

Abstract
The Na+-Cl− cotransporter (NCC) in distal convoluted tubule (DCT) plays important roles in renal NaCl reabsorption. The current hypothesis for the mechanism of regulation of NCC focuses on WNK4 and intracellular Cl− concentration ([Cl−]i). WNK kinases bind Cl−, and Cl− binding decreases the catalytic activity. It is believed that hypokalemia under low K+ intake decreases [Cl−]i to activate WNK4, which thereby phosphorylates and stimulates NCC through activation of SPAK. However, increased NCC activity and apical NaCl entry would mitigate the fall in [Cl−]i. Whether [Cl−]i in DCT under low-K+ diet is sufficiently low to activate WNK4 is unknown. Furthermore, increased luminal NaCl delivery also stimulates NCC and causes upregulation of the transporter. Unlike low K+ intake, increased luminal NaCl delivery would tend to increase [Cl−]i. Thus we investigated the role of WNK4 and [Cl−]i in regulating NCC. We generated Wnk4-knockout mice and examined regulation of NCC by low K+ intake and by increased luminal NaCl delivery in knockout (KO) and wild-type mice. Wnk4-KO mice have marked reduction in the abundance, phosphorylation, and functional activity of NCC vs. wild type. Low K+ intake increases NCC phosphorylation and functional activity in wild-type mice, but not in Wnk4-KO mice. Increased luminal NaCl delivery similarly upregulates NCC, which, contrary to low K+ intake, is not abolished in Wnk4-KO mice. The results reveal that modulation of WNK4 activity by [Cl−]i is not the sole mechanism for regulating NCC. Increased luminal NaCl delivery upregulates NCC via yet unknown mechanism(s) that may override inhibition of WNK4 by high [Cl−]i.
Keywords: DCT, dietary potassium, intracellular chloride, NCC, WNK1, WNK4
INTRODUCTION
The thiazide-sensitive Na+-Cl− cotransporter (NCC) in the distal convoluted tubule (DCT) is responsible for reabsorbing 5–10% of the filtered Na+ load and plays an important role in regulating total body Na+ and extracellular fluid homeostasis. Recent studies show that NCC-mediated NaCl reabsorption in DCT also plays a critical role in renal K+ excretion and the total body K+ homeostasis (16, 25). Renal K+ excretion occurs predominantly via transcellular K+ secretion in the late DCT, CNT, and early CCD (28). The processes include K+ efflux from cell into lumen through apical K+ channels. Na+ reabsorption via the epithelial Na+ channel ENaC depolarizes the apical membrane potential and provides the electrical driving force for cellular K+ exit. The activity of NCC in DCT thus regulates K+ secretion by altering Na+ delivery to ENaC in the downstream DCT, CNT, and CCD.
To maintain K+ homeostasis, the kidney adjusts urinary K+ excretion to match dietary intake. Dietary K+ intake alters distal K+ secretion via both aldosterone-dependent and -independent mechanisms (29, 31). With respect to aldosterone-dependent mechanisms, K+ loading increases circulating levels of aldosterone to increase the activity of ENaC and Na+-K+-ATPase. These effects increase the electrical and chemical driving force for K+ secretion through K+ channels. The effect of dietary K+ on NCC, however, is independent of aldosterone. Dietary K+ loading downregulates NCC to increase Na+ delivery to ENaC to enhance K+ secretion (8, 26, 27). Unlike the effect on ENaC and Na+-K+-ATPase, downregulation of NCC by K+ loading is not mediated by aldosterone. Downregulation of NCC by K+ loading persists in mice with defective synthesis of aldosterone and in mice with knockout of SGK1, a downstream mediator of aldosterone (21, 26). Conversely to the effect of K+ loading, dietary K+ restriction stimulates NCC to diminish Na+ entry via ENaC and to decrease K+ secretion.
Recent studies have suggested that WNK (with-no-lysine [K]) kinases likely mediate aldosterone-independent mechanism of regulation of NCC by dietary K+. The serine-threonine WNK kinase family consists of four members in mammals (32). Mutations of two members, WNK1 and WNK4, in humans cause a hyperkalemic hypertension disease (30). WNK1 and WNK4 activate intermediate kinases, ste20-related proline/alanine-rich kinase (SPAK) or oxidative-stress response kinase-1 (OSR1), to phosphorylate NCC to increase its abundance and the overall activity in DCT (9). WNK kinases bind Cl−, and Cl− binding decreases their catalytic activity (18). It is believed that decreased serum [K+] resulting from dietary K+ deficiency enhances K+ and Cl− exit in DCT cells, lowering the intracellular [Cl−] ([Cl−]i) to activate WNK4 and NCC (1, 24). Results supporting this hypothesis are primarily based on studies in vitro and in cultured cells under nontransporting conditions. Also, in vivo evidence using Wnk4-knockout mice to support the hypothesis remains unsettled. One difficulty is that baseline NCC activity is very low in Wnk4-KO mice (4). Under the condition of low baseline activity, a 100% increase in activity may still be within experimental variations and preclude detection of significance by statistical analysis.
DCT undergoes adaptation in response to changes in the transport activity (6, 11, 12, 17). An increase in NaCl delivery to NCC, such as in the setting of inhibition of Na+ reabsorption in the thick ascending limb by furosemide, induces hypertrophy of DCT and upregulation of NCC. Increased luminal NaCl delivery, unlike low K+ intake, would be expected to increase [Cl−]i. Conversely, inhibition of NCC activity by thiazide results in atrophy of DCT and downregulation of NCC transporter. How activity causes adaptive changes of NCC and DCT is unknown.
It should be emphasized that in DCT cells with active transcellular NaCl flux, a decrease in [Cl−]i under low-K+ diet would create a favorable driving force to increase NaCl entry via NCC, which would mitigate the magnitude of fall in [Cl−]i. Whether the level of [Cl−]i under low-K+ diet is sufficiently low to activate WNK4 to stimulate NCC is unknown. Conceivably, the upregulation of NCC under low-K+ diet could be an adaptive response to increased activity from favorable driving force. Thus the hypothesis that modulation of WNK4 activity by [Cl−]i is important for regulation of NCC in vivo remains unclear. Furthermore, increased luminal NaCl delivery and low K+ intake, conditions expected to increase and decrease [Cl−]i, respectively, both cause upregulation of NCC. These issues prompt us to conduct the current study to ask whether WNK4-[ Cl−]i is the sole mechanism of regulation of NCC.
MATERIALS AND METHODS
Animals and genotyping.
All animal care, maintenance, and experiments were conducted in accordance with the Guide for the Use and Care of Laboratory Animals and approved by IACUC of the University of Texas Southwestern Medical Center at Dallas. Wild-type (WT), Ncc-KO (obtained from mutant mouse regional center) (20), and Wnk4-KO mice used in these studies are in a C57BL/6 and 129 mixed background. For genotyping, mice tail clips were digested overnight in Viagen DirectPCR reagents with 0.2 mg/ml proteinase K at 55°C, heat-inactivated by boiling in water bath for 5 min or at 85°C for 45 min. Genomic DNA was analyzed by using PCR using primers as described below. All genotyping PCR were performed using GoTaq Green Master Mix (Promega, Madison, WI) and with the setting of hot start at 95°C for 4 min, followed with 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s. For genotyping of Wnk4-KO mice, forward and reverse primer sequences were 5′-ACATAGCTTGGGGACAGGC-3′ and 5′-ATAGTCGACGTCTGGGTCGGAAAGAAACT-3′, respectively. For genotyping of Ncc-KO mice, forward primer sequence was 5′-AGGGTCAAGGGCACGGTTGGC-3′; reverse primer sequences were 5′-GCATGCTCCAGACTGCCTTG-3′ and 5′-GGTAAAGGGAGCGGGTCCGAGG-3′ for mutant and wild-type allele, respectively.
Metabolic cage studies.
Before setting up for physiological studies, mice had ad libitum access to water and normal rodent chow (control K diet, CK; Harlan TD.88238) containing 0.3% (g/100 g food) Na+, and 1% K+. In K+-deficient diet (15–30 ppm K+, LK; Harlan TD.95006) studies, mice were fed with LK diets for 7 days before being placed in metabolic cages for urine collection. During diuretic studies, mice were pair-fed with 2 g of food daily for 2 days before the drug administration at time 0, and then continuously pair-fed for another day after the treatment. Mice had free access to water at all times. Hydrochlorothiazide [HCTZ, Sigma-Aldrich (St. Louis, MO), 50 mg/kg body wt] and furosemide (Sigma-Aldrich, 5 mg/kg body wt) were prepared in 0.9% NaCl with 2.5% DMSO solution, sterilized by filtering, and delivered to mice via subcutaneous or intraperitoneal injection as indicated. Urine samples were collected 24 h (Pre-) before, within 4 h (0–4 h) after, and 4–24 h after the drug or vehicle administration. Each mouse was removed from the metabolic cage and placed individually in a clean shoebox-sized plastic container for at least 5–10 min until a spontaneous urination occurred or were given a bladder massage to ensure an empty bladder at the end of each collection point. Urine Na+ and K+ concentration was measured using flame photometer (Jenway, PFP 7). In NaCl loading studies, 0.9% NaCl was injected to mice subcutaneously twice daily for 3 consecutive days before mice were subjected to experimentation. To avoid potential K+ depletion kaliuresis, drinking water for mice during the NaCl loading period contains 0.1% KCl.
Western blot analysis.
Kidneys were harvested from mice at indicated time points. Total kidney protein extracts were obtained by homogenizing kidney in chilled lysis buffer containing 250 mM sucrose, 10 mM triethanolamine (Sigma-Aldrich), 50 mM NaF, 1 mM EDTA, 1 mM EGTA, complete protease inhibitor cocktail (Roche, Indianapolis, IN), and PhosSTOP (Roche, Indianapolis, IN). Homogenates were centrifuged at 16,000 g at 4°C for 20 min to remove nuclei and cell debris. Protein concentration in supernatant was determined using Bio-Rad DC protein assay (Bio-Rad, Hercules, CA). Total 40 μg lysates in Laemmli buffer were heated at 60°C for 15 min before loading onto a 6–8% SDS-PAGE gradient gel. After electrophoresis, separated proteins were transferred to polyvinylidene fluoride (PVDF) membrane (Thermo Scientific, Rockford, IL) for Western blot analysis. The membrane was blocked with 10% BSA in TBS buffer containing 0.1% Tween-20 before probing with various antibodies. Antibodies used in Western blot studies were: T-NCC (EMD Millipore, Temecula, CA), p-NCC (T53) (33), and β-actin (Santa Cruz Biotechnology).
Quantitative real-time PCR analysis.
Mouse kidney mRNAs were extracted and then subjected to cDNA synthesis using iScript Reverse Transcription Supermix (Bio-Rad). Real-time PCR was performed with iTaq SYBR Green Supermix on CFX Connect Real-Time System machine (Bio-Rad). PCR cycles were 95°C for 10 s, 60°C for 30 s, and repeated cycles for 39 times. GAPDH was used as internal control for all samples. The primer sequences used for mouse renin analysis were as described (15). Both GAPDH and renin primers were checked for their amplification efficiency, and a single melting curve was observed to ensure a specific amplification in each reaction. The expression of renin in samples was normalized to the internal control GAPDH gene expression and their relative ratio of expression to GAPDH was analyzed and graphed.
Statistical analysis.
All data are presented as means ± SE. Statistical comparisons between two groups of data were made using a two-tailed unpaired Student’s t-test. Multiple comparisons were determined using one-way ANOVA followed by Tukey’s multiple-comparison tests. P values of <0.05 and <0.01 were considered significant for single and multiple comparisons, respectively.
RESULTS
Validation of thiazide-sensitive increases in urinary Na+ excretion as readout of NCC transporter activity in vivo.
The most direct way to assess NCC activity is to measure thiazide-sensitive Na+ reabsorption by in vivo microperfusion of DCT. Unfortunately, it is virtually impossible to perform in vivo microperfusion in mice due to the short length of the surface DCT loop accessible for perfusion. An increase in urinary Na+ excretion in response to thiazide has been established as an alternative measurement for functional NCC activity in vivo. Many of these studies have employed intraperitoneal injection of thiazide (3, 33). However, we found that intraperitoneal injection of hydrochlorothiazide (HCTZ, 50 mg/kg body wt) induces a brisk diuresis and natriuresis as expected, but does not lead to kaliuresis (not shown). A recent paper reported a similar finding of lack of kaliuresis following intraperitoneal delivery of HCTZ (10). Because K+ reabsorption in proximal tubules parallels Na+ and water reabsorption, we considered the possibility that thiazide-induced volume loss may cause compensatory K+ reabsorption in the proximal tubules. Our pilot experiments revealed that HCTZ induced a net loss of Na+ and water equivalent to that in 0.5 ml normal saline. We thus administered HCTZ along with 0.5 ml normal saline. Despite repletion of volume loss, we found that lack of kaliuresis persists (Fig. 1A). This lack of kaliuresis is not unique to HCTZ: it also occurs during intraperitoneal injection of furosemide (Fig. 1B). Knowing that mice receiving intraperitoneal injection of saline vehicle have a rapid rise of an injury marker NGAL (human neutrophil gelatinase-associated lipocalin) (5), and to avoid this potentially untoward effect as the cause of lack of kaliuresis, we designed a protocol for administration of HCTZ along with volume replacement via subcutaneous route.
Fig. 1.

Comparison of natriuretic and kaliuretic response to hydrochlorothiazide (HCTZ) and furosemide administered via intraperitoneal (ip) vs. subcutaneous (sq) route. A and B: HCTZ or furosemide was administered to wild-type mice through ip injection. Vehicle contains 0.5 ml normal saline and 12.5 μl dimethyl sulfoxide (DSMO; solvent for diuretics) to mice (~25 g). The hourly rate of Na+ and K+ excretion during 24-h before the injection (Pre-), 0–4 h, and 4–24 h after the injection is shown; n = 4 each. C: the rate of urinary Na+ excretion in mice administered with vehicle alone (n = 4) or HCTZ (n = 4) through subcutaneous route. The total amounts of Na+ excretion in the urine during each collection period and the daily sums are shown. D: the rate of Na+ and K+ excretion during each collection period from mice injected subcutaneously with HCTZ; n = 8. Mice were pair-fed with a normal-Na+ diet from −2 to +1 days. Diuretics were administered at time 0. *P < 0.05 vs. “Pre” period.
As shown in Fig. 1C (set of 3 open bars on the left), subcutaneous administration of vehicle (0.5 ml NS + DMSO) caused a sustained increase in urinary Na+ excretion over 24 h postadministration. The total Na+ excretion was 257 μmol for the 0–4 h period and 4–24 h period combined. The 24-h Na+ excretion before vehicle (labeled “Pre”) was 149 μmol. Note that Na+ contained in the 0.5 ml NS (77 μmol) was essentially excreted over 24 h (149 + 77 = 226 μmol, roughly ≈ 257 μmol). In contrast, administration of HCTZ in 0.5 ml NS induced a brisk natriuresis within 4 h (0–4 h period), which subsided to the pre-HCTZ level during 4–24 h period (Fig. 1C, set of 3 gray bars on the right). Interestingly, the total 24-h Na+ excretion with HCTZ (165 μmol for period 0–4 h plus 80 μmol for period 4–24 h = 245 μmol) was essentially identical to that with vehicle (257 μmol). Thus these results indicate that coadministration with 0.5 ml NS prevented HCTZ-induced volume contraction; HCTZ induced natriuresis by excreting coadministered NaCl within the first 4 h instead of slowly excreting over 24 h as in vehicle-treated. Using this protocol, we found that subcutaneously administered HCTZ stimulated kaliuresis alone with natriuresis reproducibly (Fig. 1D).
A previous paper by Leviel et al. (13) reported that HCTZ also inhibits Na+-driven Cl−/- exchanger (NDCBE/SLC4A8). In the paper, authors demonstrated that HCTZ (given by ip injection at 50 mg/kg) caused an increase in urinary Na+ excretion in Ncc-KO mice. The effect, however, occurs in 6–12 h after HCTZ, but not in the period 0–6 h after HCTZ, indicating that the effect on NCC is separate from on NBCDE. To validate the fidelity of HCTZ-sensitive urinary Na+ excretion truly reflecting NCC activity in vivo, we examined urinary Na+ excretion rate during the acute phase (0–4 h) of HCTZ administration in wild-type vs. Ncc-KO mice (Fig. 2) (20). Genotyping and Western blot analysis confirmed deletion of Ncc and lack of NCC protein expression in Ncc-KO mice (Fig. 2, A–C). As shown in Fig. 2D, HCTZ administrated using our protocol did not cause natriuresis in Ncc-KO mice within 0–4 h while inducing brisk natriuresis in wild type. Thus HCTZ-induced Na+ excretion within 4 h after HCTZ is a good assessment of functional NCC activity in our experimental system.
Fig. 2.

Blunting of HCTZ-induced Na+ excretion in Ncc-knockout mice. A: primer position for genotyping for the targeted Ncc (Na+- Cl− cotransporter) gene. A Neo cassette was inserted into exon 12 of Ncc to disrupt gene function. B: genotyping of WT vs. Ncc-KO mice. C: Western blot analysis of NCC protein in kidney extracts from WT and Ncc-KO mice. D: HCTZ-induced Na+ excretion during the acute phase (HCTZ, from 0 to 4 h) after the drug administration was detected in WT (n = 5) mice, but not in the Ncc-KO (n = 5) mice. In Ncc-KO mice, the level of Na+ excretion after HCTZ was equivalent to vehicle-treated WT mice (n = 6) (indicated by the horizontal line).
NCC activity is markedly reduced in Wnk4-null mice.
To examine the role of WNK4 in the regulation of NCC, we generated global Wnk4-KO (Wnk4−/−) mice by deleting exons 1 and 2 of Wnk4. We removed both exons because exon 2 contains an in-frame ATG. We generated ES cells containing the Wnk4-floxed allele using standard recombineering techniques (14) and a BAC clone containing the mouse Wnk4 gene (Fig. 3A). Global Wnk4-KO mice were generated using ES cells in which floxed regions were excised in the ES cell stage, and confirmed by genotyping using primer sets that detect mutant allele (“CD-IJ”) or wild-type allele (“GH-IJ”) (Fig. 3B).
Fig. 3.

Blunting of HCTZ-induced Na+ excretion in Wnk4-KO mice. A: gene targeting strategy and primer position for genotyping. B: genotyping of Wnk4-KO mice. The “CD-IJ” primer set generates ~500 bp PCR product from the mutant allele, in which the large (>5 kb) DNA fragment flanked by the loxP sites is deleted. This primer set does not generate PCR product from WT allele due to its size. The “GH-IJ” primer set generates PCR product from WT, but not the mutant allele. C: Western blot analysis of total NCC (T-NCC) and phospho-NCC recognized by antibody specific to threonine-53 phosphorylated NCC peptide [p-NCC (T53)] in kidney extracts from WT and Wnk4-KO mice. β-Actin is for loading control. D: HCTZ-induced Na+ excretion during the acute phase (HCTZ) after the drug administration was detected in WT mice, but not in the Wnk4-KO mice. The level of HCTZ-induced Na+ excretion in Wnk4-KO mice was not significantly different from that in vehicle-treated Wnk4-KO or WT mice. n = 15–17 each.
The abundance of total NCC (T-NCC) as well as threonine-53 phosphorylated NCC [p-NCC (T53)] were markedly reduced in Wnk4-KO mice relative to WT littermate (Fig. 3C). A small amount of T-NCC was detectable in the kidney extracts of Wnk4-KO at prolonged exposure. Functional NCC activity in WT and KO mice were compared. HCTZ induced a large increase in urinary Na+ excretion in WT mice above the level of that observed in by vehicle-treated controls (Fig. 3D). The HCTZ-induced increase in Na+ excretion was mostly eliminated in Wnk4-KO mice. Whole kidney WNK1 abundance (analyzed by Western blot) was not significantly different between WT and Wnk4-KO mice (measured by densitometry and normalized to WT: 1.01 ± 0.05 vs. 1.15 ± 0.09, n = 4 each; NS). Susa et al. (22) found a slight compensatory upregulation of WNK1 abundance in Wnk4-KO mice, but concluded that it does not contribute significantly to NCC activities. Of note, despite the marked reduction of NCC activity in Wnk4-KO mice, baseline urinary Na+ excretion rate was not significantly different between WT and Wnk4-KO mice (“Pre” in WT vs. “Pre” in Wnk4-KO, Fig. 3D). This finding is likely due to compensatory upregulation of Na+ transporters in upstream and downstream nephron segments, such as NKCC2 and ENaC, in the knockout mice (2). Overall, serum Na+ levels are slightly lower and BUN higher in Wnk4-KO mice relative to WT mice (Table 1), consistent with the notion of mild and compensated hypovolemia in Wnk4-KO mice. These results agree with previous studies using Wnk4-null mice reporting that WNK4 stimulates NCC by increased transporter abundance and phosphorylation (23).
Table 1.
Blood chemistry in WT vs. Wnk4-KO mice
| Na, mmol/l | K, mmol/l | Cl, mmol/l | Ca, mmol/l | Mg, mmol/l | BUN, mg/dl | |
|---|---|---|---|---|---|---|
| WT (n = 10) | 141.5 ± 1.3 | 5.62 ± 0.29 | 117.2 ± 0.76 | 0.59 ± 0.04 | 0.28 ± 0.01 | 13.8 ± 1.4 |
| Wnk4-KO (n = 11) | 137.4 ± 0.9 | 5.02 ± 0.20 | 109.7 ± 0.93* | 0.53 ± 0.01 | 0.24 ± 0.01* | 19.1 ± 2.09 |
Values are means ± SE.
P < 0.05 WT vs. Wnk4-KO.
WNK4 is essential for upregulation of NCC stimulated by low dietary potassium intake.
Mice fed a K+-deficient diet have increased abundance of the total and phosphorylated NCC in DCT (24). We examined the role of WNK4 in the upregulation of NCC by low K+ intake. Mice were pair-fed on a control K+ diet (“CK”; 1% K+ in g/100 g food) or K+-deficient diet (“LK”; ~0% K+) for 7 days. Feeding a K+-deficient diet caused hypokalemia in WT mice (Fig. 4A; 5.03 ± 0.14 mM control-K+ vs. 4.44 ± 0.21 mM low-K+), which was more pronounced in Wnk4-KO mice (4.76 ± 0.13 vs. 4.01 ± 0.24). Serum Na+ levels were not different in WT and Wnk4-KO mice under either control-K+ or K+-deficient diet (Fig. 4B). K+-deficient diets increased the abundance of both T-NCC and p-NCC (T53) in WT mice (Fig. 4C). The K+ deficiency-induced increase in abundance of T-NCC and p-NCC was absent in Wnk4-KO mice (Fig. 4D). Consistent with the results of analysis of protein abundance, K+-deficient diets enhanced functional NCC activity reflected by HCTZ-sensitive increases in urinary Na+ excretion in WT, but not in Wnk4-KO mice (Fig. 4E).
Fig. 4.

Potassium-deficient (LK) diet induces NCC upregulation in WT mice, but not in Wnk4-KO mice. A and B: serum K+ and Na+ levels in WT and Wnk4-KO mice under control K+ (CK) or low-K+ (LK) diet. Mice on normal rodent chow were placed on CK or LK diet under pair-feeding for 7 days. *P < 0.05 LK vs. CK; # P < 0.05 between indicated. C and D: kidney extracts were analyzed for the abundance of T-NCC and p-NCC (T53) by Western blotting. Please note that cropped images of control K (CK) and Low K (LK) shown in C are from Western blotting of kidney tissues analyzed in the same SDS-PAGE gel electrophoresis. E: HCTZ-sensitive Na+ excretion was increased by LK diet in WT, but not in Wnk4-KO mice. HCTZ-sensitive Na+ excretion is calculated by subtracting excretion at “Pre” period from excretion at 0–4 h period after HCTZ. n = 16 for WT mice on CK diet, n = 19 for WT on LK diet, n = 17 for Wnk4-KO on CK, n = 19 for Wnk4-KO on LK. *P < 0.05.
WNK4 is not required for upregulation of NCC stimulated by increasing luminal NaCl delivery.
Sustained increases in luminal NaCl delivery to NCC leads to adaptive hypertrophy of DCT and upregulation of NCC (6, 11, 12). One experimental approach to increase luminal NaCl delivery to NCC is inhibition of NaCl reabsorption in the thick ascending limb by furosemide. This approach requires careful volume replacement to prevent potential compensatory upregulation of NCC resulting from furosemide-induced diuresis and volume loss. To simplify the experimental approach for increasing luminal NaCl delivery to NCC, we experimented with a protocol that involves injection of 0.5 ml normal saline subcutaneously twice daily for 3 days. We found that by the end of 3 days of mild NaCl loading, mice reached a new steady-state Na+ balance, and excreted the entire injected Na+ in urine (Fig. 5A: 7.4 μmol/h more Na+ excretion in bar 3 vs. bar 1; 7.4 × 24 h = 177 μmol/day ≈ 155 μmol contained in 1 ml normal saline). At this new steady state, no significant volume expansion occurs as evidenced by comparable levels of renin expression between control and NaCl-loaded mice (Fig. 6). Hematocrit was not significantly different between vehicle and NaCl-loaded mice (47 ± 1 vs. 46.1 ± 0.9, n = 15 each; NS), further supporting that no significant volume expansion by mild NaCl loading. Please note that the amount of NaCl in 1 ml NS is relatively very small compared with high-salt diet (3% Na+ per 100 g diet) commonly used to induce volume expansion in experimental animals (155 vs. ~3,000 μmol/day). As expected from volume contraction as the result of reduced NCC activity, renin expression was upregulated in Wnk4-KO mice vs. WT mice. This finding also confirms the validity of renin expression as an index of volume status. Urinary K+ excretion slightly increased in NaCl-loaded vs. control mice (Fig. 5B). In part due to addition of KCl (0.1%) in the injected normal saline to compensate for mild kaliuresis in NaCl loading, serum K+ levels were not significantly different between NaCl-loaded and control mice (Fig. 5C; see also Table 1).
Fig. 5.

High NaCl loading stimulates NCC activity in both WT and Wnk4-KO mice. A: HCTZ-induced Na+ excretion in WT mice under control or NaCl loading (0.5 ml normal saline per 25 g body wt via subcutaneous injection, twice a day for 3 consecutive days). n = 8 each. B: urinary K+ excretion in WT and Wnk4-KO mice that received no (control) or subcutaneous injection of NaCl. Number inside bars indicates number of mice for each condition. C: serum K+ levels of mice from experiments in B. D: HCTZ-induced Na+ excretion in Wnk4-KO mice under control or NaCl loading. n = 5 each. E: HCTZ-sensitive Na+ excretion rate (0–4 h post-HCTZ administration minus Pre-HCTZ) in WT and Wnk4-KO mice under control or NaCl loading. *P < 0.05 NaCl vs. control for respective strain. #P < 0.05, Wnk4-KO vs. WT. F: Na+ excretion rate 0–4 h post-amiloride administration and pre-amiloride in WT mice under control or NaCl loading. *P < 0.05 Post vs. Pre for respective condition. #P < 0.05 between indicated. Open bar (labeled “∆”) indicates amiloride-sensitive Na+ excretion rate (0–4 h post-administration minus Pre).
Fig. 6.
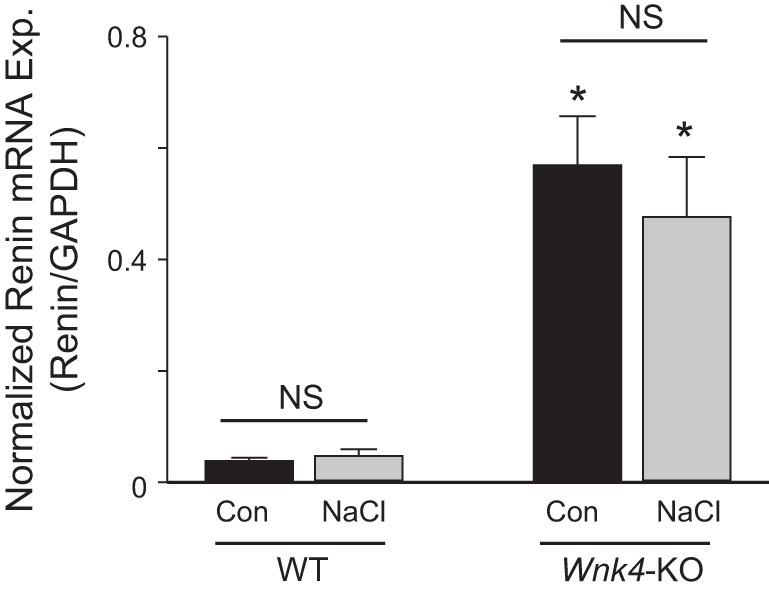
Renin expression in WT and Wnk4-KO mice under control or NaCl loading. Renin mRNA expression was quantified by real-time PCR. The mRNA expression was normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase), an internal housekeeping gene control. n = 5 each group. *P < 0.05, Wnk4-KO vs. WT for respective condition.
Indeed, we found that the NaCl loading protocol induced increases in HCTZ-sensitive Na+ excretion (Fig. 5, A and D). Interestingly, NaCl loading also induced a large increase in HCTZ-sensitive Na+ excretion in Wnk4-KO mice (Fig. 5, D and E). Because of a lower baseline HCTZ-sensitive Na+ excretion in Wnk4-KO, the level of HCTZ-sensitive Na+ excretion during NaCl loading was slightly lower in the Wnk4-KO than in WT mice (Fig. 5E). However, NaCl loading-induced increases in HCTZ-sensitive Na+ excretion were comparable between WT and Wnk4-KO mice. The fact that NaCl loading could enhance HCTZ-sensitive Na+ excretion in Wnk4-KO mice suggests that the lack of stimulation of NCC by low K+ intake in KO mice is due to a specific requirement for the WNK4 signaling cascade, rather than intrinsic defects in NCC preventing it from upregulation. The upregulation of functional NCC activity was parallel with an increase in the abundance of the activated form of NCC, p-NCC (T53), in both WT and Wnk4-KO mice (Fig. 7A). Semiquantitative analysis of the p-NCC (T53) signal intensity showed significant increases upon NaCl loading in both strains of mice (Fig. 7B).
Fig. 7.
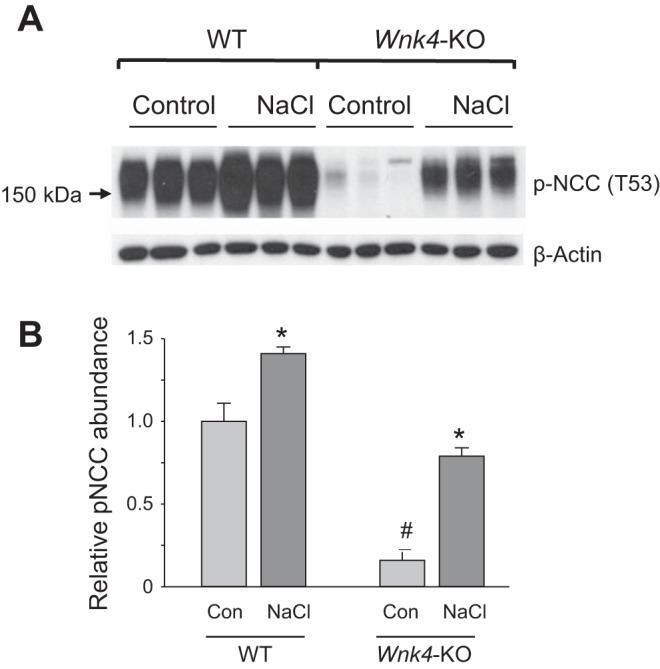
NaCl loading upregulates the abundance of phospho-NCC in WT as well as Wnk4-KO mice. A: Western blot analysis of p-NCC (T53) from kidney extracts of WT and Wnk4-KO mice under control or NaCl loading. B: semiquantitative analysis of the p-NCC (T53) signal intensity normalized to β-actin signal intensity in each lane. *P < 0.05, NaCl vs. control for respective strain. #P < 0.05, Wnk4-KO vs. WT.
Changes in NCC activity in DCT is frequently accompanied by reciprocal changes in epithelial Na+ channel (ENaC) activity in the downstream CNT and CCD segments (20). Consistent with this notion, we found that mild NaCl loading induced a decrease in amiloride-sensitive Na+ excretion rate (Fig. 5F). This adaptive decrease in ENaC activity probably contributes to the overall outcome of minimal volume in mildly NaCl loaded mice.
DISCUSSION
Downregulation and upregulation of NCC are important renal adaptive responses to high K+ intake and deficiency, respectively (16, 25). How dietary K+ intake regulates NCC remains incompletely understood. Recent studies strongly suggest that WNK kinases are involved. Piala et al. (18) reported that two key leucine residues, L369 and L371 in the DLG motif within the kinase domain of WNK1, form a chloride-binding site, and that WNK1 kinase activity is inhibited when chloride is bound to the site. Subsequently, Terker et al. (24) and Bazúa-Valenti et al. (1) demonstrated that changes in extracellular [K+] alter membrane potential and Cl− efflux, leading to changes in [Cl−]i, WNK kinase activity, and phosphorylation and activity of NCC. These studies used cultured cells under nontransporting conditions. Whether changes in [Cl−]i in DCT cells under the low- or high-K+ diet are sufficient to account for observed differences in WNK kinase activity is unknown. Under active transcellular NaCl transport in DCT, coupling between apical NaCl entry to basolateral exit will almost certainly blunt the changes in [Cl−]i compared with nontransporting conditions.
DCT and NCC undergo adaptation in response to changes in NCC transport activity. Sustained activation of NCC-mediated NaCl reabsorption leads to hypertrophy of DCT and upregulation of NCC whereas decreased activity has the opposite effect (6, 11, 12, 17). The molecular mechanism of the activity-associated adaptation of DCT and NCC is unknown. The model of transcellular NaCl reabsorption in DCT predicts that increasing luminal Na+ delivery will have a similar effect of stimulating NCC activity as lowering basolateral [K+] by low-K+ diet (Fig. 8A). Interestingly, although both stimulations increase NCC activity, they have opposite effects on DCT [Cl−]i. Enhanced NaCl entry by increasing luminal NaCl delivery will increase [Cl−]i, whereas lowering basolateral [K+] will decrease [Cl−]i (Fig. 8, B and C). These facts, together with that changes in [Cl−]i under active transport conditions may be too small to affect WNK kinase activity, beg the question whether mechanism(s) other than [Cl−]i regulates WNK kinase activity to affect NCC activity in vivo. Therefore, we set out to examine the effect of increasing luminal NaCl delivery on NCC in Wnk4-KO mice. These experiments allow us to investigate the role of WNK4 in mediating the general mechanism of use hypertrophy as well as to investigate whether low baseline NCC activity in Wnk4-KO mice prevents detection of upregulation of NCC by other stimulations. Our results show that deletion of Wnk4 in mice eliminated NCC upregulation by dietary K+ restriction, but that WNK4 is dispensable for upregulation of NCC stimulated by increasing luminal NaCl delivery. The results provide compelling support for the notion that WNK4 is critical for upregulation of NCC by low K+ intake. The results appear to support the notion that low dietary K+ intake decreases [Cl−]i to activate WNK4. Yet, the fact that increasing luminal NaCl delivery, which if anything is expected to increase [Cl−]i, also induces upregulation of NCC indicates that other additional, WNK4- and [Cl−]i-independent mechanism(s) regulates NCC as well. The additional mechanism can override the effect of inhibition of WNK4 by high [Cl−]i. The result that Wnk4-KO does not affect the additional mechanism of regulation is consistent with the notion.
Fig. 8.

A working model of mechanism of regulation of NaCl reabsorption in distal convoluted tubule (DCT). A: Na+ and Cl− enter the cells via the apical NCC. Na+ is extruded out of cells into circulation by basolateral membrane Na+-K+-ATPase in exchange for K+. K+ is recycled across the basolateral membrane through inward rectifier K+ channel Kir4.1, which provides the electric driving force for parallel Cl− exit via Cl− channel ClC. B and C: both increasing luminal NaCl delivery and lowering basolateral [K+] ([K+]bl) by low-K+ diet stimulate NaCl entry via NCC, but have opposite effects on DCT [Cl−]i. Enhanced NaCl entry by increasing luminal NaCl delivery will increase [Cl−]i, whereas lowering [K+] will decrease [Cl−]i. Enhanced NaCl entry by increasing luminal NaCl delivery will also increase [Na+]i. The effect of low-K+ diet on [Na+]i is less clear, depending on whether the effect of [K+]i on Na+-K+-ATPase to extrude Na+ or effect of [Cl−]i on Na+ entry via NCC is more dominant. Our results apparently support the hypothesis that [Cl−]i-dependent regulation of WNK4 is important for upregulation of NCC by low K+ intake. In the case of enhanced NaCl entry by increasing luminal NaCl delivery, WNK4 is inhibited by high [Cl−]i. Yet NCC is upregulated via unknown mechanism(s).
Previously, Castañeda-Bueno et al. (4) showed that dietary K+-intake induced regulation of NCC (based on analysis of phospho-NCC) is absent in Wnk4-KO mice. However, they found that phosphorylation of SPAK is unaltered in Wnk4-KO. Because of a very low baseline NCC protein in Wnk4-KO mice, they reasoned that phosphorylation of SPAK, the downstream target of WNK4, might be a better readout for WNK4 activity and concluded that WNK4 is not required for dietary K+ regulation of NCC. To address the question whether low baseline NCC activity may confound the interpretation of results, we examined the upregulation of NCC in Wnk4-KO mice by increasing luminal NaCl delivery. We show that increasing luminal NaCl induces upregulation of NCC in Wnk4-KO mice, suggesting that failure for upregulation of NCC by dietary K+ restriction in these mice is specific and due to defects in the WNK4 kinase cascade. Of note, double knockout of Spak and Osr1 in mice abolishes dietary K+ regulation of NCC (7). Phospho-SPAK from the whole kidney extract likely does not reliably reflect the activity of the WNK4 kinase cascade in DCT.
With respect to the identity of WNK kinases in mediating [Cl−]i-sensitive regulation of NCC, Terker et al. (25) showed that in vitro WNK1 is less sensitive to [Cl−]i than WNK4, and suggested that WNK4, but not WNK1, is the principal mediator of regulation of NCC by low-K+ diet. Our finding is consistent with the hypothesis. WNK1 is also present in DCT, particularly the alternative splice variant lacking exon 11 (19, 28). The role of WNK1 and splice variants in the regulation of NCC remains largely unknown. WNK1 splice variants may mediate upregulation of NCC stimulated by increasing luminal NaCl delivery via [Cl−]i-independent mechanism(s) and/or by participating in the regulation by dietary K+ in formation of heteromers with WNK4. In the case of obligatory WNK1 and WNK4 heteromers, deletion of WNK4 will result in a complete loss of function of heteromers. Overall, our present findings support the hypothesis that WNK4 is essential for regulation of NCC by dietary K+, potentially through the mechanism of [Cl−]i-dependent regulation of kinase activity. Definitive evidence for the role of [Cl−]i-dependent regulation of WNK kinases in vivo requires direct measurement of DCT [Cl−]i under active transport condition and/or studies using knockin mice carrying Cl−-insensitive WNK mutants. In vitro, WNK kinase activity is also modulated by changes in [K+], although appears less so when compared with modulation with changes in [Cl−] (18). The possibility that [K+]i contributes to WNK kinase regulation of NCC by K+ diets or activity remains to be investigated. Finally, our study indicates additional aldosterone- and [Cl−]i-independent mechanism(s) for regulating NCC yet to be uncovered.
GRANTS
The study is supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-109887, DK-079328, DK-111542, and DK-59530 and by the National Science Council of Taiwan (100–2314-B-016-018-MY3). C.-L. Huang formerly held the Jacob Lemann Professorship in Calcium Transport and Ruth W. and Milton P. Levy, Sr. Chair in Molecular Nephrology at the University of Texas Southwestern Medical Center, and currently holds the Roy J. Carver Chair in Internal Medicine in the University of Iowa Carver College of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.-S.Y., J.X., S.-S.Y., and C.-L.H. conceived and designed research; Y.-S.Y., J.X., and S.-S.Y. performed experiments; Y.-S.Y., J.X., S.-S.Y., and C.-L.H. analyzed data; Y.-S.Y., J.X., S.-S.Y., S.-H.L., and C.-L.H. interpreted results of experiments; Y.-S.Y., J.X., and C.-L.H. prepared figures; Y.-S.Y., J.X., and S.-S.Y. drafted manuscript; Y.-S.Y., J.X., S.-S.Y., S.-H.L., and C.-L.H. approved final version of manuscript; S.-H.L. and C.-L.H. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Aylin Rodan for comments.
Present address of J. Xie and C.-L. Huang: Div. of Nephrology and Hypertension, Dept. of Medicine, University of Iowa Carver College of Medicine, Iowa City, IA.
REFERENCES
- 1.Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl− cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks HL, Sorensen AM, Terris J, Schultheis PJ, Lorenz JN, Shull GE, Knepper MA. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J Physiol 530: 359–366, 2001. doi: 10.1111/j.1469-7793.2001.0359k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantone A, Yang X, Yan Q, Giebisch G, Hebert SC, Wang T. Mouse model of type II Bartter’s syndrome. I. Upregulation of thiazide-sensitive Na-Cl cotransport activity. Am J Physiol Renal Physiol 294: F1366–F1372, 2008. doi: 10.1152/ajprenal.00608.2007. [DOI] [PubMed] [Google Scholar]
- 4.Castañeda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vázquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christov M, Waikar SS, Pereira RC, Havasi A, Leaf DE, Goltzman D, Pajevic PD, Wolf M, Jüppner H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int 84: 776–785, 2013. doi: 10.1038/ki.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellison DH, Velázquez H, Wright FS. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest 83: 113–126, 1989. doi: 10.1172/JCI113847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferdaus MZ, Barber KW, López-Cayuqueo KI, Terker AS, Argaiz ER, Gassaway BM, Chambrey R, Gamba G, Rinehart J, McCormick JA. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol 594: 4945–4966, 2016. doi: 10.1113/JP272311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang CL, Cheng CJ. A unifying mechanism for WNK kinase regulation of sodium-chloride cotransporter. Pflugers Arch 467: 2235–2241, 2015. doi: 10.1007/s00424-015-1708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter RW, Craigie E, Homer NZ, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 306: F457–F467, 2014. doi: 10.1152/ajprenal.00339.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaissling B, Bachmann S, Kriz W. Structural adaptation of the distal convoluted tubule to prolonged furosemide treatment. Am J Physiol Renal Fluid Electrolyte Physiol 248: F374–F381, 1985. doi: 10.1152/ajprenal.1985.248.3.F374. [DOI] [PubMed] [Google Scholar]
- 12.Kaissling B, Stanton BA. Adaptation of distal tubule and collecting duct to increased sodium delivery. I. Ultrastructure. Am J Physiol Renal Fluid Electrolyte Physiol 255: F1256–F1268, 1988. doi: 10.1152/ajprenal.1988.255.6.F1256. [DOI] [PubMed] [Google Scholar]
- 13.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13: 476–484, 2003. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lübkemeier I, Machura K, Kurtz L, Neubauer B, Dobrowolski R, Schweda F, Wagner C, Willecke K, Kurtz A. The connexin 40 A96S mutation causes renin-dependent hypertension. J Am Soc Nephrol 22: 1031–1040, 2011. doi: 10.1681/ASN.2010101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonough AA, Youn JH. Need to quickly excrete K+? Turn off NCC. Kidney Int 83: 779–782, 2013. doi: 10.1038/ki.2012.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nijenhuis T, Hoenderop JG, Loffing J, van der Kemp AW, van Os CH, Bindels RJ. Thiazide-induced hypocalciuria is accompanied by a decreased expression of Ca2+ transport proteins in kidney. Kidney Int 64: 555–564, 2003. doi: 10.1046/j.1523-1755.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 18.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YP, Butterworth MB, Pastor-Soler NM, Hallows KR, Staub O, Subramanya AR. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015. doi: 10.1172/JCI75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J Biol Chem 273: 29150–29155, 1998. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- 21.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 22.Susa K, Sohara E, Takahashi D, Okado T, Rai T, Uchida S. WNK4 is indispensable for the pathogenesis of pseudohypoaldosteronism type II caused by mutant KLHL3. Biochem Biophys Res Commun 491: 727–732, 2017. doi: 10.1016/j.bbrc.2017.07.121. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi D, Mori T, Nomura N, Khan MZ, Araki Y, Zeniya M, Sohara E, Rai T, Sasaki S, Uchida S. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep 34: e00107, 2014. doi: 10.1042/BSR20140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl− cotransporter. Am J Physiol Renal Physiol 305: F1177–F1188, 2013. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 28.Vidal-Petiot E, Cheval L, Faugeroux J, Malard T, Doucet A, Jeunemaitre X, Hadchouel J. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 7: e37751, 2012. doi: 10.1371/journal.pone.0037751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W. Regulation of renal K transport by dietary K intake. Annu Rev Physiol 66: 547–569, 2004. doi: 10.1146/annurev.physiol.66.032102.112025. [DOI] [PubMed] [Google Scholar]
- 30.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 31.Wingo CS, Seldin DW, Kokko JP, Jacobson HR. Dietary modulation of active potassium secretion in the cortical collecting tubule of adrenalectomized rabbits. J Clin Invest 70: 579–586, 1982. doi: 10.1172/JCI110650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 275: 16795–16801, 2000. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 33.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]