematoxylin–eosin staining.
Tissue microarray construction and immunohistochemistry
Tissue microarray (TMA) construction was performed on 38 tumour samples from the postsurgical residual tumour xenograft nude mice. Nine tissue samples were obtained from the control group and 29 from the high‐dosage groups (20 mg·kg−1). Two cores of formalin‐fixed, paraffin‐embedded tumour tissues were taken, and the diameters of the punched cores ranged from 1.5 to 2.0 mm. TMA sections (4 mm) from 76 sites were obtained using standard techniques for immunohistochemistry (IHC) or in situ hybridization. Slides were scanned by an AperioScanScope® slide scanner (Leica Biosystems Inc., Buffalo Grove, IL, USA).
For IHC, heat‐induced epitope retrieval was performed by use of Citrate Antigen Retrieval solution (Solarbio, Beijing, China) for 40 min for vimentin, or by EDTA Antigen Retrieval solution (ZSGB‐Bio, Beijing, China) for E‐cadherin and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1633. The tissue preparations were incubated with the primary antibodies for E‐cadherin (1:500; #14472S; Cell Signaling Technology, Danvers, MA, USA), vimentin (1:250; HPA001762; Sigma‐Aldrich) or MMP9 (1:200; ab76003; Abcam) at 4°C overnight, followed by incubation with the secondary antibody EnVision™+/HRP mouse (rabbit) polymer (Dako, Glostrup, Denmark) at room temperature for 30 min. Secondary antibody detection was performed by using the SIGMAFAST™ 3,3′‐diaminobenzidine tablets (DAB Peroxidase Substrate Tablet Set) (D4168; Sigma‐Aldrich). Slides were counterstained with haematoxylin for 2 min following colour separation by 1% acetic acid for 30 s. The reaction products were visualized by the EnVision Detection System (Dako).
In situ hybridization
The slides were deparaffinized in xylene and then pre‐digested with 15 μg·mL−1 proteinase K (for 5–15 min at 37°C). Subsequently, they were prehybridized with 5′‐DIG and 3′‐DIG labelled miRCURY LNA™ Detection probe (has‐miR‐146b‐5p, 5′‐AGCCTATGGAATTCAGTTCTCA‐3′; Exiqon) in hybridization buffer at 50°C for 1 h. Then the slides were washed thoroughly in SCC buffers, followed by incubation with anti‐DIG‐AP Fab fragments. Finally, the slides were counterstained with nuclear fast red and mounted using ProLong Gold solution (Invitrogen).
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). In general, statistical comparisons between two groups were conducted with Student's t‐test, and between multiple groups were determined by one‐way ANOVA with Dunnett's post hoc test. A probability value of 0.05 was considered to be significant. Results are expressed as mean ± SEM.
For miRNA microarray analysis, results are presented as mean ± SD. Data were analysed by one‐way ANOVA, Student's unpaired two‐tailed t‐test or by two‐way ANOVA among multiple groups at a point in time with Bonferroni correction.
Survival curves were plotted by use of the Kaplan–Meier technique and compared with the log‐rank test. Results are expressed as the mean of at least five or more determinations. Statistical analysis was performed by using GraphPad Software 5.0 (GraphPad Software, San Diego, CA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
PEP06 possesses similar activity to endostatin
It is well known that angiogenesis is vital event for tumour growth and metastasis (Folkman, 1971). PEP06 was designed based on the effective motif of endostatin, and as such, it should possess similar anti‐angiogenic activity to the latter. HUVECs can spontaneously form a three‐dimensional tubular capillary‐like network on Matrigel culture. To examine the effect of PEP06 on tubule formation of HUVECs, we performed a tube formation assay in the presence of PEP06 at a concentration of 200 μg·mL−1 and compared its effects with that of the RGD‐sequences‐deleted PEP06 (namely, 24 amino acid or 24aa in this study). PEP06 produced greater inhibition of tube formation than 24aa (Supporting Information Figure S1C, D). Numerous studies have demonstrated that the expression level of VEGF is critical for tumour angiogenesis (Goel and Mercurio, 2013). Therefore, we next examined whether PEP06 affects VEGF expression. As shown in Supporting Information Figure S1E, F, PEP06 significantly inhibited VEGF expression in HUVECs. Compared with PEP06, 24aa also down‐regulated VEGF expression but with much a weaker effect. These results suggest that PEP06 and 24aa both regulated angiogenesis to exert their antitumour activities in vitro. We then further examined the inhibitory effect of PEP06 on tumour angiogenesis using a postoperative recurrent tumour model depicted in Figure 6. Immunohistochemical staining of VEGF with the tumour sections from PEP06‐treated mice showed significantly decreased microvessel density compared with the vehicle group (Supporting Information Figure S1G, H), indicating that PEP06 inhibited tumour growth by inhibiting angiogenesis in xenograft mice.
Figure 6.
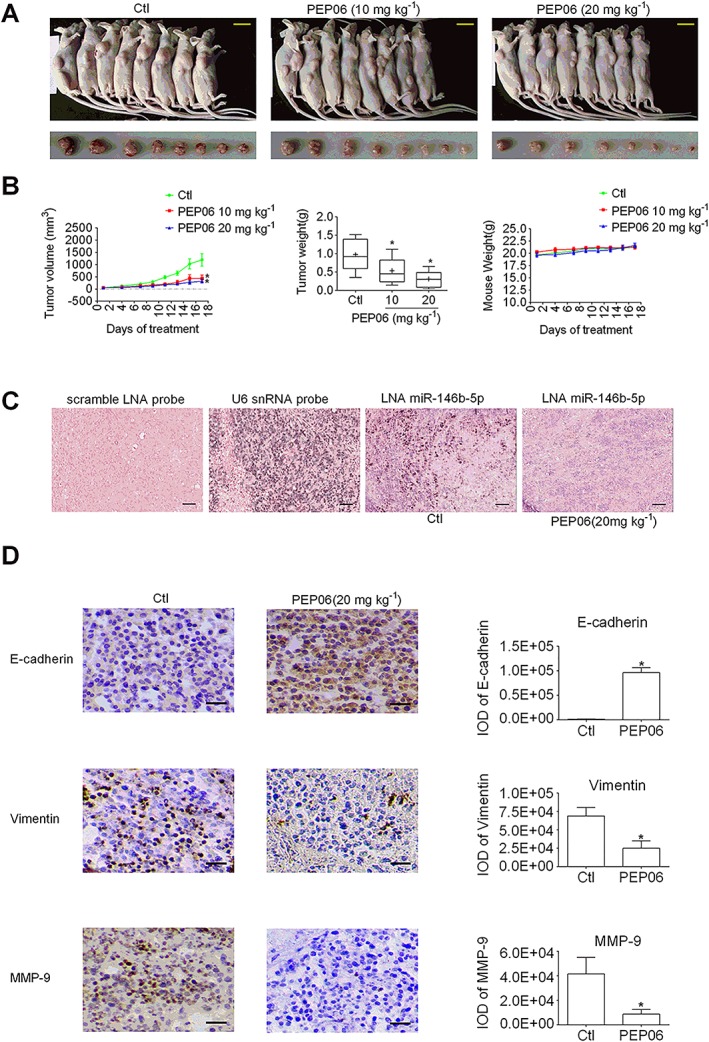
PEP06 inhibits CRC growth by inhibiting miR‐146b‐5p in vivo. (A) Photographs of LOVO residual tumour xenografts treated with 5% glucose or PEP06 (10 and 20 mg·mL−1). Scale bar: 1 cm. (B) Tumour growth curve showing the suppression of growth of SW620 residual tumour xenograft tumours by PEP06. The average tumour weights in the PEP06 groups were reduced, and the mouse weight was unaffected by PEP06. * P < 0.05 by one‐way ANOVA, Dunnett's test: compared with Ctl. (C) Representative in situ hybridization images showing the inverse correlation between PEP06 and the cellular expression of miR‐146b‐5p (dark blue staining). Scale bar: 50 μm. (D) Representative images of tumour sections stained for the EMT‐related and migration markers. Paraffin‐embedded tumour sections were stained for E‐cadherin, vimentin and MMP9 using immunohistochemistry (IHC; brown) on sections to identify tumour cells. Scale bar: 50 μm. IHC results showed that PEP06 can inhibit the expression of vimentin and MMP9 while increasing the level of E‐cadherin. The integral optical density (IOD) of E‐cadherin, vimentin and MMP9 was calculated by IPP. * P < 0.05 by unpaired t‐test: compared with Ctl.
PEP06 suppresses CRC via inhibiting metastasis
Impairing cell viability due to decreased cell proliferation, increased cellular apoptosis and G2/M cell‐cycle arrest are the most common mechanisms for anticancer drugs. As the first step towards understanding the actions of PEP06 in CRC, the effects of PEP06 on cell proliferation and/or death were assessed by measuring the viability of colon cancer cells and HUVECs using the MTT assay. As shown in Figure 1 and Supporting Information Figure S1A, there were no significant differences in the number of viable cells among the PEP06‐treated group, the vehicle‐treated (glucose) negative control group and the 5‐FU (12.5 μg·mL−1)‐treated or ATO (5 nM)‐treated positive control group. Consistently, in vitro experiments demonstrated that PEP06 at concentrations up to 200 μg·mL−1 failed to affect the viability of SW620, HCT116, LOVO and HUVECs cells 24 and 48 h after treatment (Figure 1 and Supporting Information Figure S1A, B). Subsequently, a TUNEL assay was performed to detect apoptotic cells. The TUNEL positive cells (red fluorescence) did not significantly decrease after PEP06 treatment as compared with the control SW620 cells (Supporting Information Figure S2A). Furthermore, we examined whether PEP06 can block cell‐cycle progression in SW620 cells. Flow cytometry analysis showed a similar percentage of cell‐cycle arrest at the G2/M‐phase in PEP06‐treated SW620 cells to that in control cells (Supporting Information Figure S2B). The results indicate that PEP06 does not produce any net effects on cell proliferation, apoptosis or cell‐cycle arrest at the G2/M phase at the concentrations tested in our models. It is possible that PEP06 suppresses CRC by an alternative mechanism; specifically, PEP06 might act on metastasis.
Figure 1.
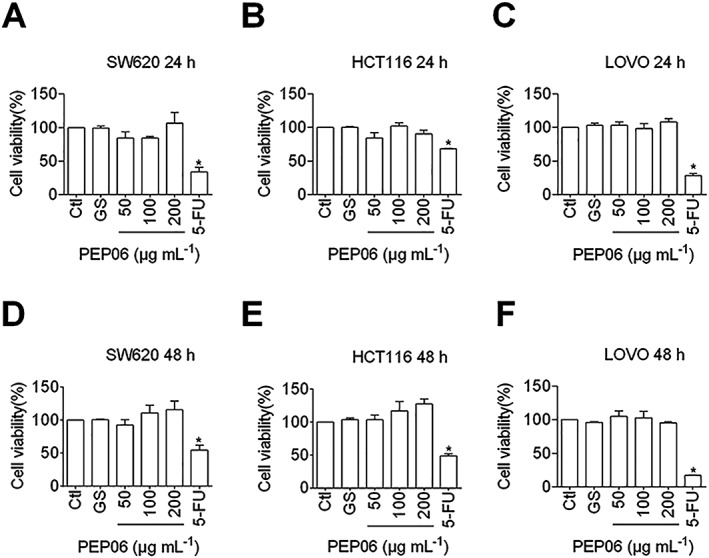
Lack of effects of PEP06 on the viability of SW620, HCT116 and LOVO cells. Relative or % cell viability was determined by MTT assay with the OD values from the PEP06 groups normalized to those from the control groups. The measurements were taken 24 or 48 h after PEP06 treatment. The data show the lack of significant effect of varying concentrations of PEP06 (50, 100 and 200 μg·mL−1) on viability in SW620, HCT116 and LOVO cells at 24 h (A–C) and 48 h (D–F) after treatment. Glucose (GS) solution was used as a vehicle control and 5‐FU (12.5 μg·mL−1) as a positive control. Averaged data (mean ± SEM, n = 5) of OD in 570 nm. * P < 0.05 by one‐way ANOVA; Dunnett's test for comparison with Ctl.
To examine this point, we investigated the effects of PEP06 on the migration of CRC cells by wound‐healing and transwell migration assays. As illustrated in Figure 2A, B, wound‐healing experiments revealed that PEP06 treatments (50, 100 and 200 μg·mL−1) for 24 h substantially inhibited the migration of both SW620 and HCT116 cells (Figure 2A, B, E). Similar results were observed with the transwell migration assay (Figure 2C, D, F). In particular, PEP06 at a concentration of 200 μg·mL−1 led to 87.2 and 92.1% inhibition of migration in SW620 and HCT116 cells, respectively.
Figure 2.
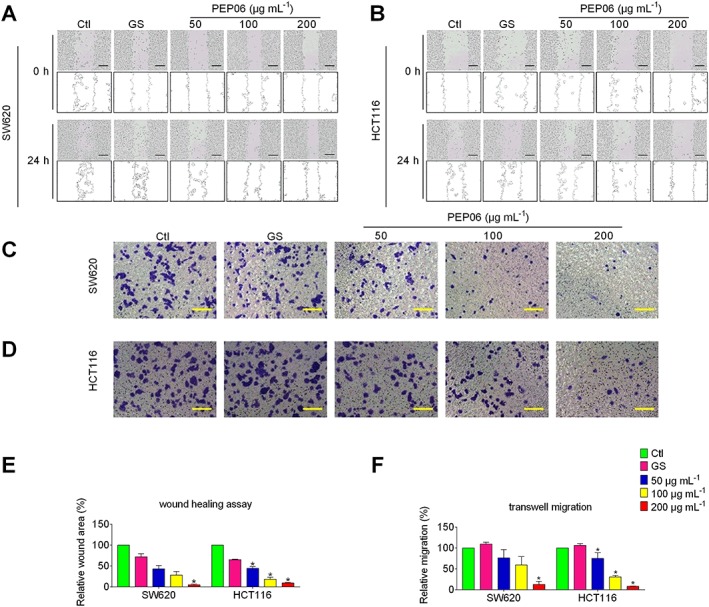
Inhibitory effects of PEP06 on migratory capacity of colon cancer cells SW620 and HCT116. PEP06 significantly inhibited the migration of SW620 cells (A) and HCT116 cells (B), as demonstrated in a wound healing assay. Scale bar: 100 μm. PEP06 significantly inhibited the migration of SW620 cells (C) and HCT116 cells (D), as demonstrated in a transwell migration assay. Scale bar: 50 μm. (E) Averaged data (mean ± SEM, n = 5) from the wound healing assay showing the concentration‐dependent suppression of migration. * P < 0.05 by one‐way ANOVA, Dunnett's test: compared with Ctl. (F) Averaged data (mean ± SEM, n = 5) from transwell migration assay showing the concentration‐dependent suppression of migration. * P < 0.05, by one‐way ANOVA, Dunnett's test: compared with Ctl. GS, glucose.
PEP06 inhibits EMT in CRC cells
EMT is a critical process for aggressive metastatic dissemination of carcinomas (Polyak and Weinberg, 2009), which undergoes multiple and dynamic transitional states from epithelial to mesenchymal phenotypes. Decreases in mesenchymal markers N‐cadherin and vimentin and/or increases in epithelial markers E‐cadherin and α‐catenin indicate dampening of EMT, and vice versa (Nieto et al., 2016). We explored whether EMT plays a role in mediating the anti‐metastasis property of PEP06 by measuring the expression changes of these biomarkers using Western blot analysis. As depicted in Figure 3A, B, PEP06 significantly inhibited EMT at all three concentrations tested, as indicated by the down‐regulation of N‐cadherin and vimentin expression and concomitant up‐regulation of E‐cadherin and α‐cadherin expression in SW620 and HCT116 cells. It was noted that the effects were not concentration‐ dependent (Figure 3D, E). Consistently, immunofluorescent staining demonstrated that PEP06 (200 μg·mL−1) treatment for 24 h resulted in a significant decrease in vimentin expression and an increase in E‐cadherin expression relative to control as well (Figure 3C).
Figure 3.
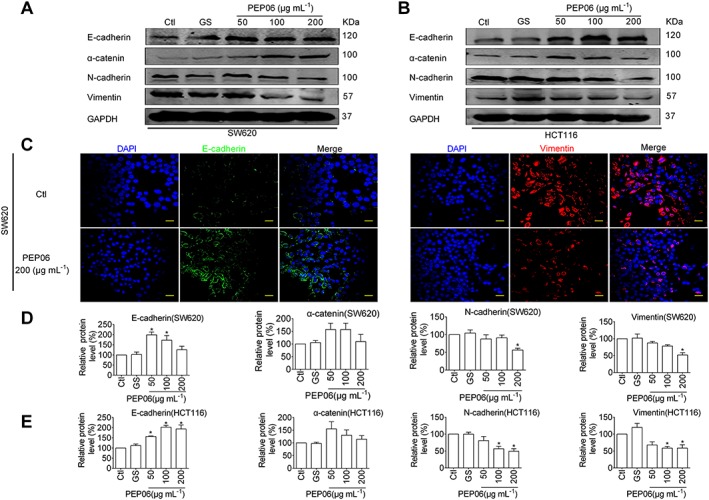
Inhibitory effects of PEP06 on EMT in CRC cells. (A, B) Effects of PEP06 (50, 100 and 200 μg·mL−1) on the protein levels of EMT marker genes in SW620 and HCT116 cells observed by immunoblotting analysis. GAPDH was used as a sample loading control. (C) Effects of PEP06 (200 μg·mL−1) on the protein expression of EMT marker genes in SW620 and HCT116 cells observed by immunofluorescent staining. Blue: DAPI; green: E‐cadherin; and red: vimentin. Scale bar: 20 μm. (D, E) PEP06 increased E‐cadherin and α‐cantenin protein levels and decreased N‐cadherin and vimentin protein levels compared with the negative controls. Averaged data (mean ± SEM) of (A, B) from five independent experiments. * P < 0.05 by one‐way ANOVA, Dunnett's test: compared with Ctl. GS, glucose.
PEP06 decreases the expression of miR‐146b‐5p by direct binding to integrin αvβ3
To elaborate the potential mechanisms by which PEP06 inhibits metastasis of CRC cells and EMT, we set up the following experiments looking at the effects of PEP06 on miRNAs. We first employed miRNA microarray analysis to identify the differentially expressed miRNAs in both SW620 cells and HCT116 cells pretreated with PEP06 at a concentration of 200 μg·mL−1 for 24 h. According to the criteria with a cut‐off fold change >1.5 and P‐value ≤0.05, we found miR‐146b‐5p to be a differentially expressed miRNA (Figure 4A). qRT‐PCR confirmed that the expression level of miR‐146b‐5p was considerably reduced in both SW620 and HCT116 cells exposed to PEP06 of varying concentrations (50, 100 and 200 μg·mL−1; Figure 4B, C). The data are summarized in Table 1.
Figure 4.
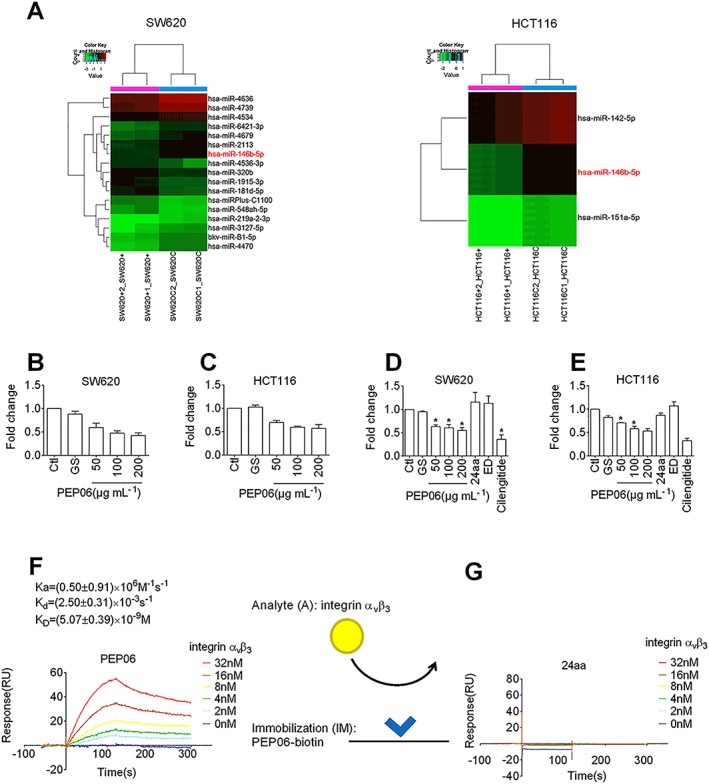
PEP06 down‐regulated the expression of miR‐146b‐5p by forming high‐affinity interactions with αvβ3 integrin in CRC cells. (A) Heat map of microRNA microarray analysis showing the alterations in the expression of miRNAs in SW620 and HCT116 cells exposed to 200 μg·mL−1 PEP06 for 24 h relative to control cells. SW620C1, SW620C2, HCT116C1 and HCT116C2 represent control samples in duplicate, and SW620 + 1, SW620 + 2, HCT116 + 1 and HCT116 + 2 represent PEP06‐treated samples in duplicate. (B, C) qRT‐PCR verification of miR‐146b‐5p down‐regulation in SW620 and HCT116 cells treated with varying concentrations of PEP06. Data (mean ± SEM) were from eight independent experiments. * P < 0.05 by one‐way ANOVA, Dunnett's test: compared with Ctl. (D, E) qRT‐PCR verification of miR‐146b‐5p down‐regulation in SW620 and HCT116 cells treated with PEP06 compared with 24aa, Endostar and cilengitide. Data (mean ± SEM) were from five independent experiments. * P < 0.05 by one‐way ANOVA, Dunnett's test: compared with Ctl. (F) Sensorgrams of αvβ3 integrin (1–32 nM) binding to immobilized PEP06 (50 nM) and K D values calculated. The kinetic parameters were measured by Biacore evaluation software. (G) SPR sensorgrams showing no binding of αvβ3 integrin (1–32 nM) to immobilized 24aa (50 nM).
Table 1.
The differentially expressed miRNAs in SW620 and HCT116 cells following PEP06 treatment (PEP06 treatment group vs. control group)
| ID | Name | Fold change | P‐value | FDR |
|---|---|---|---|---|
| SW620 cells | ||||
| 46324 | hsa‐miR‐320b | 1.73529 | 0.02482 | 0.97188 |
| 169408 | hsa‐miR‐181d‐5p | 1.71659 | 0.0496 | 0.97188 |
| 168616 | hsa‐miR‐4536‐3p | 1.91075 | 0.04687 | 0.97188 |
| 146068 | hsa‐miR‐1915‐3p | 1.67705 | 0.0262 | 0.97188 |
| 169383 | hsa‐miR‐548ah‐5p | 1.77938 | 0.04526 | 0.97188 |
| 17841 | hsa‐miRPlus‐C1100 | 1.88811 | 0.01636 | 0.97188 |
| 10306 | hsa‐miR‐146b‐5p | 0.65933 | 0.00734 | 0.97188 |
| 168942 | hsa‐miR‐4636 | 0.46846 | 0.0124 | 0.97188 |
| 42834 | hsa‐miR‐219a‐2‐3p | 0.59956 | 0.0328 | 0.97188 |
| 169026 | hsa‐miR‐4679 | 0.56884 | 0.0493 | 0.97188 |
| 146179 | hsa‐miR‐2113 | 0.60185 | 0.03082 | 0.97188 |
| 169089 | hsa‐miR‐4470 | 0.58255 | 0.0374 | 0.97188 |
| 146185 | bkv‐miR‐B1‐5p | 0.65649 | 0.00858 | 0.97188 |
| 147996 | hsa‐miR‐3127‐5p | 0.5274 | 0.04515 | 0.97188 |
| 169322 | hsa‐miR‐4534 | 0.63109 | 0.02625 | 0.97188 |
| 168696 | hsa‐miR‐4739 | 0.64513 | 0.01853 | 0.97188 |
| 169059 | hsa‐miR‐642a‐3p | 0.59162 | 0.00809 | 0.97188 |
| HCT116 cells | ||||
| 11260 | hsa‐miR‐151a‐5p | 0.62914 | 0.013 | 0.99974 |
| 10306 | hsa‐miR‐146b‐5p | 0.5499 | 0.01213 | 0.99974 |
| 145798 | hsa‐miR‐142‐5p | 0.65826 | 0.01725 | 0.99974 |
Glucose was added as a control.
We then examined the effects of another two endostatin‐derived short peptides Endostar and the 24aa. The results showed that neither Endostar nor 24aa inhibited the expression of miR‐146b‐5p. By comparison, cilengitide, a cyclic RGD peptide‐targeting integrin αvβ3 and αvβ5, significantly reduced the expression of miR‐146b‐5p (Figure 4D, E). SPR revealed that the biotinylated PEP06 had an appreciable binding affinity for integrin αvβ3. Moreover, PEP06 exhibited a high affinity for integrin αvβ3, which was dose‐dependent, with the mean K d value of 5.07 ± 0.39 nM (Figure 4F), while 24aa failed to bind to integrin αvβ3 (Figure 4G). These results suggest that the regulation of miR‐146b‐5p by PEP06 might be due to the direct binding of PEP06 to integrin αvβ3.
MiR‐146b‐5p mediates the inhibitory effect of PEP06 on EMT in CRC cells
Since miR‐146b‐5p has been shown to be involved in promoting metastasis of CRC (Zhu et al., 2017), we determined whether or not miR‐146b‐5p can regulate the EMT in CRC cells. We overexpressed miR‐146b‐5p in SW620 with lentivirus infection and plasmid vector transfection. Successful introduction of miR‐146b‐5p into the cells was confirmed by qRT‐PCR (Supporting Information Figure S3A). Normal SW620 cells and empty lentivirus (miR‐Ctl) were used as negative controls. Transwell migration assay demonstrated that miR‐146b‐5p overexpression (miR‐146b‐OE) increased the number of SW620 cells passing through the membrane (Figure 5A, B). Meanwhile, miR‐146b‐OE significantly accelerated the migration of these cells (Figure 5D, E). To further understand the impact of miR‐146b‐5p on the CRC metastasis, SW620 cells with or without stable expression of miR‐146b were inoculated into the tail vein of nude mice. Overexpression of miR‐146b‐5p markedly increased the visible pulmonary metastasis compared with the negative control (Supporting Information Figure S3B) and decreased the expression levels of epithelial markers while increasing the levels of mesenchymal markers (Figure 5E, F and Supporting Information Figure S3D). Immunofluorescent staining showed that miR‐146b‐5p overexpression decreased the expression of the epithelial marker E‐cadherin and increased the expression of the mesenchymal marker vimentin in SW620 cells (Figure 5G). However, the EMT‐promoting effect of miR‐146b‐5p was partially reversed by co‐transfection with AMO‐146b (Figure 5E, D). Furthermore, Smad4 protein level was increased by PEP06, and miR‐146b‐5p reversed this alteration (Supporting Information Figure S3E, F).
Figure 5.
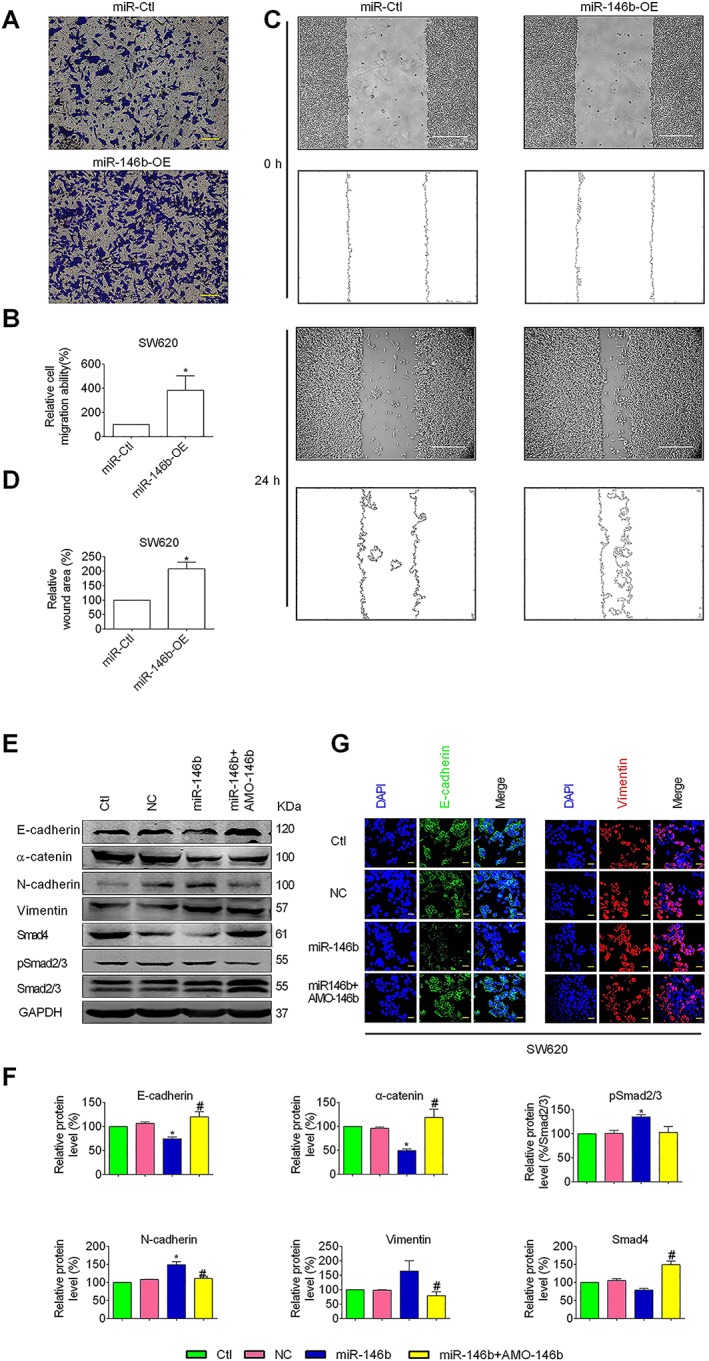
Role of miR‐146b‐5p in mediating the effects of PEP06 on EMT in CRC cells. (A) MiR‐146‐5p and miR‐control expression vectors were each transfected into SW620 cells. The transwell migration assay showing the migration ability of SW620 cells with enforced expression of miR‐146b‐5p (miR‐146b‐OE) compared with the miR‐Ctl. (B) Averaged data (mean ± SEM, n = 5) from the transwell migration assay showing the acceleration of migration by miR‐146b‐5p. * P < 0.05 by unpaired t‐test, compared with miR‐Ctl. (C) The wound healing assay showed that the overexpression of miR‐146b (miR‐146b‐OE) enhanced the migratory capacity of SW620 cells compared with the negative controls (miR‐Ctl). (D) Averaged data (mean ± SEM, n = 5) from the wound‐healing assay showing the acceleration of migration by miR‐146b‐5p. * P < 0.05 by unpaired t‐test, compared with miR‐Ctl. (E) Effects of ectopic expression of miR‐146b‐5p on the protein levels of EMT markers, Smad4 and pSmad2/3 in SW620 cells. Cells were transfected with miR‐146b‐5p mimic, negative control mimic or miR‐146b‐5p mimic together with its antisense inhibitor AMO‐146b. (F) Western blot analysis showed that the level of E‐cadherin, α‐catenin and Smad4 was significantly decreased by the ectopic expression of miR‐146b‐5p, and the level of vimentin, N‐cadherin and pSmad2/3 (% Smad2/3) was increased. Whereas co‐transfection with miR‐146b mimic and AMO‐146b reversed this trend. The values of the protein band densities were normalized to those of the blank control group. Averaged values (mean ± SEM, n = 5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the Ctl groups. * P < 0.05 by one‐way ANOVA, Tukey's multiple comparison test: compared with Ctl. # P < 0.05, by one‐way ANOVA, Tukey's multiple comparison test: compared with miR‐146b. (G) Immunofluorescent staining of EMT marker proteins E‐cadherin (green) was decreased by miR‐146b mimic, and vimentin was increased (red) and DAPI was used to stain (blue) nuclei. Scale bar: 20 μm.
Since Smad4 protein has been reported to be a target for miR‐146b‐5p, we subsequently investigated the effects miR‐146b‐5p on Smad4 in SW620 cells to further understand how miR‐146b‐5p could affect the expression of the EMT marker proteins. Indeed, compared with the control group, Smad4 protein level was decreased in SW620 cells infected or transfected with the miR‐146b‐5p construct, an effect that was prevented by co‐transfection with AMO‐146b. Concomitantly, the levels of pSmad2/3 were elevated by miR‐146b‐5p, which was reversed by AMO‐146b (Figure 5E, F and Supporting Information Figure S3D). These results indicate that miR‐146b‐5p promoted EMT in CRC cells by down‐regulating the expression of Smad4. To further verify the relationship between miR‐146b‐5p and Smad4 in CRC, Smad4 was restored in SW620 cells stably overexpressing miR‐146b‐5p (Supporting Information Figure S3G, H).
PEP06 suppresses CRC tumour growth by inhibiting miR‐146b‐5p in vivo
It is well known that surgical resection accelerates the growth of residual tumours. We next evaluated the antitumour activity of PEP06 in a postoperative recurrent tumour model and its effects on the expression of miR‐146b‐5p. The primary colon tumours were surgically removed leaving a portion of the tumours, 60 mm3, to mimic the postoperative recurrence in the clinic. The average tumour volume, tumour weight and body weights were measured twice a week. Following a single dosage of 10 or 20 mg·kg−1 (body weight) by injection through the tail vein, PEP06, but not 5% glucose, effectively suppressed the growth of residual tumours after surgical resection, with 20 mg·kg−1 showing greater antitumour activity (tumour weight inhibitory rate % = 68.2% and tumour volume inhibitory rate % = 73.3%) than 10 mg·kg−1 (tumour weight inhibitory rate % = 44.7% and tumour volume inhibitory rate % = 64.1%; Figure 6A, B). The antitumour activities of PEP06 are summarized in Table 2. The body weight of nude mice remained stable after PEP06 treatment.
Table 2.
Antitumour activity of PEP06 on postsurgical residual tumour xenografts of human LOVO colon cancer in nude mice
| Drug administration | Toxicity | Anticancer activity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Dose (mg·kg−1) | Schedule | Route | Average body weight (g, x ± s) | Death | Tumour weight (g) | IR (%) | Tumour volume (mm3) | IR (%) | |
| Start | Stop | |||||||||
| Control | – | QD × 17 | i.v. | 19.66 ± 0.67 | 21.26 ± 1.33 | 0/8 | 0.97 ± 0.43 | – | 1197.81 ± 726.72 | – |
| PEP06 | 20 | QD × 17 | i.v. | 19.65 ± 1.29 | 21.61 ± 1.31 | 0/8 | 0.31 ± 0.22 * | 68.2 | 319.36 ± 200.97 * | 73.3 |
| PEP06 | 10 | QD × 17 | i.v. | 20.25 ± 0.96 | 21.28 ± 1.28 | 0/8 | 0.54 ± 0.36 * | 44.7 | 430.30 ± 390.66 * | 64.1 |
The significance of differences (vs. control) was determined by one‐way ANOVA with t‐test. QD, every day.
P < 0.05 (n = 8, x ± s)
Accordingly the antitumour activity of PEP06 was verified in xenograft mice. TMA was performed using the samples from the high‐dosage group. The in situ hybridization results demonstrated that the expression of miR‐146b‐5p was down‐regulated by PEP06 (20 mg·mL−1; Figure 6C). In a follow‐up IHC study, the epithelial marker E‐cadherin was found to be up‐regulated while the mesenchymal marker N‐cadherin down‐regulated by PEP06 (200 mg·kg−1). MMPs, especially MMP2 and MMP9, have been demonstrated to promote tumour progression and are implicated in EMT (Kessenbrock et al., 2010; Vinnakota et al., 2017). In this study, as demonstrated by IHC, the degradation of MMP9 was inhibited by PEP06 in the tumours from the high‐dosage group (Figure 6D).
PEP06 inhibits metastatic capacity in vivo
To demonstrate the efficacy of PEP06 (20 mg·kg−1) at inhibiting the metastasis of colon cancer in vivo, BLI was used to monitor and compare metastatic tumour growth of the CLY mouse model. Five minutes after i.v. injection of GFP‐Luc2‐transfected CLY cells (1.5 × 106 cells per mouse), BLI images were acquired to confirm the accumulation of cells in mouse lungs (Figure 7A, B).
Figure 7.
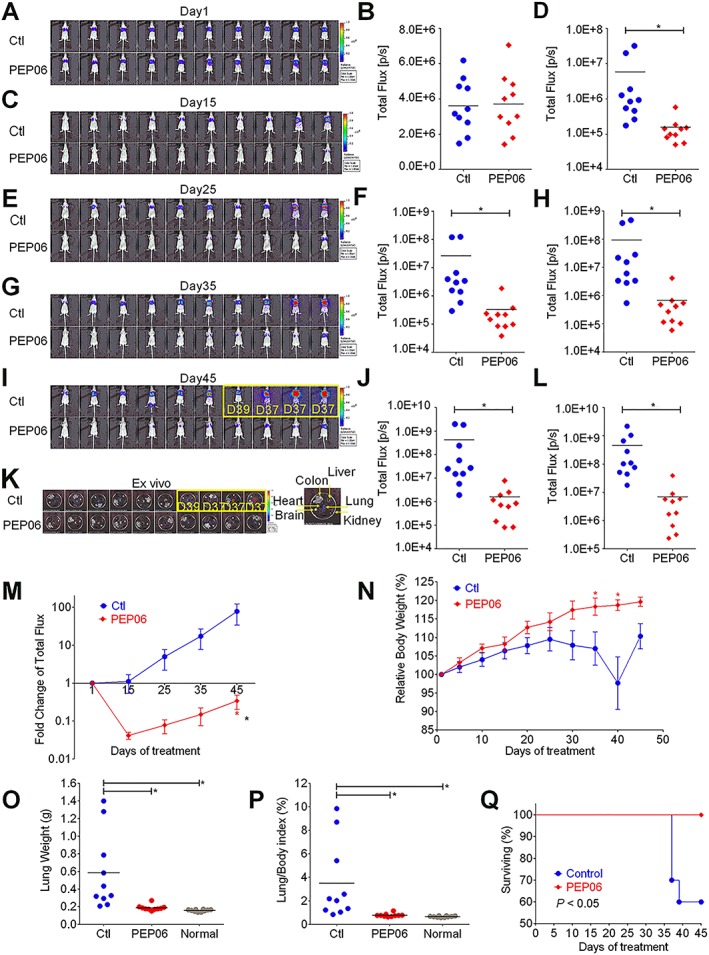
PEP06 inhibits lung metastasis in CRC and increases mouse survival. (A–I) Bioluminescence (BLI) of tumour progression in mice after injection of CLY cells pretreated with glucose (GS) and PEP06 at day 1 (A), day 15 (C), day 25 (E), day 35 (G) and day 45 (I). The statistical charts of the BLI signal intensities at day 1 (B), day 15 (D), day 25 (F), day 35 (H) and day 45 (J). Measurements were automatically generated as an integrated flux of photons (photons s−1; p·s−1) by the LivingImage® processing software. The images framed by a yellow box show the dead mice. Three mice died at day 37, and one died at day 39. During follow‐up, the lung metastasis of CLY cells was significantly hampered in the PEP06‐treated group when compared with that in the Ctl group (GS). * P < 0.05 by unpaired t‐test. (K) Treatment stopped on day 45 for all mice, and ex vivo fluorescence images of the main internal organs were captured on the IVIS® system. More pulmonary metastases were observed in the Ctl group than the PEP06‐treated group. Dissection of all mice did not show metastases in other organs. (L) Bioluminescent signals were quantified by using the IVIS imaging system. * P < 0.05 by unpaired t‐test: compared with Ctl. (M) Line graph showing the fold changes in the total flux of photons for the mice during the treatment. * P < 0.05 by two‐way ANOVA. (N) The relative mouse body weight curve during the treatment with PEP06 compared with the control group. * P < 0.05 by two‐way ANOVA. (O) Quantitative analysis of PEP06 treatment affecting the weight of lung and (P) the lung/body weight ratio. * P < 0.05 by one‐way ANOVA. (Q) Animal survival curve showing the number of live animals during the study and the survival times of mice.
The mice were divided into two groups (n = 10) with equal mean BLI signal intensities. During the first 7 days, most of these signals disappeared over time in both groups compared with day 1 (data not shown), indicating that cells that failed to metastasize did not survive. However, on day 15, all control mice developed obvious bioluminescent signals in the lung. In contrast, only two PEP06‐treated mice showed visible signals (Figure 7C, D). From days 25 to 45, PEP06 significantly attenuated the development of bioluminescent foci in the lung, showing lower BLI signals by two orders of magnitude (Figure 7E–G). At the end of the animal experiment (day 45), the mean in vivo BLI signal intensity of the control group was 77.5‐fold higher than that on day 1, while the PEP06 groups were 2.9‐fold lower (Figure 7I, J, M). The ex vivo images also supported the finding that PEP06 inhibited lung metastasis formation (Figure 7K, L) with inhibition amounting to 98.5%. The lung/body weight ratio is an indirect measure of pulmonary metastatic burden. Quantitative analysis of lung weight (Figure 7O) and lung/body weight ratio (Figure 7P) further confirmed the efficacy of PEP06 in inhibiting cancer metastasis. In addition, PEP06 had no obvious side effects in PEP06‐treated mice, which showed stable increases in body weight during the whole course, while control mice experienced significant body weight loss in the later stage (Figure 7N). Of note, three mice in the control group died on day 37, and one died on day 39, while the mice in the PEP06 groups all survived. The survival time of the mice in the PEP06 groups was significantly prolonged (Figure 7Q). Taken together, PEP06 treatment dramatically delays and attenuates lung metastasis formation in our CLY mouse model.
Discussion
In the present study, we investigated the possible mechanisms of the antitumour activity of PEP06 in in vitro and in vivo models. Our results indicate that as an RGD‐fusion endostatin polypeptide, PEP06 exerted anti‐metastasis effects in CRC via inhibiting EMT as a subcellular mechanism. Mechanistically, PEP06 directly bound to integrin αvβ3 to down‐regulate miR‐146b‐5p and its direct target gene Smad4 to elicit anti‐EMT and anti‐metastasis activities in colon cancer. To our knowledge, PEP06 represents the first endostatin‐derived RGD motif‐including polypeptide that possesses strong miRNA‐targeting EMT‐suppressing properties, resulting in anti‐angiogenic effects in colon cancer.
The present study demonstrated that PEP06 had significant antitumour effects in both a CRC cell model and ex vivo xenografts from a nude mouse model. A modified endostatin with a 9‐amino‐acid‐hanging sequence to form an additional his‐tag structure acts as an angiogenesis inhibitor known as Endostar (Ling et al., 2007), which holds potential to inhibit angiogenesis and lymphangiogenesis in CRC (Jia et al., 2012). A synthetic recombinant endostatin‐derived 27 peptide also elicits strong antitumour, anti‐migration and anti‐permeability activities in lung cancer (Tjin Tham Sjin et al., 2005). Interestingly, PEP06 did not affect cell viability (Figure 1), which was consistent with Endostar exerting small inhibitory effects on the growth of human oesophageal squamous cell carcinoma (Chen et al., 2016a). As an RGD motif was introduced, we assumed that the effect of PEP06 was not simply due to direct tumour cell killing but additional mechanisms are involved, such as anti‐metastasis. The PEP06 dosage used in this study was within the effective range reported by Wu et al. (2008).
Other than the modifications mentioned above, some studies aimed to assemble endostatin with some functional components. In this study, we inserted a consecutive RGD motif into the basic fragment of endostatin. Integrins are key regulators in communication between cells and their micro‐environment (Nieberler et al., 2017). Certain integrins (namely, αIIbβ3, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α5β1 and α8β1) can recognize the RGD motif and affect cell adhesion with strikingly distinct profiles (Nieberler et al., 2017). The importance of αvβ3 integrin in mediating colon cancer growth and metastasis has been demonstrated previously (Reinmuth et al., 2003), as the RGD motif can also be used for tumour treatment (Wang et al., 2013), and several RGD‐modified endostatins have been synthesized in other laboratories. Li et al. (2011) showed that the RGD‐modified endostatin polypeptide 30 produces strong antitumour activities in vitro possibly through the αvβ3 integrin pathway. In another study, RGD‐P125A‐endostatin‐Fc fusion proteins were constructed to increase the half‐life of endostatin and to enhance its antiangiogenic and antitumour activities (Jing et al., 2011). The RGD sequence has also been added to the N‐ or C‐terminus of EDSM (a synthetic polypeptide contains the amino acids 6–48 of endostatin) to enhance the interaction of endostatin with integrin αvβ3, thereby improving its antiangiogenic activity (Pu et al., 2012). An introduction of one RGD motif could be a drug carrier specifically targeting tumour angiogenesis (Mitra et al., 2005; Pu et al., 2012). RGD sequences alone are also able to inhibit cancer metastasis in vitro and in vivo (Saiki et al., 1990; Li et al., 2011) by cross‐talking with the integrin superfamily that mediates cell migration and invasion. Metastasis is a bottleneck in cancer therapy including colon cancer (Ganguly et al., 2013), a process that is associated with EMT (Nieberler et al., 2017). EMT has been implicated in the processes of tumour metastasizing to distant areas and in the acquisition of therapeutic resistance (Polyak and Weinberg, 2009). Shah et al. (2012) reported that a pituitary tumour‐transforming gene has effects on cell migration and the induction of EMT through the regulation of the integrin αvβ3‐FAK signalling pathway in lung cancer. The RGD motif also contributes to TGFβ1‐induced EMT suppression in renal cell carcinoma (Feldkoren et al., 2017). In agreement with these published studies, the present study showed that PEP06 inhibited CRC metastasis in a dose‐dependent manner and significantly restricted the migration of CRC cells (Figure 2). During the follow‐up in the postoperative residual tumour models (An et al., 2014), we found the weight of mice was kept within a relatively stable range during the course of PEP06 treatment, indicating the toxicity of PEP06 is mild (Figure 6). In the present study, PEP06 increased the expression of mesenchymal markers and concomitantly decreased the expression of epithelial markers, which is consistent with the view that EMT is critical to the metastasis of colon cancers (Chen et al., 2016a). Moreover, our xenograft model of LOVO cells clearly demonstrated that PEP06 markedly decreased CRC pulmonary metastasis. Collectively, we have demonstrated that PEP06 can inhibit CRC metastasis by, at least in part, suppressing EMT both in vitro and in vivo.
To our knowledge, there has not been any RGD‐modified drug applied in the clinic until now. The tumour‐homing peptide RGD selectively targets both tumour endothelial cells and tumour cells (Zitzmann et al., 2002) by specifically binding to αvβ3 integrin, which is commonly overexpressed in many cancer cells and can increase tumour targeting efficiency (Park et al., 2016). And integrins (such as αvβ3) recognize RGD sequences in ECM proteins. It has been reported that the amino acids 60–70 of endostatin including surface‐exposed arginine residues can interact with integrins (Wickstrom et al., 2004). We also demonstrated that the RGD motif included in PEP06 possessed αvβ3 integrin‐binding affinity and, thereby, leads to changes in the expression of miR‐146b‐5p (Figure 4F). Although previous studies have demonstrated that the RGD motif can recognize integrins, we are the first to verify the direct binding of an RGD‐modified peptide with integrin. Drugs designed based on the RGD structure may also provide a new treatment for targeting tumours with minimal toxicity (Rubtsov et al., 2016). This assumption was validated by the MTT assay in vitro (Figure 1) and postsurgical residual tumour xenografts animal models in vivo (Figure 6B). Possibly due to the introduction of RGD, PEP06 could effectively target tumour cells and inhibit the growth of colon cancer. Our results support the notion that RGD‐modified endostatin might open up a new and more effective regimen for cancer therapy.
Our study further revealed that miR‐146b‐5p is a key regulator for the underlying mechanisms of anti‐EMT‐mediated metastasis of PEP06 as demonstrated by the significant down‐regulation of miR‐146b‐5p after PEP06 treatment (Figure 4A, B). This regulation of PEP06 tightly relied on the existence of the RGD motif as well as the expression of integrin αvβ3 in CRC cells, as the treatment with Endostar or 24aa showed no effects on the level of miR‐146b‐5p, while the RGD peptides cilengitide inhibited miR‐146b‐5p to a greater extent relative to the peptides without an RGD motif (Figure 4). This suggests that RGD sequences mediate the effect of PEP06 on miR‐146b‐5p; however, the detailed mechanisms by which the RGD sequence down‐regulates miR‐146b‐5p are still not clear. A number of studies have shown that miR‐146b‐5p plays an important role in the initiation and development of colon cancer (Kanaan et al., 2012; Ranjha et al., 2015) through either regulating pyruvate dehydrogenase B (Zhu et al., 2017) or inducing EMT (Deng et al., 2015). In our study, miR‐146b‐5p significantly promoted EMT phenotypes in cells, which is consistent with the research of Zhu et al. (2017), indicating that at least in CRC, miR‐146b‐5p has an oncogenic role. Furthermore, our findings suggest Smad4 as a molecular basis for the modulating effect of miR‐146b‐5p on EMT, as revealed by Geraldo's group (Geraldo et al., 2011). Smad4 associates with the phosphorylated receptor‐regulated Smads (pSmads) and mediates TGFβ signal transduction, a pathway that plays a key role in regulating EMT of cancer cells (Bardeesy et al., 2006). As demonstrated by the elevated level of pSamd2/3 after miR‐146b‐5p treatment, Smad4 increased the responsiveness of the TGFβ pathway (Figure 5 and Supporting Information Figure S3).
In conclusion, this study elucidated a mechanism by which PEP06 exerts its anti‐metastasis effects in CRC; it appears to inhibit EMT through down‐regulating miR‐146b‐5p, revealing an important role of miR‐146b‐5p in the regulation of Smad4 and thereby TGFβ. A schematic diagram is provided in Figure 8 for explaining our findings. PEP06 also appeared to have an ameliorative effect, combating the recurrence of post‐resection colon cancer. We speculate that PEP06 could improve the life quality of late‐stage CRC patients with few and tolerable side effects.
Figure 8.
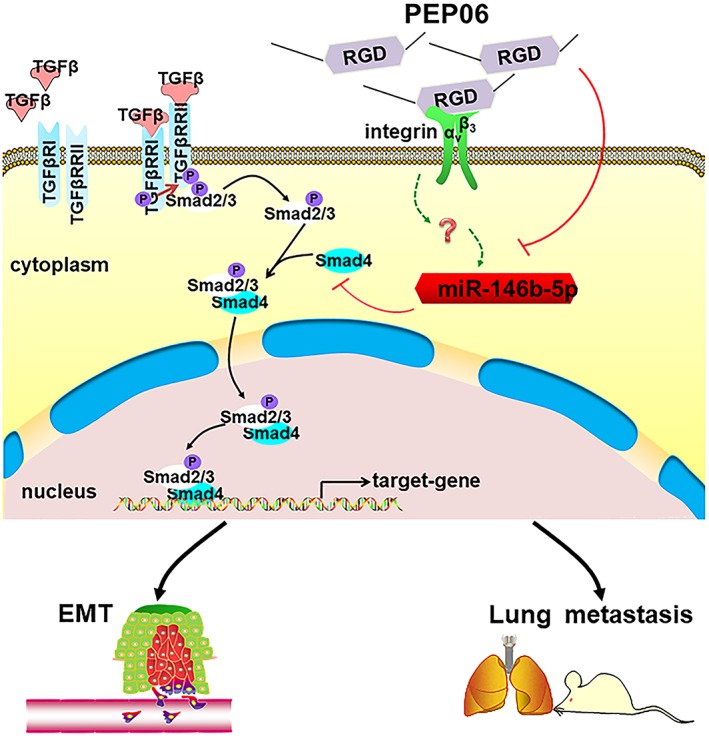
Schematic diagram illustrating the mechanism for the anticancer effect of PEP06. PEP06 exerts anti‐metastasis effects in CRC via inhibiting EMT and increases mouse survival times. This was possibly mediated by the ability of PEP06 to inhibit miR‐146b‐5p, indicating an important role of miR‐146b‐5p in the regulation of Smad4 and TGFβ.
Author contributions
B.Y. designed the experiments. S.Y. composed the draft and analysed the data. W.‐F.C. and Z.D. revised the manuscript. X.L. and X.W. assisted in the technical work of synthesizing PEP06. S.Y., W.T. and W.M. conducted the most experiments. L.L. and F.Q. performed the animal models about PEP06. D.N. were responsible for HUVECs culture. The SPR studies were performed by Y.L.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Oligonucleotide and primer sequences.
Figure S1 The anti‐angiogenic property of PEP06. Lack of effects of PEP06 on viability of HUVEC cells. Relative or percent cell viability was determined by MTT assay with the OD values from the PEP06 groups normalized to those from the control groups. The measurements were taken 24 h or 48 h after PEP06 treatment. Lack of effect of different concentrations of PEP06 (50, 100, and 200 μg mL‐1) on viability in HUVECs at 24 h (A) and 48 h (B) after treatment. Glucose solution was used as a vehicle control and ATO (5 nM) was employed as a positive control. *P<0.05 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (C) Representative photomicrographs of tubule formation in the control, vehicle, PEP06‐treated and 24aa‐treated groups were shown. Cells were plated in 24‐ well plates previously coated with Matrigel and incubated for 6 h at 37°C in the absence of 200 μg mL‐1 peptides. Scale bar: 50 μm. (D) Tubular structures were counted. PEP06 inhibited HUVEC tubule formation with a greater efficacy than 24aa. ‘Percentage (%) of control’ is the mean length of tube or mean number of nodes formed as a proportion of that in the control group. ** P<0.01; ***, P<0.001 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (E) HUVECs were treated with a concentration of 200 μg mL‐1 PEP06 or 24aa for 24 h and subjected to Western blot analysis with anti‐VEGF antibody. (F) Western blot analysis showed that the level of VEGF was significantly decreased by both PEP06 and 24aa. PEP06 produced stringer inhibitory effects on VEGF expression than 24aa in HUVECs. The values of protein band densities were normalized to those of control group. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the Ctl groups. ** P<0.01; *** P<0.001 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (G) PEP06 significantly inhibited tumour vessels in LOVO tumour xenografts. Paraffin sections of LOVO tumour tissue were tested by immunohistochemical analysis with anti‐ VEGF antibody. Representative tumour vasculature (brown) from vehicle‐ or PEP06‐ treated mice (20 mg kg‐1) was shown. Scale bar: 50 μm. (H) The Integral Optical Density (IOD) of VEGF was calculated by IPP. *P<0.05, by unpaired t test: compared with Ctl.
Figure S2 Lack of effects of PEP06 on cell apoptosis and cell cycle on SW620 cells. (A) SW620 cells were treated with 5% GS, 200 μg mL‐1 PEP06, 12.5 μg mL‐1 5‐FU (positive control) for 24 h and then stained with the TUNEL assay. PEP06‐treated cells showed no more TUNEL‐positive cells (red fluorescence) in comparison to Ctl cells. Scale bar: 50 μm. (B) Cell cycle analysis in SW620 cells after treated with 200 μg mL‐1 PEP06 for 24 h by flow cytometry, showing the average percentage of cells in G0/G1, S and G2/M phase. (C) There was no significant effect of each cell population in the presence of PEP06. Averaged data (mean ± SEM, n=5) of cell population in PI‐A channels.
Figure S3 MiR‐146b‐5p re‐expression partly abrogates the tumour suppressive effect of PEP06 in CRC. (A) Successful construction of stable expression of miR‐146b‐5p in SW620. SW620 cells (mock), SW620 cells that stably expressing control lentiviral plasmid (miR‐Ctl), and miR‐146b‐5p lentiviral plasmid (miR‐146b‐OE) were analysed by qRT‐PCR. RNU6 was used as an internal control. Data (mean ± SEM) were from five independent experiments. ***P<0.001 by one‐way ANOVA, Dunnett: compared with mock. (B) The SW620 cells stably expressing miR‐146b‐5p (miR‐146‐OE) and negative control (miR‐Ctl) were injected intravenous on the tails of BALB/c mice and the lungs of the mice were dissected at Day 40. The miR‐146‐OE group induced pulmonary metastasis in 6 out of 8 mice (6/8), including 3 cases visible metastatic nodules in the lungs and 3 cases of microscopic nodules. However, miR‐Ctl group induced pulmonary metastasis in 1 cases of microscopic nodules out of 8 mice (1/8). n=8, use one as example. (C) Representative H & E‐stained sections of the lung tissues collected from the miR‐Ctl and miR‐146b‐OE groups. Scale bar: 50 μm (upper), 500 μm (lower). (D) Forced expression of miR‐146b‐5p through stably transfection cell sublines decreased E‐cadherin protein levels and Smad4 protein levels, and increased vimentin levels compared with the blank control group by western blot analysis. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the mock groups. **P<0.01, ***P<0.001 by one‐way ANOVA, Dunnett: compared with mock. (E) The expression of Smad4 was elevated by PEP06 and was abrogated in partial by miR‐146b‐5p mimic. (F)Averaged values (mean ± SEM, n=5) of western blot band density normalized to the internal control GAPDH and to the data obtained from the Ctl groups. **P<0.01 by one‐way ANOVA, Dunnett: compared with Ctl; #P<0.05, by unpaired t test: compared with the PEP06 group. (G) Western blot analysis of Smad4 and EMT‐related markers in the Smad4 rescue experiments in miR‐146b‐5p stable overexpressing SW620 cells. (H) Smad4 and E‐cadherin were upregulated and vimentin was downregulated after Smad4 overexpression. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the LV‐NC groups. *P<0.05, ***P<0.001 by one‐way ANOVA, Dunnett: compared with LV‐NC.
Acknowledgements
This study was supported by the National Key R&D Program of China (2017YFC1307403) and the National Natural Science Foundation of China (nos. 81730012, 81672827, 81472687 and 81773761).
Yu, S. , Li, L. , Tian, W. , Nie, D. , Mu, W. , Qiu, F. , Liu, Y. , Liu, X. , Wang, X. , Du, Z. , Chu, W.‐F. , and Yang, B. (2018) PEP06 polypeptide 30 exerts antitumour effect in colorectal carcinoma via inhibiting epithelial–mesenchymal transition. British Journal of Pharmacology, 175: 3111–3130. https://doi.org/10.1111/bph.14352.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y, Li L, Yang D, Jia N, Xu C, Wang Q et al (2014). Anticancer activity of tuftsin‐derived T peptide in postoperative residual tumors. Anticancer Drugs 25: 857–867. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P et al (2006). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20: 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken CP, Khew‐Goodall Y, Goodall GJ (2015). Network‐based approaches to understand the roles of miR‐200 and other microRNAs in cancer. Cancer Res 75: 2594–2599. [DOI] [PubMed] [Google Scholar]
- Cantini L, Isella C, Petti C, Picco G, Chiola S, Ficarra E et al (2015). MicroRNA–mRNA interactions underlying colorectal cancer molecular subtypes. Nat Commun 6: 8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhang H, Zhu H, Yang X, Yang Y, Yang Y et al (2016a). Endostatin combined with radiotherapy suppresses vasculogenic mimicry formation through inhibition of epithelial–mesenchymal transition in esophageal cancer. Tumour Biol 37: 4679–4688. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Zheng R, Baade PD, Zhang S, Zeng H, Bray F et al (2016b). Cancer statistics in China, 2015. CA Cancer J Clin 66: 115–132. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Wu B, Xiao K, Kang J, Xie J, Zhang X et al (2015). MiR‐146b‐5p promotes metastasis and induces epithelial–mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell Physiol Biochem 35: 71–82. [DOI] [PubMed] [Google Scholar]
- Dkhissi F, Lu H, Soria C, Opolon P, Griscelli F, Liu H et al (2003). Endostatin exhibits a direct antitumor effect in addition to its antiangiogenic activity in colon cancer cells. Hum Gene Ther 14: 997–1008. [DOI] [PubMed] [Google Scholar]
- Feldkoren B, Hutchinson R, Rapaport Y, Mahajan A, Margulis V (2017). Integrin signaling potentiates transforming growth factor‐beta 1 (TGF‐beta1) dependent down‐regulation of E‐cadherin expression – important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp Cell Res 355: 57–66. [DOI] [PubMed] [Google Scholar]
- Folkman J (1971). Tumor angiogenesis: therapeutic implications. N Engl J Med 285: 1182–1186. [DOI] [PubMed] [Google Scholar]
- Folkman J (2006). Antiangiogenesis in cancer therapy – endostatin and its mechanisms of action. Exp Cell Res 312: 594–607. [DOI] [PubMed] [Google Scholar]
- Ganguly KK, Pal S, Moulik S, Chatterjee A (2013). Integrins and metastasis. Cell Adh Migr 7: 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldo MV, Yamashita AS, Kimura E (2011). MicroRNA miR‐146b‐5p regulates signal transduction of TGF‐β by repressing SMAD4 in thyroid cancer. Oncogene 318: 1–13. [DOI] [PubMed] [Google Scholar]
- Goel HL, Mercurio AM (2013). VEGF targets the tumour cell. Nat Rev Cancer 13: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massague J (2006). Cancer metastasis: building a framework. Cell 127: 679–695. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL (1998). Are angiostatin and endostatin cures for cancer? Lancet 351: 1598–1599. [DOI] [PubMed] [Google Scholar]
- Jia Y, Liu M, Huang W, Wang Z, He Y, Wu J et al (2012). Recombinant human endostatin endostar inhibits tumor growth and metastasis in a mouse xenograft model of colon cancer. Pathol Oncol Res 18: 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Lu H, Wu K, Subramanian IV, Ramakrishnan S (2011). Inhibition of ovarian cancer by RGD‐P125A‐endostatin‐Fc fusion proteins. Int J Cancer 129: 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA (2009). The basics of epithelial–mesenchymal transition. J Clin Invest 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan Z, Rai SN, Eichenberger MR, Barnes C, Dworkin AM, Weller C et al (2012). Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease‐associated colorectal cancer. Hum Mutat 33: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141: 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Lee Y, Kim NY, Hwang Y, Hwang B, Min JK et al (2013). Pancreatic adenocarcinoma upregulated factor, a novel endothelial activator, promotes angiogenesis and vascular permeability. Oncogene 32: 3638–3647. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang H, Yuan S, Tian Z, Sun Z (2007). Establishment and characterization of a novel human colorectal cancer cell line (CLY) metastasizing spontaneously to the liver in nude mice. Oncol Rep 17: 835–840. [PubMed] [Google Scholar]
- Li S, Wei J, Yuan L, Sun H, Liu Y, Zhang Y et al (2011). RGD‐modified endostatin peptide 30 derived from endostatin suppresses invasion and migration of HepG2 cells through the alphavbeta3 pathway. Cancer Biother Radiopharm 26: 529–538. [DOI] [PubMed] [Google Scholar]
- Ling Y, Yang Y, Lu N, You QD, Wang S, Gao Y et al (2007). Endostar, a novel recombinant human endostatin, exerts antiangiogenic effect via blocking VEGF‐induced tyrosine phosphorylation of KDR/Flk‐1 of endothelial cells. Biochem Biophys Res Commun 361: 79–84. [DOI] [PubMed] [Google Scholar]
- Marshall E (2002). Cancer therapy setbacks for endostatin. Science 295: 2198–2199. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Mulholland J, Nan A, McNeill E, Ghandehari H, Line BR (2005). Targeting tumor angiogenic vasculature using polymer–RGD conjugates. J Control Release 102: 191–201. [DOI] [PubMed] [Google Scholar]
- Nieberler M, Reuning U, Reichart F, Notni J, Wester HJ, Schwaiger M et al (2017). Exploring the role of RGD‐recognizing integrins in cancer. Cancers (Basel) 9: pii: E116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP (2016). Emt: 2016. Cell 166: 21–45. [DOI] [PubMed] [Google Scholar]
- O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS et al (1997). Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88: 277–285. [DOI] [PubMed] [Google Scholar]
- Paolillo M, Serra M, Schinelli S (2016). Integrins in glioblastoma: still an attractive target? Pharmacol Res 113: 55–61. [DOI] [PubMed] [Google Scholar]
- Park SH, Zheng JH, Nguyen VH, Jiang SN, Kim DY, Szardenings M et al (2016). RGD peptide cell‐surface display enhances the targeting and therapeutic efficacy of attenuated Salmonella‐mediated cancer therapy. Theranostics 6: 1672–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E (1984). Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 309: 30–32. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9: 265–273. [DOI] [PubMed] [Google Scholar]
- Pu CY, Xu HM, Hu JL, Zheng H, Huang XF, Zhang C et al (2012). RGD‐modified endostatin fragments showed an antitumor effect through antiangiogenesis. Anticancer Drugs 23: 788–802. [DOI] [PubMed] [Google Scholar]
- Radisky DC (2005). Epithelial–mesenchymal transition. J Cell Sci 118: 4325–4326. [DOI] [PubMed] [Google Scholar]
- Ranjha R, Aggarwal S, Bopanna S, Ahuja V, Paul J (2015). Site‐specific microRNA expression may lead to different subtypes in ulcerative colitis. PloS one 10: e0142869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinmuth N, Liu W, Ahmad SA, Fan F, Stoeltzing O, Parikh AA et al (2003). Alphavbeta 3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 63: 2079–2087. [PubMed] [Google Scholar]
- Rubtsov MA, Syrkina MS, Aliev G (2016). RGD‐based therapy: principles of selectivity. Curr Pharm Des 22: 925–932. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E (1996). RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12: 697–715. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Pierschbacher MD (1987). New perspectives in cell adhesion: RGD and integrins. Science (New York, NY) 238: 491–497. [DOI] [PubMed] [Google Scholar]
- Saiki I, Murata J, Matsuno K, Ogawa R, Nishim N, Tokura S et al (1990). Anti‐metastatic and anti‐invasive effect of polymeric Arg‐Gly‐Asp (RGD) peptide, poly(RGD), and its analogues. Jpn J Cancer Res 81: 660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Fong MY, Kakar SS (2012). PTTG induces EMT through integrin alphaVbeta3‐focal adhesion kinase signaling in lung cancer cells. Oncogene 31: 3124–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R (2003). Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A 100: 4766–4771. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Tang J, Li Y, Wang J, Wen Z, Lai M, Zhang H (2016). Molecular mechanisms of microRNAs in regulating epithelial–mesenchymal transitions in human cancers. Cancer Lett 371: 301–313. [DOI] [PubMed] [Google Scholar]
- Tjin Tham Sjin RM, Satchi‐Fainaro R, Birsner AE, Sadagopa Ramanujam VM, Folkman J, Javaherian K (2005). A 27‐amino‐acid synthetic peptide corresponding to the NH2‐terminal zinc‐binding domain of endostatin is responsible for its antitumor activity. Cancer Res 65: 3656–3663. [DOI] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A (2015). Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108. [DOI] [PubMed] [Google Scholar]
- Valeri N, Braconi C, Gasparini P, Murgia C, Lampis A, Paulus‐Hock V et al (2014). MicroRNA‐135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 25: 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinnakota K, Zhang Y, Selvanesan BC, Topi G, Salim T, Sand‐Dejmek J et al (2017). M2‐like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J Cell Physiol 232: 3468–3480. [DOI] [PubMed] [Google Scholar]
- Wang F, Li Y, Shen Y, Wang A, Wang S, Xie T (2013). The functions and applications of RGD in tumor therapy and tissue engineering. Int J Mol Sci 14: 13447–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstrom SA, Alitalo K, Keski‐Oja J (2004). An endostatin‐derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J Biol Chem 279: 20178–20185. [DOI] [PubMed] [Google Scholar]
- Wu G, Zhang R, Ren J, Sun Y (2008). Autophagic cell death of human hepatoma cells induced by endostar, a recombinant human endostatin. Cancer Biother Radiopharm 23: 735–740. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Gao R, Wu W, Qin H (2013). Epithelial–mesenchymal transition and its role in the pathogenesis of colorectal cancer. Asian Pac J Cancer Prev 14: 2689–2698. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wu G, Yan W, Zhan H, Sun P (2017). miR‐146b‐5p regulates cell growth, invasion, and metabolism by targeting PDHB in colorectal cancer. Am J Cancer Res 7: 1136–1150. [PMC free article] [PubMed] [Google Scholar]
- Zitzmann S, Ehemann V, Schwab M (2002). Arginine‐glycine‐aspartic acid (RGD)‐peptide binds to both tumor and tumor‐endothelial cells in vivo. Cancer Res 62: 5139–5143. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Oligonucleotide and primer sequences.
Figure S1 The anti‐angiogenic property of PEP06. Lack of effects of PEP06 on viability of HUVEC cells. Relative or percent cell viability was determined by MTT assay with the OD values from the PEP06 groups normalized to those from the control groups. The measurements were taken 24 h or 48 h after PEP06 treatment. Lack of effect of different concentrations of PEP06 (50, 100, and 200 μg mL‐1) on viability in HUVECs at 24 h (A) and 48 h (B) after treatment. Glucose solution was used as a vehicle control and ATO (5 nM) was employed as a positive control. *P<0.05 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (C) Representative photomicrographs of tubule formation in the control, vehicle, PEP06‐treated and 24aa‐treated groups were shown. Cells were plated in 24‐ well plates previously coated with Matrigel and incubated for 6 h at 37°C in the absence of 200 μg mL‐1 peptides. Scale bar: 50 μm. (D) Tubular structures were counted. PEP06 inhibited HUVEC tubule formation with a greater efficacy than 24aa. ‘Percentage (%) of control’ is the mean length of tube or mean number of nodes formed as a proportion of that in the control group. ** P<0.01; ***, P<0.001 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (E) HUVECs were treated with a concentration of 200 μg mL‐1 PEP06 or 24aa for 24 h and subjected to Western blot analysis with anti‐VEGF antibody. (F) Western blot analysis showed that the level of VEGF was significantly decreased by both PEP06 and 24aa. PEP06 produced stringer inhibitory effects on VEGF expression than 24aa in HUVECs. The values of protein band densities were normalized to those of control group. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the Ctl groups. ** P<0.01; *** P<0.001 by one‐way ANOVA; Dunnett for comparison with controls (Ctl). (G) PEP06 significantly inhibited tumour vessels in LOVO tumour xenografts. Paraffin sections of LOVO tumour tissue were tested by immunohistochemical analysis with anti‐ VEGF antibody. Representative tumour vasculature (brown) from vehicle‐ or PEP06‐ treated mice (20 mg kg‐1) was shown. Scale bar: 50 μm. (H) The Integral Optical Density (IOD) of VEGF was calculated by IPP. *P<0.05, by unpaired t test: compared with Ctl.
Figure S2 Lack of effects of PEP06 on cell apoptosis and cell cycle on SW620 cells. (A) SW620 cells were treated with 5% GS, 200 μg mL‐1 PEP06, 12.5 μg mL‐1 5‐FU (positive control) for 24 h and then stained with the TUNEL assay. PEP06‐treated cells showed no more TUNEL‐positive cells (red fluorescence) in comparison to Ctl cells. Scale bar: 50 μm. (B) Cell cycle analysis in SW620 cells after treated with 200 μg mL‐1 PEP06 for 24 h by flow cytometry, showing the average percentage of cells in G0/G1, S and G2/M phase. (C) There was no significant effect of each cell population in the presence of PEP06. Averaged data (mean ± SEM, n=5) of cell population in PI‐A channels.
Figure S3 MiR‐146b‐5p re‐expression partly abrogates the tumour suppressive effect of PEP06 in CRC. (A) Successful construction of stable expression of miR‐146b‐5p in SW620. SW620 cells (mock), SW620 cells that stably expressing control lentiviral plasmid (miR‐Ctl), and miR‐146b‐5p lentiviral plasmid (miR‐146b‐OE) were analysed by qRT‐PCR. RNU6 was used as an internal control. Data (mean ± SEM) were from five independent experiments. ***P<0.001 by one‐way ANOVA, Dunnett: compared with mock. (B) The SW620 cells stably expressing miR‐146b‐5p (miR‐146‐OE) and negative control (miR‐Ctl) were injected intravenous on the tails of BALB/c mice and the lungs of the mice were dissected at Day 40. The miR‐146‐OE group induced pulmonary metastasis in 6 out of 8 mice (6/8), including 3 cases visible metastatic nodules in the lungs and 3 cases of microscopic nodules. However, miR‐Ctl group induced pulmonary metastasis in 1 cases of microscopic nodules out of 8 mice (1/8). n=8, use one as example. (C) Representative H & E‐stained sections of the lung tissues collected from the miR‐Ctl and miR‐146b‐OE groups. Scale bar: 50 μm (upper), 500 μm (lower). (D) Forced expression of miR‐146b‐5p through stably transfection cell sublines decreased E‐cadherin protein levels and Smad4 protein levels, and increased vimentin levels compared with the blank control group by western blot analysis. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the mock groups. **P<0.01, ***P<0.001 by one‐way ANOVA, Dunnett: compared with mock. (E) The expression of Smad4 was elevated by PEP06 and was abrogated in partial by miR‐146b‐5p mimic. (F)Averaged values (mean ± SEM, n=5) of western blot band density normalized to the internal control GAPDH and to the data obtained from the Ctl groups. **P<0.01 by one‐way ANOVA, Dunnett: compared with Ctl; #P<0.05, by unpaired t test: compared with the PEP06 group. (G) Western blot analysis of Smad4 and EMT‐related markers in the Smad4 rescue experiments in miR‐146b‐5p stable overexpressing SW620 cells. (H) Smad4 and E‐cadherin were upregulated and vimentin was downregulated after Smad4 overexpression. Averaged values (mean ± SEM, n=5) of Western blot band density normalized to the internal control GAPDH and to the data obtained from the LV‐NC groups. *P<0.05, ***P<0.001 by one‐way ANOVA, Dunnett: compared with LV‐NC.