

Abstract
Destabilized heme proteins release heme, and free heme is toxic. Heme is now recognized as an agonist for the Toll-like receptor-4 (TLR4) receptor. This study examined whether the TLR4 receptor mediates the nephrotoxicity of heme, specifically, the effects of heme on renal blood flow and inflammatory responses. We blocked TLR4 signaling by the specific antagonist TAK-242. Intravenous administration of heme to mice promptly reduced renal blood flow, an effect attenuated by TAK-242. In vitro, TAK-242 reduced heme-elicited activation of NF-κB and its downstream gene monocyte chemoattractant protein-1(MCP-1); in contrast, TAK-242 failed to reduce heme-induced activation of the anti-inflammatory transcription factor Nrf2 and its downstream gene heme oxygenase-1 (HO-1). TAK-242 did not reduce heme-induced renal MCP-1 upregulation in vivo. TAK-242 did not reduce dysfunction and histological injury in the glycerol model of heme protein-induced acute kidney injury (AKI), findings corroborated by studies in TLR4+/+ and TLR4−/− mice. We conclude that 1) acute heme-mediated renal vasoconstriction occurs through TLR4 signaling; 2) proinflammatory effects of heme in renal epithelial cells involve TLR4 signaling, whereas the anti-inflammatory effects of heme do not; 3) TLR4 signaling does not mediate the proinflammatory effects of heme in the kidney; and 4) major mechanisms underlying glycerol-induced, heme protein-mediated AKI do not involve TLR4 signaling. These findings in the glycerol model are in stark contrast with findings in virtually all other AKI models studied to date and emphasize the importance of TLR4-independent pathways of heme protein-mediated injury in this model. Finally, these studies urge caution when using observations derived in vitro to predict what occurs in vivo.
Keywords: heme, heme oxygenase-1, MCP-1, sickle cell disease, TLR4 receptor
INTRODUCTION
The tetrapyrrole heme ring is biochemically versatile as it possesses, among other properties, the capacity to transfer electrons, to engage in redox cycling, and to bind gases such as oxygen, nitric oxide, and carbon monoxide (3, 17, 35). As a “free” species, the tetrapyrrole heme ring is also potentially cytotoxic because it can generate oxidants, oxidize lipids, destabilize proteins, perturb cell membranes, induce proinflammatory genes, and damage organelles such as mitochondria (3, 4, 23, 35, 38, 39, 43). Accordingly, in health, cellular concentrations of free heme are very low, the vast bulk of heme being liganded with diverse and distinct protein moieties (3, 17, 35). Members of this heterogeneous heme protein family are diffusely arrayed and functionally diverse; indeed, they are found in all cellular compartments and are involved, in aggregate, in essentially all cellular processes. Heme proteins thus capitalize on the biochemical versatility of heme in enabling their broad range of function, while concomitantly ensuring that heme is entirely contained and not free.
The strength of the ligand between heme and the protein moieties varies substantially among family members, being relatively weak for cytochrome P450 members and robust for cytochrome c. The relative weakness of this ligand in cytochrome P450 proteins is pathophysiologically significant; for example, following ischemic, hypoxic, nephrotoxic, and other insults, destabilization of cytochrome P450 proteins in the intracellular compartment readily occurs, with the attendant release of “free” heme (3, 26, 35, 39). Such increased availability of free heme in the intracellular compartment of the kidney provides one of the common pathways for cellular damage irrespective of the original insult.
Heme proteins in the extracellular compartment may also cause renal injury via heme. In the widely employed glycerol model of heme protein-mediated acute kidney injury (AKI), the intramuscular injection of hypertonic glycerol immediately causes hemolysis and myolysis, with the attendant appearance of hemoglobin and myoglobin in plasma (21, 42). Filtered into the urinary space, these proteins can be destabilized as they are progressively oxidized by hydrogen peroxide present in urine, leading to release of free heme. Filtered heme proteins may also undergo intracellular uptake by the proximal tubule where they are degraded to release heme (21, 42). Interestingly, in addition to the glycerol model of AKI, diverse AKI models (ischemia, sepsis, and cisplatin) exhibit elevated plasma heme levels in the course of AKI (44). Finally, certain mutant heme proteins (for example, hemoglobin S) are inherently unstable and can release heme directly into plasma (2, 3). In sickle cell disease, for example, ongoing hemolysis releases hemoglobin S into plasma where the mutant hemoglobin is autoxidized and denatured, with the ensuing accumulation of heme in plasma (2, 3).
Understanding the basis for the toxicity of increased amounts of extracellular and intracellular heme is thus an important issue in the pathogenesis of tissue injury. In this regard, an important insight was the demonstration that free heme is a ligand for the TLR4 receptor in macrophages; by activating this receptor, heme may thus elicit the innate immune response (13, 14). Such TLR4 receptor activation by heme has been shown for other cells such as microglial cells (18) and endothelial cells (2, 15). Activation of TLR4 receptors on endothelial cells by heme is especially relevant to sickle cell disease where this pathways is involved as a mechanism for acute vasoocclusive disease and the acute chest syndrome (2, 15).
With the use of an in vitro and in vivo approach, the present study was undertaken to determine the role of TLR4 signaling in mediating the toxic effects of heme in the kidney. To interrupt TLR4 signaling, the selective inhibitor of the TLR4 receptor TAK-242 was employed. This compound is widely employed in examining the involvement of TLR4-dependent pathways, and in numerous disease models involving the kidney and other organs, TAK-242 has been shown to reduce such signaling and confer protective effects (1, 2, 7, 11, 12, 16, 33, 41, 46, 47). Because of such efficacy in preclinical studies, TAK-242 was identified as a therapeutic agent meriting clinical evaluation in the treatment of human sepsis (40). However, in a Phase III clinical trial in humans, TAK-242 failed to demonstrate efficacy (29), an outcome that possibly reflects the multifactorial and multiphasic nature of human sepsis and the narrow window in the clinical course of sepsis wherein key events are instigated by TLR4-dependent signaling.
MATERIALS AND METHODS
Studies In Vitro
Rat renal tubular epithelial cells (NRK-52E; ATCC, Manassas, VA) were cultured at 37°C in 95% air–5% CO2 in DMEM (Mediatech, Manassas, VA) containing 4.5 g/l glucose, 10% heat-inactivated FBS (Atlanta Biologicals, Lawrenceville, GA), and the following supplements: 20 mM HEPES, 1 mM sodium pyruvate, 100 U/ml of penicillin, and 100 μg/ml streptomycin (20). For experiments, test medium consisting of the culture medium with reduced FBS content (0.5%) and without sodium pyruvate was used. In studies involving the specific TLR4 signaling inhibitor TAK-242 (Catalog No. CLI-095; Invivogen, San Diego, CA), an initial stock solution was dissolved in DMSO at a concentration of 1 mg/ml and subsequently diluted in test medium to a final concentration of 1 μg/ml. Confluent monolayers of cells were incubated with TAK-242 or an equivalent concentration of DMSO for 30 min before and during exposure to heme (5 μM hemin; Frontier Scientific, Logan UT) for 3, 4, or 8 h. At these time points, cells were harvested for analysis of transcription factor activation, or gene and protein expression, as described below. Similarly, NRK-52E cells were incubated with hemin with or without TAK-242 for 8 h and the production of monocyte chemoattractant protein-1 (MCP-1) in the conditioned media was determined by ELISA as described below.
Studies In Vivo
Mouse studies described here were conducted as approved by the Institutional Animal Care and Use Committee of Mayo Clinic and according to National Institutes of Health guidelines. All mice employed in these studies were obtained from The Jackson Laboratory (Bar Harbor, ME). Male C57BL/6J mice (10–15 wk of age), male C57BL/10ScNJ mice carrying a spontaneous deletion of the TLR4 gene (TLR4−/−, 6 mo of age), and male C57BL/10ScSnJ (TLR4+/+, age-matched control mice for C57BL/10ScNJ strain) were used in studies examining the role of TLR4 signaling in renal responses to heme protein-mediated injury, as described below.
Studies of the role of TLR4 signaling on renal responses to the administration of intravenous heme.
C57BL/6J mice were anesthetized with pentobarbital sodium (50 mg/kg ip) and placed on a heated surface to maintain body temperature. The left kidney and left renal artery were exposed via a midline abdominal incision, and a perivascular flow probe (Transonic Systems, Ithaca, NY) was placed on the renal artery to measure renal blood flow (RBF). After a 30-min equilibration period, a baseline RBF measurement was made over 5 min. Hemin (32 μmol/kg; Frontier Scientific) or saline vehicle was then administered via tail vein injection. RBF was monitored over 30 min following hemin/vehicle administration. A specific inhibitor of TLR4 signaling, TAK-242 (2 mg/kg ip), or vehicle (10% DMSO in saline) was also administered at the time of anesthesia (2).
In other studies C57BL/6J mice were injected with hemin (32 μmol/kg iv) or vehicle via tail vein, with intraperitoneal administration of TAK-242 (2 mg/kg) or vehicle 30 min before and 2 h after hemin injection. Mice were euthanized at 8 h after hemin treatment for assessment of renal gene expression.
Studies of the role of TLR4 signaling in renal responses to heme protein-mediated injury.
TLR4+/+ and TLR4−/− mice were subjected to the glycerol model of acute heme protein-mediated injury as we have previously described (24). Briefly, after overnight dehydration, the mice were anesthetized with an intraperitoneal injection of ketamine and xylazine (90 and 10 mg/kg, respectively) and given intramuscular injection of glycerol (50% in water, 7.5 ml/kg), one-half of the dose into each anterior thigh muscle. Measurements of plasma blood urea nitrogen (BUN) levels, measured using a commercially available kit (Pointe Scientific, Canton, MI), and plasma creatinine (Creatinine Analyzer 2; Beckman/Coulter, Brea, CA) were made at 1 and 2 days after injection. The mice were euthanized for renal histological assessment at day 2 after injection. In complementary studies, C57BL/6J mice were pretreated with TAK-242 (2 mg/kg ip) 30 min before the induction of the glycerol model as described above, employing a dose of glycerol of 5 ml/kg. Additional doses of TAK-242 were administered at 3 h and 1 day after the administration of glycerol. Plasma concentrations of BUN and creatinine were made at 1 and 2 days after the administration of glycerol, and the mice were euthanized at day 2 for assessment of renal histology.
ELISA for MCP-1 Protein Expression
MCP-1 protein production by NRK-52E cells after exposure to hemin was measured in conditioned media using an ELISA kit (BD PharMingen, San Diego, CA), according to the manufacturer’s instructions, and as we have previously described (20). MCP-1 production was normalized for cellular protein measured by the Lowry method (Bio-Rad Laboratories, Hercules, CA).
mRNA Expression by Quantitative Real-Time RT-PCR
Gene expression in mouse kidney and NRK-52E cells was assessed by a two-step quantitative real-time RT-PCR method, as described in our previous study (28). Total RNA, extracted using the TRIzol method (Invitrogen, Carlsbad, CA) and purified with an RNeasy Mini kit (Qiagen, Valencia, CA), was used for cDNA synthesis (Transcriptor First Strand cDNA Synthesis kit; Roche Applied Science, Indianapolis, IN). Quantitative real-time PCR analysis was performed on an ABI Prism 7900HT (Applied Biosystems, Foster City, CA) using TaqMan Gene Expression Assays (Applied Biosystems) for target and housekeeping genes. Expression of 18S rRNA was used for normalization of the expression of each target gene.
Electrophoretic Mobility Shift Assay
Nuclear extracts from NRK-52E cells were prepared using a kit from Active Motif (Cat. No. 40010; Carlsbad, CA). Gel shift analysis for the activation of NF-κB was performed as described for our previous studies (25, 36, 37). Briefly, extracted nuclear protein (10 µg) was used in the binding reaction with a double-stranded consensus oligonucleotide for nuclear NF-κB (Cat. No. E329A; Promega, Madison, WI), which was labeled with [γ-32P]ATP for detection of transcription factor binding. Bound and unbound oligonucleotide were separated on a 5% Tris-boric acid-EDTA nondenaturing polyacrylamide gel (Bio-Rad) and visualized by autoradiography. Supershift analysis was performed with an anti-p65 rabbit polyclonal antibody (Cat. No. sc-109; Santa Cruz Biotechnology, Dallas, TX).
Western Analysis
Western analysis of heme oxygenase-1 (HO-1) protein in whole cell extracts from NRK-52E cells was performed as described in our earlier studies (28, 36, 37). Primary antibodies directed against HO-1 (Cat. no. ADI-SPA-895; Enzo Life Sciences, Farmingdale, NY) and β-Actin (Cat. No. 612656; BD Biosciences, San Jose, CA) were used in overnight 4°C incubations. After incubation with a peroxidase-conjugated secondary antibody, bands were visualized using an enhanced chemiluminescence method. Western analysis of expression of Nrf2 in nuclear extracts from NRK-52E cells was used to assess the activation of this transcription factor in response to heme incubation. Nuclear extracts were prepared as described above, and primary antibodies directed against Nrf2 (Cat. No. sc-722; Santa Cruz Biotechnology) and HDAC1 (Cat. No. 5356, Cell Signaling Technology, Danvers, MA) were employed for these studies.
Statistical Analysis
Results are expressed as means ± SE, and P < 0.05 was considered statistically significant. The Student’s t-test was used for parametric data, and the Mann-Whitney U-test was employed for nonparametric data.
RESULTS
Renal Blood Flow
The administration of heme led to an immediate and significant reduction in RBF that was sustained for 30 min (Fig. 1). Pretreatment with TAK-242 markedly blunted the fall in RBF and significantly so for all time points between 7.5 and 30 min.
Fig. 1.
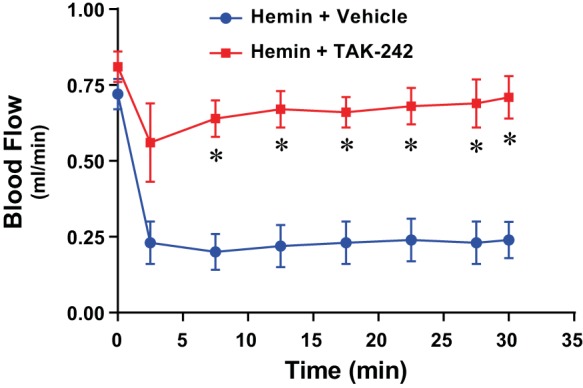
Effect of inhibition of Toll-like receptor-4 (TLR4) signaling on renal blood flow following the intravenous administration of hemin. With the use of a perivascular flow probe, blood flow rate in the renal artery was measured for 30 min after the administration of hemin (32 μmol/kg iv) with pretreatment with the TLR4 signaling inhibitor TAK-242 (2 mg/kg ip) or vehicle; n = 4 in each group. *P < 0.05 vs. vehicle treatment at that time point.
Cellular Production of MCP-1 and NF-κB Activation In Vitro
In addition to renal hemodynamics, renal inflammatory responses elicited by heme were examined as a potential pathophysiological pathway that may be interrupted by TAK-242. We employed MCP-1 expression as a readout for renal inflammation (19, 45). Renal proximal tubular epithelial cells in culture were exposed to heme in the presence and absence of TAK-242. As shown in Figs. 2 and 3, respectively, heme markedly increased expression of MCP-1 mRNA and MCP-1 protein content in the supernatant of heme-exposed cells, and both these effects were significantly reduced by TAK-242.
Fig. 2.
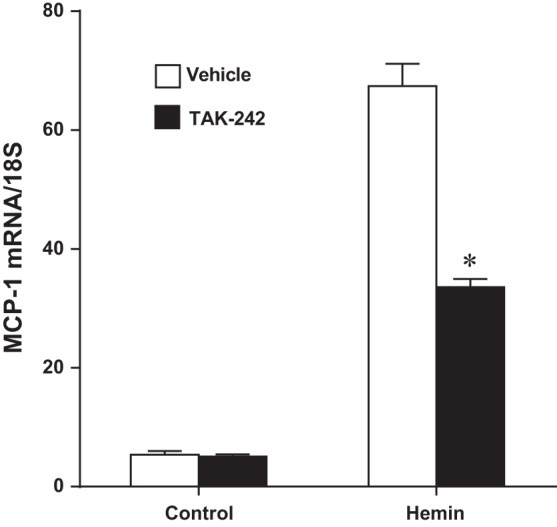
Effect of inhibition of TLR4 signaling on monocyte chemoattractant protein-1(MCP-1) mRNA expression in NRK-52E cells exposed to hemin. MCP-1 gene expression was assessed by quantitative real-time RT-PCR after 8 h of exposure to control media or media containing 5 μM hemin, with or without inhibition of TLR4 signaling with TAK-242; n = 4 in each group. *P < 0.05 vs. corresponding vehicle-treated group.
Fig. 3.
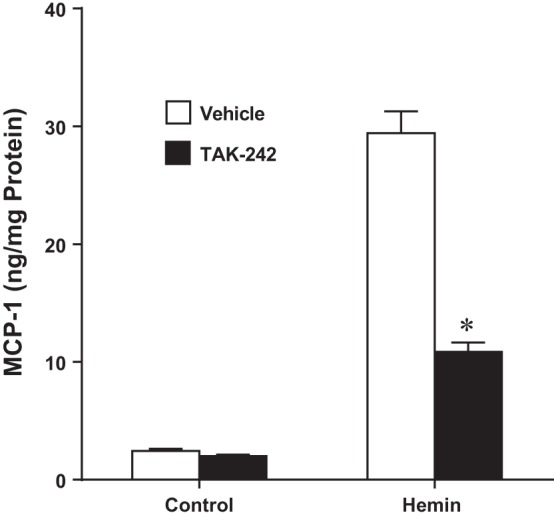
Effect of inhibition of TLR4 signaling on MCP-1 protein production in NRK-52E cells exposed to hemin. Assessment of MCP-1 protein measured in the conditioned media by ELISA after 8 h of exposure to control media or media containing 5 μM hemin, with or without inhibition of TLR4 signaling with TAK-242; n = 4 in each group. *P < 0.05 vs. corresponding vehicle-treated group.
We also undertook studies to confirm that the effects of heme did not reflect contamination with endotoxin. We measured cellular production of MCP-1 protein in response to heme in the absence or presence of polymyxin, a compound that binds endotoxin with high affinity. Cellular production of MCP-1 protein induced by heme was not significantly different in the absence of polymyxin (23.4 ± 1.7 vs. 3.2 ± 0.3 ng/mg protein, n = 4 in each group, P < 0.05) as compared with the production of MCP-1 in the presence of polymyxin (26.4 ± 2.6 vs. 5.1 ± 0.3 ng/mg protein, n = 4 in each group, P < 0.05). Thus the observed effects of heme are not due to contamination with endotoxin.
The main transcription factor that regulates MCP-1 is NF-κB. Studies were thus undertaken to determine whether heme induces NF-κB through a TAK-242-inhibitable pathway. As demonstrated in Fig. 4, heme activates NF-κB in renal epithelial cells, and such activation was completely blocked by TAK-242.
Fig. 4.
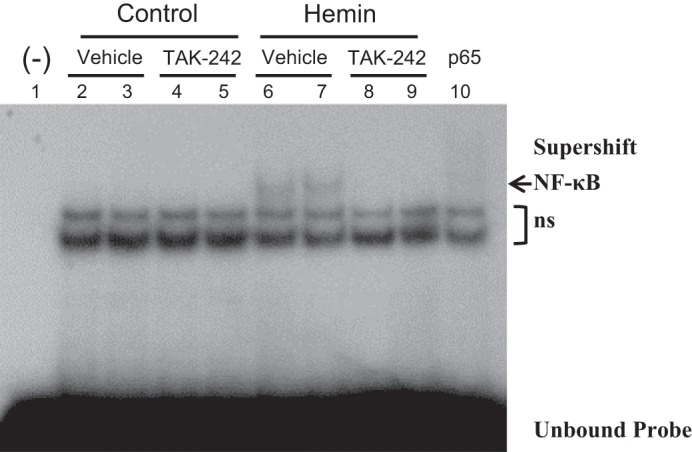
Effect of inhibition of TLR4 signaling on NF-κB activation in NRK-52E cells exposed to hemin. EMSA for the expression of NF-κB was performed using nuclear extracts from NRK-52E cells exposed to control media or media containing 5 μM hemin for 3 h, with or without inhibition of TLR4 signaling with TAK-242. A negative control consisting of a binding reaction in the absence of nuclear extract is displayed in lane 1. Supershifting (lane 10) of the nuclear extract from lane 6 was conducted using a p65 antibody; ns, nonspecific bands.
HO-1 and Nrf2 Expression
Heme robustly induces HO-1. Such HO-1 induction usually confers an anti-inflammatory, cytoprotective response that opposes the proinflammatory response, reflected, for example, by MCP-1 induction (5, 21, 22). Studies were thus undertaken to determine whether induction of HO-1 by heme occurs through a TLR4-dependent pathway. As demonstrated in Fig. 5, TAK-242 prevented neither the upregulation of HO-1 mRNA nor the increased expression of HO-1 protein in heme-exposed cells.
Fig. 5.

Effect of inhibition of TLR4 signaling on heme oxygenase-1 (HO-1) expression in NRK-52E cells exposed to hemin. HO-1 mRNA expression (A) and HO-1 protein expression (B) after 4 h of exposure to control media or media containing 5 μM hemin, with or without inhibition of TLR4 signaling with TAK-242; n = 5 in each group for mRNA analysis in A. Immunoblotting for β-actin was performed for the assessment of equivalency of protein loading for Western analysis in B.
Expression of HO-1 is largely regulated by Nrf2. We thus determined whether heme induced Nrf2 and the extent to which this can be interrupted by TAK-242. As shown in Fig. 6, heme induced nuclear Nrf2 expression, but such expression was not influenced by prior treatment with TAK-242.
Fig. 6.
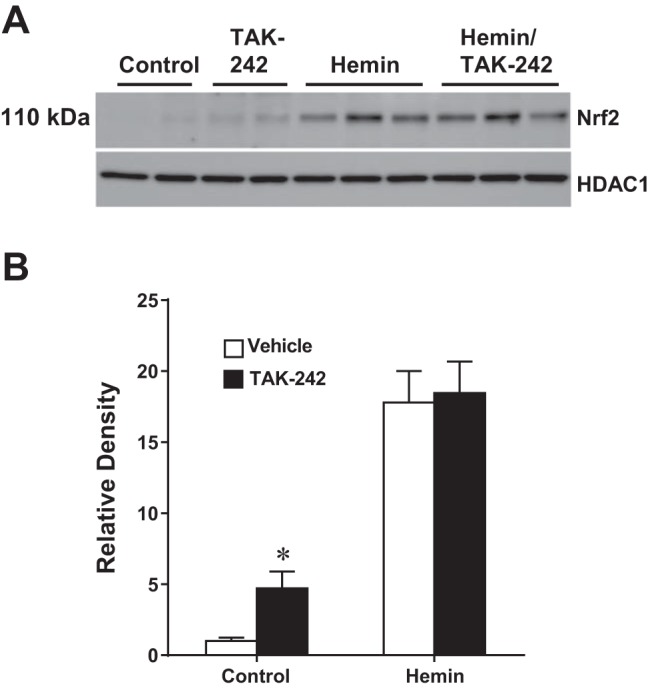
Effect of inhibition of TLR4 signaling on the activation of Nrf2 in NRK-52E cells exposed to hemin. A: representative Western analysis of the nuclear expression of Nrf2 after 3 h of exposure to control media or media containing 5 μM hemin, with or without inhibition of TLR4 signaling with TAK-242. Equivalency of protein loading was assessed by immunoblotting for HDAC1. B: densitometric assessment of the Western analysis of Nrf2 expression; n = 4 in each of the control groups and n = 6 in each of the hemin-treated groups. *P < 0.05 vs. corresponding vehicle-treated group.
Expression of MCP-1 In Vivo
We next determined whether the capacity of TAK-242 to interrupt heme-induced MCP-1 expression in vitro also occurs in vivo. Administration of heme significantly induced renal expression of MCP-1 mRNA at 8 h, the latter entirely unchanged in mice pretreated with TAK-242 (Fig. 7).
Fig. 7.
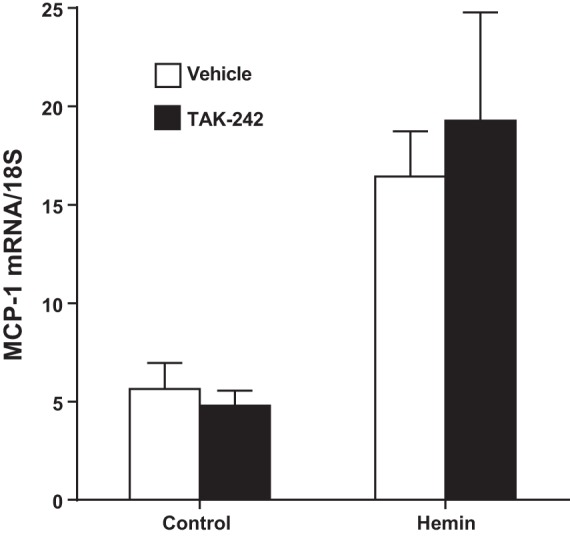
Effect of inhibition of TLR4 signaling on renal MCP-1 mRNA expression in mice subjected to intravenous administration of hemin. Renal MCP-1 gene expression was assessed by quantitative real-time RT-PCR 8 h after the administration of hemin (32 μmol/kg iv) with treatment with either the TLR4 signaling inhibitor TAK-242 or vehicle; n = 5 in each group.
Effect of TAK-242 on Glycerol-Induced, Heme Protein-Mediated AKI
Assessment of renal function by serum creatinine and BUN on 2 successive days after the administration of intramuscular glycerol failed to reveal any functional effect of TAK-242 as compared with vehicle (Fig. 8). Assessment of renal histology 2 days after glycerol-induced AKI showed comparable tubular necrosis, sublethal injury, tubular dilatation, epithelial sloughing, and cast formation in mice treated with TAK-242 or vehicle. Quantitative analysis of these renal histological changes showed no significant difference between the vehicle-treated and TAK-242-treated mice as regards either percent cortical necrosis (33 ± 3 vs. 28 ± 3%, n = 7 in each group, P = NS) or percent extension into the cortex from the cortico-medullary junction (77 ± 9 vs. 69 ± 12%, n = 7 in each group, P = NS).
Fig. 8.

Effect of inhibition of TLR4 signaling on renal function in mice subjected to the glycerol model of heme protein-mediated injury. Plasma levels of creatinine (A) and blood urea nitrgoen (BUN; B) in mice 1 and 2 days after the induction of the glycerol model with treatment with the TLR4 signaling inhibitor TAK-242 or vehicle; n = 5 in each saline-treated control group and n = 8 in each group subjected to the glycerol model.
Glycerol-Induced, Heme Protein-Mediated AKI in TLR4+/+ and TLR4−/− Mice
To corroborate or refute the findings with TAK-242, we examined glycerol-induced, heme protein-mediated AKI in TLR4+/+ and TLR4−/− mice. Assessment of renal function by serum creatinine and BUN on 2 successive days after the administration of intramuscular glycerol showed no significant differences between the groups (Fig. 9); three TLR4−/− mice died between the first and second day after the administration of glycerol, while no deaths occurred in TLR4+/+ mice. Qualitative assessment of renal histological injury at day 2 showed the presence of tubular necrosis, sublethal injury, tubular dilatation, epithelial sloughing, and cast formation in both the TLR4+/+ and TLR4−/− mice. Shown in Fig. 10 are representative histological sections of the kidney at day 2 after the administration of intramuscular injection of glycerol in TLR4+/+ and TLR4−/− mice; tubular necrosis, sublethal injury, epithelial sloughing, and cast formation qualitatively seemed more severe in TLR4−/− mice. Quantitative analysis of these renal histological changes revealed that following the glycerol-induced AKI, TLR4−/− mice, as compared with TLR4+/+ mice, exhibited numerically higher but not significantly different percent cortical necrosis (65 ± 7 vs. 47 ± 6%, n = 4 and n = 7, respectively, P = NS) and percent extension of necrosis from the cortico-medullary junction into the cortex (93 ± 7 vs. 79 ± 8%, n = 4 and n = 7, respectively, P = NS).
Fig. 9.

Renal function in TLR4+/+ and TLR4−/− mice subjected to the glycerol model of heme protein-mediated injury. Plasma levels of creatinine (A) and BUN (B) in TLR4+/+ mice and TLR4−/− mice 1 and 2 days after the induction of the glycerol model; n = 7 in each group at day 1; n = 7 for TLR4+/+ mice and n = 4 for TLR4−/− mice at day 2. Dashed lines in A and B represent the control values for mice with intact kidney function.
Fig. 10.

Renal histological changes in TLR4+/+ and TLR4−/− mice subjected to the glycerol model of heme protein-mediated injury. Renal histological sections 2 days after the induction of the glycerol model in TLR4+/+ mice (A and C) and TLR4−/− mice (B and D) at lower magnification (×100, A and B) and higher magnification (×200, C and D). Tubular necrosis, sublethal injury, epithelial sloughing, and cast formation were present in both groups, such changes seeming more severe in the TLR4−/− mice. Scale bars = 200 µm for A and B and 100 µm for C and D.
DISCUSSION
To the best of our knowledge, the present studies are the first to show that the acute administration of heme leads to an immediate and pronounced reduction in RBF, and this reduction is markedly attenuated by blockade of the TLR4 receptor. Our prior studies in endothelial cells demonstrate that heme leads to the discharge of Weibel-Palade bodies present in endothelial cells and that the engagement of the TLR4 receptor on the endothelial cell surface by heme is critical in eliciting this response (2). Weibel Palade bodies are rich in endothelin-1 (and other vasoconstricting species), and such release of endothelin-1 is implicated in vasoconstriction induced by certain agonists (8, 31, 32). We thus suggest that the immediate reduction in RBF induced by heme reflects the activation of the TLR4 receptor and the discharge of endothelial Weibel Palade bodies and that the vasoconstriction so induced may be endothelin-1 dependent. Heme has a high affinity for nitric oxide and thereby may siphon off nitric oxide from the vasculature with attendant vasoconstriction. This mechanism involves the diffusion of nitric oxide down its concentration gradient and its binding to heme in plasma; it does not require the engagement of the endothelial TLR4 receptor by heme. As much of the observed heme-induced renal vasoconstriction was reduced by TAK-242, we suggest that the binding of nitric oxide by heme does not substantially contribute to this heme-induced renal vasoconstrictive response.
Prior studies examining the renal hemodynamic effects of heme using different time points and conditions have demonstrated a vasodilatory response in the rat kidney. For example, chronic daily administration of heme for 4 days to rats increases RBF and upregulates renal HO-1, the latter representing a system with pronounced vasodilatory effects (6). The acute intravenous administration of heme in anesthetized rats leads to increased RBF, and, based on studies employing an inhibitor of cyclooxygenase and measurements of urinary 6-keto-PGF1α, this study concluded that heme-induced renal vasodilation results from induction of cyclooxygenase activity in the kidney (30). In these studies it should be noted that, in addition to differences in the dose of heme and the species studied, RBF was assessed beyond the 30 min time frame employed in our studies; additionally, the vasodilating effect of heme necessitated a time delay for the needed induction of cyclooxygenase activity. Integrating these studies, the renal hemodynamic effects of heme are complex and time dependent: first, an immediate TLR4-dependent vasoconstriction; second, a cyclooxygenase-dependent vasodilation (30); and third, a HO-dependent vasodilation (6). Interestingly, when heme is intermittently and chronically administered over weeks, RBF diminishes because of heme-induced renal injury (34).
TLR4 was also involved in mediating heme-elicited inflammatory responses in renal epithelial cells in vitro. TAK-242 substantially reduced heme-induced MCP-1 mRNA expression and production of MCP-1 protein; TAK-242 also markedly attenuated the activation of NF-κB, the transcription factor that promotes MCP-1 mRNA expression. These findings indicate that, at least in vitro, the inflammatory effects of heme, as reflected by MCP-1 induction, are channeled through the TLR4 receptor. Interestingly, the same is not true for the anti-inflammatory effects of heme as reflected by induction of HO-1: namely, while heme strongly activated Nrf2 and induced HO-1 mRNA and protein expression, none of these heme-induced effects were reduced by TAK-242. Extrapolating from these data, at least in vitro, the injurious effects of heme, as reflected by induction of NF-κB/MCP-1, are mediated through TLR4, whereas the countervailing potentially cytoprotective effects of heme, as reflected by activation of Nrf2 and induction of HO-1, are not. TLR4-dependent signaling thus accounts for some but not all of the heme-elicited cellular effects in vitro. Interestingly, heme differs from endotoxin in the way the TLR4 receptor is engaged (27), and these observations may be germane to the current finding that the proinflammatory effects, but not the anti-inflammatory effects, are linked to heme-induced TLR4-dependent cell signaling.
Studies were undertaken to determine whether these in vitro findings are also evinced in vivo. We demonstrate that heme robustly induces MCP-1 mRNA expression in the kidney in vivo and prior treatment with TAK-242 entirely failed to blunt such MCP-1 induction. We suggest that these findings in vivo, quite discordant from those obtained in vitro, may be explained in the following way. When heme is administered in vivo it is increasingly bound by hemopexin in plasma. How much of the heme-hemopexin complex transgresses the glomerular filtration barrier and escapes into the urinary space is uncertain because the molecular mass of hemopexin (80–85 kDa) predicts little passage of the heme-hemopexin complex through the glomerular filtration barrier. However, it is quite possible that the heme-hemopexin complex may injure the podocyte and the glomerular filtration barrier such that glomerular size permselectivity is impaired, with the attendant appearance of heme-hemopexin in the urinary space. Any heme-hemopexin appearing in the urinary space would be readily incorporated in proximal tubular epithelial cells via the apical megalin-cubilin receptors. These receptors have very high affinity for albumin and numerous other proteins and serve as a reclamation pathway for proteins leaked into the urinary pathway. Heme administered in vivo, as it becomes bound to hemopexin (and thus not free) and to the extent it appears in the urinary space, would enter tubular epithelial cells via the apical megalin/cubilin receptors, thereby sidestepping the neighboring TLR4 receptor on proximal renal tubular epithelial cells. Blocking the TLR4 receptor in vivo with TAK-242 would thus not interrupt the intracellular incorporation of heme in proximal tubular epithelial cells and thus would not reduce heme-driven induction of MCP-1.
The administration of TAK-242 did not reduce renal dysfunction or renal histological injury in the glycerol-induced, heme protein-mediated model of AKI; these findings were corroborated by the failure of TLR4−/− mice, as compared with TLR4+/+ mice to exhibit resistance to AKI so induced. The present findings, the first to address the role of TLR4 in this AKI model, are in stark contrast to findings in virtually all other models of AKI studied to-date, which demonstrate an involvement of TLR4-signaling in causing AKI. Based on our “negative” findings in conjunction with findings in the literature regarding the pathogenesis of the glycerol model of heme protein-induced AKI, the involvement of heme (either bound or free) in the glycerol model includes the following (3, 35, 42): 1) the scavenging of nitric oxide by extracellular heme bound in intact hemoglobin/myoglobin present in plasma; 2) the redox cycling supported by heme bound in intact extracellular heme proteins with the attendant generation of superoxide anion (and scavenging of nitric oxide in the vasculature) and oxidant-induced tubular injury; and 3) the toxicity of increased intracellular levels of “free” heme in tubular epithelial cells accrued either from the “Trojan horse” entry of heme proteins via the megalin/cubilin receptors on tubular epithelial cells or from destabilized intracellular cytochrome P450 heme proteins. Additionally, intact heme proteins in the urinary space readily interact with Tamm-Horsfall protein to form obstructing urinary casts. Intact heme protein requires the proper liganding of the heme and protein moieties, and to this extent and in this context, extracellular bound heme, as part of heme proteins, contributes to AKI in this model, thereby providing a fourth mechanism whereby heme is involved.
In contrast, renal vasoconstriction and attendant ischemia may be mediated, at least in part, via a TLR4 receptor-mediated pathway in pathophysiological states in which free plasma heme is elevated. These states include 1) sickle cell disease and other hemoglobinopathies in which unstable mutant hemoglobins release large amounts of heme into plasma (2, 3, 35); 2) thrombotic microangiopathies attended by hemolysis (3, 17); 3) sepsis, a syndrome now recognized as exhibiting elevated plasma heme levels (3, 17); 4) malaria and other infectious diseases (10); and 5) AKI caused by the use of inordinate amounts of heme used to abort episodes of acute intermittent porphyria (9).
In summary, we demonstrate that while the TLR4 receptor mediates the acute renal vasoconstrictive effects of heme in vivo and the proinflammatory effects on renal epithelial cells in vitro, the TLR4 receptor does not mediate heme-induced anti-inflammatory responses in vitro or heme-induced proinflammatory effects in vivo. Notably, TLR4-dependent signaling does not contribute to renal functional and structural derangement in the glycerol model of heme protein-induced AKI. As heme engages the TLR4 receptor in vitro and heme is assigned a significant position in the pathogenesis of the glycerol-induced AKI, there is a certain irony in that this is the one model of AKI studied to date wherein blocking TLR4-dependent signaling fails to attenuate AKI. Finally, the present observations provide a notable example wherein findings in renal epithelial cells in vitro do not predict what occurs in vivo.
GRANTS
This work was supported by National Institutes of Health Grants DK-47060 (to K. A. Nath, Z. S. Katusic, and J. P. Grande), AI-00911 (to J. P. Grande and K. A. Nath), and HL-114567 (to J. D. Belcher and G. M. Vercellotti).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.A.N., J.D.B., M.C.N., J.P.G., A.J.C., Z.S.K., and G.M.V. conceived and designed research; K.A.N., J.D.B., M.C.N., J.P.G., A.J.C., A.W.A., and G.M.V. analyzed data; K.A.N., J.D.B., M.C.N., J.P.G., A.J.C., A.W.A., Z.S.K., and G.M.V. interpreted results of experiments; K.A.N., M.C.N., A.J.C., and A.W.A. prepared figures; K.A.N. and A.J.C. drafted manuscript; K.A.N., J.D.B., M.C.N., J.P.G., A.J.C., A.W.A., Z.S.K., and G.M.V. edited and revised manuscript; K.A.N., J.D.B., M.C.N., J.P.G., A.J.C., A.W.A., Z.S.K., and G.M.V. approved final version of manuscript; M.C.N., A.J.C., and A.W.A. performed experiments.
ACKNOWLEDGMENTS
We thank Qurratulaine Jadran for secretarial expertise in the preparation of this manuscript.
REFERENCES
- 1.Anderberg SB, Luther T, Frithiof R. Physiological aspects of Toll-like receptor 4 activation in sepsis-induced acute kidney injury. Acta Physiol (Oxf) 219: 573–588, 2017. doi: 10.1111/apha.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123: 377–390, 2014. doi: 10.1182/blood-2013-04-495887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belcher JD, Nath KA, Vercellotti GM. Vasculotoxic and proinflammatory effects of plasma heme: Cell signaling and cytoprotective responses. ISRN Oxidative Med 2013: 831596, 2013. doi: 10.1155/2013/831596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolisetty S, Traylor A, Zarjou A, Johnson MS, Benavides GA, Ricart K, Boddu R, Moore RD, Landar A, Barnes S, Darley-Usmar V, Agarwal A. Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells. Am J Physiol Renal Physiol 305: F255–F264, 2013. doi: 10.1152/ajprenal.00160.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolisetty S, Zarjou A, Agarwal A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am J Kidney Dis 69: 531–545, 2017. doi: 10.1053/j.ajkd.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botros FT, Dobrowolski L, Navar LG. Renal heme oxygenase-1 induction with hemin augments renal hemodynamics, renal autoregulation, and excretory function. Int J Hypertens 2012: 189512, 2012. doi: 10.1155/2012/189512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, John R, Richardson JA, Shelton JM, Zhou XJ, Wang Y, Wu QQ, Hartono JR, Winterberg PD, Lu CY. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int 79: 288–299, 2011. doi: 10.1038/ki.2010.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Mahata M, Rao F, Khandrika S, Courel M, Fung MM, Zhang K, Stridsberg M, Ziegler MG, Hamilton BA, Lipkowitz MS, Taupenot L, Nievergelt C, Mahata SK, O’Connor DT. Chromogranin A regulates renal function by triggering Weibel-Palade body exocytosis. J Am Soc Nephrol 20: 1623–1632, 2009. doi: 10.1681/ASN.2008111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhar GJ, Bossenmaier I, Cardinal R, Petryka ZJ, Watson CJ. Transitory renal failure following rapid administration of a relatively large amount of hematin in a patient with acute intermittent porphyria in clinical remission. Acta Med Scand 203: 437–443, 1978. doi: 10.1111/j.0954-6820.1978.tb14903.x. [DOI] [PubMed] [Google Scholar]
- 10.Elphinstone RE, Conroy AL, Hawkes M, Hermann L, Namasopo S, Warren HS, John CC, Liles WC, Kain KC. Alterations in systemic extracellular heme and hemopexin are associated with adverse clinical outcomes in Ugandan children with severe malaria. J Infect Dis 214: 1268–1275, 2016. doi: 10.1093/infdis/jiw357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenhammar J, Rundgren M, Forestier J, Kalman S, Eriksson S, Frithiof R. Toll-like receptor 4 inhibitor TAK-242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiology 114: 1130–1137, 2011. doi: 10.1097/ALN.0b013e31820b8b44. [DOI] [PubMed] [Google Scholar]
- 12.Fenhammar J, Rundgren M, Hultenby K, Forestier J, Taavo M, Kenne E, Weitzberg E, Eriksson S, Ozenci V, Wernerson A, Frithiof R. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit Care 18: 488, 2014. doi: 10.1186/s13054-014-0488-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graça-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282: 20221–20229, 2007. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 14.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119: 2368–2375, 2012. doi: 10.1182/blood-2011-08-375303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, Ofori-Acquah SF. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 123: 4809–4820, 2013. doi: 10.1172/JCI64578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hua F, Tang H, Wang J, Prunty MC, Hua X, Sayeed I, Stein DG. TAK-242, an antagonist for Toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab 35: 536–542, 2015. doi: 10.1038/jcbfm.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Immenschuh S, Vijayan V, Janciauskiene S, Gueler F. Heme as a target for therapeutic interventions. Front Pharmacol 8: 146, 2017. doi: 10.3389/fphar.2017.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation 9: 46, 2012. doi: 10.1186/1742-2094-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munshi R, Johnson A, Siew ED, Ikizler TA, Ware LB, Wurfel MM, Himmelfarb J, Zager RA. MCP-1 gene activation marks acute kidney injury. J Am Soc Nephrol 22: 165–175, 2011. doi: 10.1681/ASN.2010060641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murali NS, Ackerman AW, Croatt AJ, Cheng J, Grande JP, Sutor SL, Bram RJ, Bren GD, Badley AD, Alam J, Nath KA. Renal upregulation of HO-1 reduces albumin-driven MCP-1 production: implications for chronic kidney disease. Am J Physiol Renal Physiol 292: F837–F844, 2007. doi: 10.1152/ajprenal.00254.2006. [DOI] [PubMed] [Google Scholar]
- 21.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 22.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270, 1992. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nath KA, Grande JP, Croatt AJ, Likely S, Hebbel RP, Enright H. Intracellular targets in heme protein-induced renal injury. Kidney Int 53: 100–111, 1998. doi: 10.1046/j.1523-1755.1998.00731.x. [DOI] [PubMed] [Google Scholar]
- 24.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol 156: 1527–1535, 2000. doi: 10.1016/S0002-9440(10)65024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, Haggard JJ, Croatt AJ, Payne WD, Alam J. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int 59: 106–117, 2001. doi: 10.1046/j.1523-1755.2001.00471.x. [DOI] [PubMed] [Google Scholar]
- 26.Paller MS, Jacob HS. Cytochrome P-450 mediates tissue-damaging hydroxyl radical formation during reoxygenation of the kidney. Proc Natl Acad Sci USA 91: 7002–7006, 1994. doi: 10.1073/pnas.91.15.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piazza M, Damore G, Costa B, Gioannini TL, Weiss JP, Peri F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun 17: 293–301, 2011. doi: 10.1177/1753425910369020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int 68: 611–622, 2005. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- 29.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med 38: 1685–1694, 2010. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez F, Kemp R, Balazy M, Nasjletti A. Effects of exogenous heme on renal function: role of heme oxygenase and cyclooxygenase. Hypertension 42: 680–684, 2003. doi: 10.1161/01.HYP.0000085785.40581.1A. [DOI] [PubMed] [Google Scholar]
- 31.Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol 26: 1002–1007, 2006. doi: 10.1161/01.ATV.0000209501.56852.6c. [DOI] [PubMed] [Google Scholar]
- 32.Sen U, Tyagi N, Patibandla PK, Dean WL, Tyagi SC, Roberts AM, Lominadze D. Fibrinogen-induced endothelin-1 production from endothelial cells. Am J Physiol Cell Physiol 296: C840–C847, 2009. doi: 10.1152/ajpcell.00515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol 571: 231–239, 2007. doi: 10.1016/j.ejphar.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 34.Tabibzadeh N, Estournet C, Placier S, Perez J, Bilbault H, Girshovich A, Vandermeersch S, Jouanneau C, Letavernier E, Hammoudi N, Lionnet F, Haymann JP. Plasma heme-induced renal toxicity is related to a capillary rarefaction. Sci Rep 7: 40156, 2017. doi: 10.1038/srep40156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 36.Tracz MJ, Juncos JP, Grande JP, Croatt AJ, Ackerman AW, Katusic ZS, Nath KA. Induction of heme oxygenase-1 is a beneficial response in a murine model of venous thrombosis. Am J Pathol 173: 1882–1890, 2008. doi: 10.2353/ajpath.2008.080556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tracz MJ, Juncos JP, Grande JP, Croatt AJ, Ackerman AW, Rajagopalan G, Knutson KL, Badley AD, Griffin MD, Alam J, Nath KA. Renal hemodynamic, inflammatory, and apoptotic responses to lipopolysaccharide in HO-1−/− mice. Am J Pathol 170: 1820–1830, 2007. doi: 10.2353/ajpath.2007.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, Carneiro LA, Bozza MT, Travassos LH. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci USA 113: E7474–E7482, 2016. doi: 10.1073/pnas.1608928113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Shah SV, Liu H, Baliga R. Inhibition of cytochrome P450 2E1 and activation of transcription factor Nrf2 are renoprotective in myoglobinuric acute kidney injury. Kidney Int 86: 338–349, 2014. doi: 10.1038/ki.2014.65. [DOI] [PubMed] [Google Scholar]
- 40.Wittebole X, Castanares-Zapatero D, Laterre PF. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflamm 2010: 568396, 2010. doi: 10.1155/2010/568396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859, 2007. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int 49: 314–326, 1996. doi: 10.1038/ki.1996.48. [DOI] [PubMed] [Google Scholar]
- 43.Zager RA, Johnson AC. Progressive histone alterations and proinflammatory gene activation: consequences of heme protein/iron-mediated proximal tubule injury. Am J Physiol Renal Physiol 298: F827–F837, 2010. doi: 10.1152/ajprenal.00683.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zager RA, Johnson AC, Becker K. Renal cortical hemopexin accumulation in response to acute kidney injury. Am J Physiol Renal Physiol 303: F1460–F1472, 2012. doi: 10.1152/ajprenal.00426.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zager RA, Johnson AC, Lund S, Hanson S. Acute renal failure: determinants and characteristics of the injury-induced hyperinflammatory response. Am J Physiol Renal Physiol 291: F546–F556, 2006. doi: 10.1152/ajprenal.00072.2006. [DOI] [PubMed] [Google Scholar]
- 46.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 19: 923–932, 2008. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Peng W, Ao X, Dai H, Yuan L, Huang X, Zhou Q. TAK-242, a Toll-like receptor 4 antagonist, protects against aldosterone-induced cardiac and renal injury. PLoS One 10: e0142456, 2015. doi: 10.1371/journal.pone.0142456. [DOI] [PMC free article] [PubMed] [Google Scholar]